Exploring HIV-1 Transmission Dynamics by Combining Phylogenetic Analysis and Infection Timing

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Infection Timing
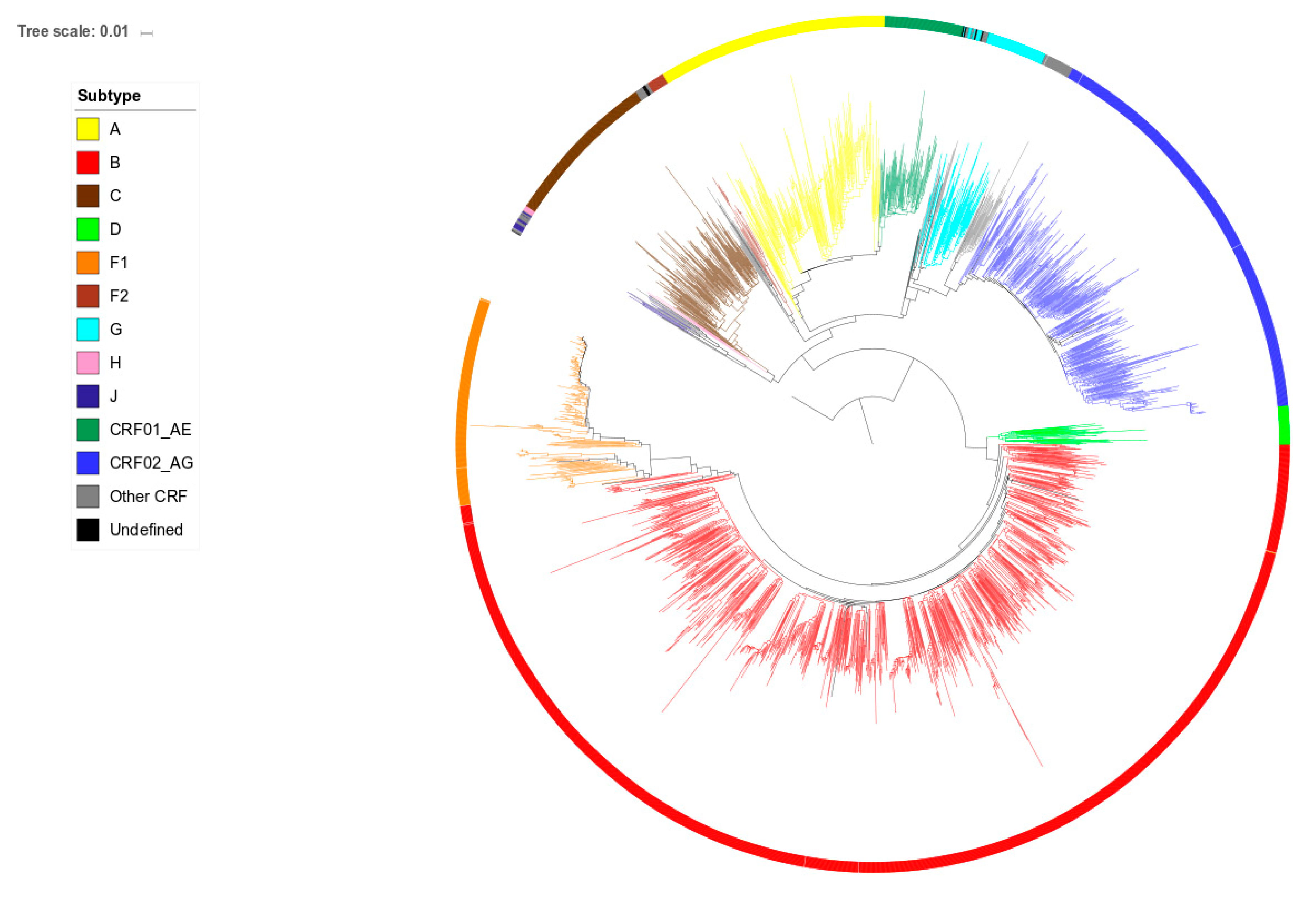
2.2. Phylogenetic Analysis
2.3. Statistical Analysis
2.4. Ethics
3. Results
3.1. Patient Characteristics
3.2. Origin and Infection Stage at Diagnosis
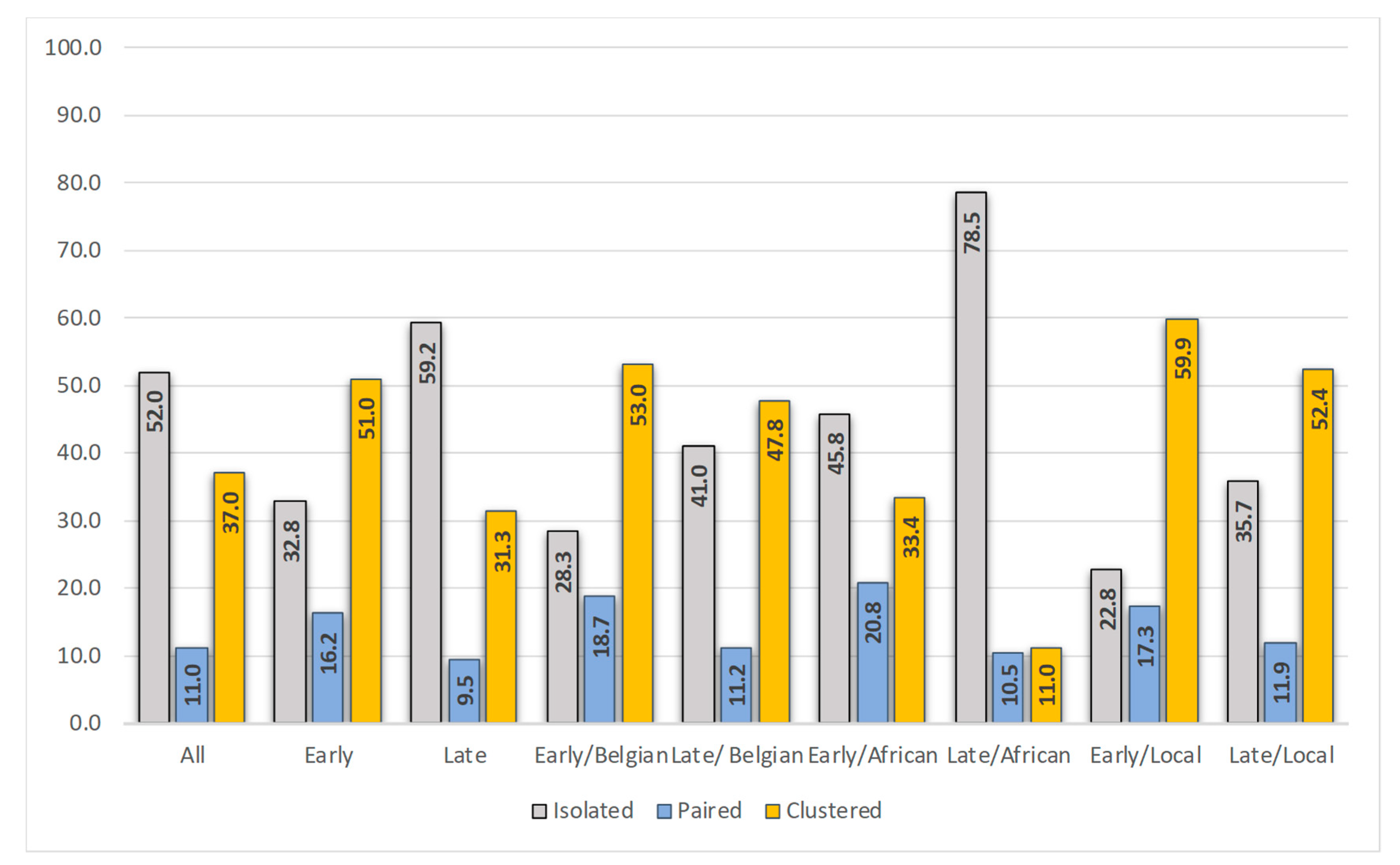
3.3. Position in the Phylogenetic Tree and Time of Diagnosis
3.4. Characteristics of the Patients Infected in Belgium
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Beckhoven, D.; Florence, E.; Ruelle, J.; Deblonde, J.; Verhofstede, C.; Callens, S.; Vancutsem, E.; Lacor, P.; Demeester, R.; Goffard, J.C.; et al. Good continuum of HIV care in Belgium despite weaknesses in retention and linkage to care among migrants. BMC Infect. Dis. 2015, 15, 496. [Google Scholar] [CrossRef] [PubMed]
- Sasse, A.; Deblonde, J.; Van Beckhoven, D. Epidemiology of AIDS and HIV in Belgium: Situation on 31 December 2017. Sciensano. Available online: http://doi.org/10.25608/NBWP-GJ70 (accessed on 25 August 2019).
- European Centre for Disease Prevention and Control, World Health Organization (WHO) Regional Office for Europe. HIV/AIDS Surveillance in Europe 2018-2017 Data. Available online: https://ecdc.europa.eu/en/publications-data/presentation-hivaids-surveillance-europe-2018-2017-data (accessed on 25 August 2019).
- Dennis, A.M.; Hue, S.; Hurt, C.B.; Napravnik, S.; Sebastian, J.; Pillay, D.; Eron, J.J. Phylogenetic insights into regional HIV transmission. AIDS 2012, 26, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Dennis, A.M.; Herbeck, J.T.; Brown, A.L.; Kellam, P.; de Oliveira, T.; Pillay, D.; Fraser, C.; Cohen, A.S. Phylogenetic Studies of Transmission Dynamics in Generalized HIV Epidemics: An Essential Tool Where the Burden is Greatest? J. Acq. Immun. Def. Synd. 2014, 67, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Lubelchek, R.J.; Hoehnen, S.C.; Hotton, A.L.; Kincaid, S.L.; Barker, D.E.; French, A.L. Transmission Clustering Among Newly Diagnosed HIV Patients in Chicago, 2008 to 2011: Using Phylogenetics to Expand Knowledge of Regional HIV Transmission Patterns. J. Acq. Immun. Def. Synd. 2015, 68, 46–54. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oster, A.M.; Wertheim, J.O.; Hernandez, A.L.; Ocfemia, M.C.B.; Saduvala, N.; Hall, H.I. Using Molecular HIV Surveillance Data to Understand Transmission Between Subpopulations in the United States. J. Acq. Immun. Def. Synd. 2015, 70, 444–451. [Google Scholar] [CrossRef]
- Fisher, M.; Pao, D.; Brown, A.E.; Sudarshi, D.; Gill, O.N.; Cane, P.; Buckton, A.J.; Parry, J.V.; Johnson, A.M.; Sabin, C.; et al. Determinants of HIV-1 transmission in men who have sex with men: A combined clinical, epidemiological and phylogenetic approach. AIDS 2010, 24, 1739–1747. [Google Scholar] [CrossRef]
- De Oliveira, T.; Kharsany, A.B.M.; Graf, T.; Cawood, C.; Khanyile, D.; Grobler, A.; Puren, A.; Madurai, S.; Baxter, C.; Karim, Q.A.; et al. Transmission networks and risk of HIV infection in KwaZulu-Natal, South Africa: A community-wide phylogenetic study. Lancet HIV 2017, 4, E41–E50. [Google Scholar] [CrossRef]
- Paraskevis, D.; Nikolopoulos, G.K.; Sypsa, V.; Psichogiou, M.; Pantavou, K.; Kostaki, E.; Karamitros, T.; Paraskeva, D.; Schneider, J.; Malliori, M.; et al. Molecular investigation of HIV-1 cross-group transmissions during an outbreak among people who inject drugs (2011-2014) in Athens, Greece. Infect. Genet. Evol. 2018, 62, 11–16. [Google Scholar] [CrossRef]
- Morgan, E.; Nyaku, A.N.; D’Aquila, R.T.; Schneider, J.A. Determinants of HIV Phylogenetic Clustering in Chicago Among Young Black Men Who Have Sex with Men From the uConnect Cohort. J. Acq. Immun. Def. Synd. 2017, 75, 265–270. [Google Scholar] [CrossRef]
- Brenner, B.G.; Ibanescu, R.I.; Hardy, I.; Roger, M. Genotypic and Phylogenetic Insights on Prevention of the Spread of HIV-1 and Drug Resistance in “Real-World” Settings. Viruses 2018, 10, 10. [Google Scholar] [CrossRef]
- Verhofstede, C.; Dauwe, K.; Fransen, K.; Van Laethem, K.; Van den Wijngaert, S.; Ruelle, J.; Delforge, M.L.; Vancutsem, E.; Vaira, D.; Stoffels, K.; et al. Phylogenetic analysis of the Belgian HIV-1 epidemic reveals that local transmission is almost exclusively driven by men having sex with men despite presence of large African migrant communities. Infect. Genet. Evol. 2018, 61, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Pilcher, C.D.; Tien, H.C.; Eron, J.J.; Vernazza, P.L.; Leu, S.Y.; Stewart, P.W.; Goh, L.E.; Cohen, M.S.; Quest, S.; Duke, U.N. Brief but efficient: Acute HIV infection and the sexual transmission of HIV. J. Infect. Dis. 2004, 189, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.C.; Wawer, M.J.; Sewankambo, N.; Serwadda, D.; Li, C.J.; Wabwire-Mangen, F.; Meehan, M.O.; Lutalo, T.; Gray, R.H. Viral load and heterosexual transmission of human immunodeficiency virus type 1. N. Engl. J. Med. 2000, 342, 921–929. [Google Scholar] [CrossRef]
- Powers, K.A.; Ghani, A.C.; Miller, W.C.; Hoffman, I.F.; Pettifor, A.E.; Kamanga, G.; Martinson, F.E.A.; Cohen, M.S. The role of acute and early HIV infection in the spread of HIV and implications for transmission prevention strategies in Lilongwe, Malawi: A modelling study. Lancet 2011, 378, 256–268. [Google Scholar] [CrossRef]
- Yerly, S.; Vora, S.; Rizzardi, P.; Chave, J.P.; Vernazza, P.L.; Flepp, M.; Telenti, A.; Battegay, M.; Veuthey, A.L.; Bru, J.P.; et al. Acute HIV infection: Impact on the spread of HIV and transmission of drug resistance. AIDS 2001, 15, 2287–2292. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.G.; Roger, M.; Routy, J.P.; Moisi, D.; Ntemgwa, M.; Matte, C.; Baril, J.G.; Thomas, R.; Rouleau, D.; Bruneau, J.; et al. High rates of forward transmission events after acute/early HIV-1 infection. J. Infect. Dis. 2007, 195, 951–959. [Google Scholar] [CrossRef]
- Pineda-Pena, A.C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gomez-Lopez, A.; Camacho, R.J.; de Oliveira, T.; Vandamme, A.M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: Performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef]
- Struck, D.; Lawyer, G.; Ternes, A.M.; Schmit, J.C.; Bercoff, D.P. COMET: Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef]
- Verhofstede, C.; Fransen, K.; Van Den Heuvel, A.; Van Laethem, K.; Ruelle, J.; Vancutsem, E.; Stoffels, K.; Van den Wijngaert, S.; Delforge, M.L.; Vaira, D.; et al. Decision tree for accurate infection timing in individuals newly diagnosed with HIV-1 infection. BMC Infect. Dis. 2017, 17, 738. [Google Scholar] [CrossRef]
- Hall, T.A. Bioedit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/INT. Nucl. Acids. Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Schlusser, K.E.; Pilcher, C.; Kallas, E.G.; Santos, B.R.; Deeks, S.G.; Facente, S.; Keating, S.M.; Busch, M.P.; Murphy, G.; Welte, A.; et al. Comparison of cross-sectional HIV incidence assay results from dried blood spots and plasma. PLoS ONE 2017, 12, e0172283. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, T.; Kennedy, S.; Pau, C.P.; McDougal, J.S.; Parekh, B.S. Performance characteristics of the immunoglobulin G-capture BED-enzyme immunoassay, an assay to detect recent human immunodeficiency virus type 1 seroconversion. J. Clin. Microbiol. 2004, 42, 2623–2628. [Google Scholar] [CrossRef]
- Hanson, D.L.; Song, R.G.; Masciotra, S.; Hernandez, A.; Dobbs, T.L.; Parekh, B.S.; Owen, S.M.; Green, T.A. Mean Recency Period for Estimation of HIV-1 Incidence with the BED-Capture EIA and Bio-Rad Avidity in Persons Diagnosed in the United States with Subtype B. PLoS ONE 2016, 11, e0152327. [Google Scholar] [CrossRef]
- Chalmet, K.; Staelens, D.; Blot, S.; Dinakis, S.; Pelgrom, J.; Plum, J.; Vogelaers, D.; Vandekerckhove, L.; Verhofstede, C. Epidemiological study of phylogenetic transmission clusters in a local HIV-1 epidemic reveals distinct differences between subtype B and non-B infections. BMC Infect. Dis. 2010, 10, 262. [Google Scholar] [CrossRef] [PubMed]
- Dauwe, K.; Mortier, V.; Schauvliege, M.; Van den Heuvel, A.; Fransen, K.; Servais, J.Y.; Bercoff, D.P.; Seguin-Devaux, C.; Verhofstede, C. Characteristics and spread to the native population of HIV-1 non-B subtypes in two European countries with high migration rate. BMC Infect. Dis. 2015, 15, 524. [Google Scholar] [CrossRef]
- Bezemer, D.; Cori, A.; Ratmann, O.; van Sighem, A.; Hermanides, H.S.; Dutilh, B.E.; Gras, L.; Faria, N.R.; van den Hengel, R.; Duits, A.J.; et al. Dispersion of the HIV-1 Epidemic in Men Who Have Sex with Men in the Netherlands: A Combined Mathematical Model and Phylogenetic Analysis. PLoS Med. 2015, 12, e1001898. [Google Scholar] [CrossRef]
- Grande, K.M.; Schumann, C.L.; Ocfemia MC, B.; Vergeront, J.M.; Wertheim, J.O.; Oster, A.M. Transmission Patterns in a Low HIV-Morbidity State-Wisconsin, 2014–2017. MMWR-Morb. Mortal. Wkly. Rep. 2019, 68, 149–152. [Google Scholar] [CrossRef]
- Whiteside, Y.O.; Song, R.G.; Wertheim, J.O.; Oster, A.M. Molecular analysis allows inference into HIV transmission among young men who have sex with men in the United States. AIDS 2015, 29, 2517–2522. [Google Scholar] [CrossRef]
- Robineau, O.; Frange, P.; Barin, F.; Cazein, F.; Girard, P.M.; Chaix, M.L.; Kreplak, G.; Boelle, P.Y.; Morand-Joubert, L. Combining the Estimated Date of HIV Infection with a Phylogenetic Cluster Study to Better Understand HIV Spread: Application in a Paris Neighbourhood. PLoS ONE 2015, 10, e0135367. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.G.; Roger, M.; Stephens, D.; Moisi, D.; Hardy, I.; Weinberg, J.; Turgel, R.; Charest, H.; Koopman, J.; Wainberg, M. Transmission Clustering Drives the Onward Spread of the HIV Epidemic Among Men Who Have Sex with Men in Quebec. J. Infect. Dis. 2011, 204, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.A.; Hogan, J.W.; Huang, A.; DeLong, A.; Salemi, M.; Mayer, K.H.; Kantor, R. Phylogenetic Investigation of a Statewide HIV-1 Epidemic Reveals Ongoing and Active Transmission Networks Among Men Who Have Sex with Men. J. Acq. Immun. Def. Synd. 2015, 70, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Ibanescu, R.I.; Roger, M.; Oliveira, M.; Hardy, I.; Wainberg, M. Phylogenetic, epidemiological and virological insights on the rise of large cluster outbreaks fuelling the HIV-1 epidemic among men having sex with men within Quebec. J. Int. Aids Soc. 2017, 20, 98–99. [Google Scholar]
- Patino-Galindo, J.A.; Torres-Puente, M.; Bracho, M.A.; Alastrue, I.; Juan, A.; Navarro, D.; Galindo, M.J.; Ocete, D.; Ortega, E.; Gimeno, C.; et al. The molecular epidemiology of HIV-1 in the Comunidad Valenciana (Spain): Analysis of transmission clusters. Sci. Rep. 2017, 7, 11584. [Google Scholar] [CrossRef]
- Ragonnet-Cronin, M.L.; Shilaih, M.; Günthard, H.F.; Hodcroft, E.B.; Böni, J.; Fearnhill, E.; Dunn, D.; Yerly, S.; Klimkait, T.; Aubert, V.; et al. A direct comparison of two densely sampled HIV epidemics: The UK and Switzerland. Sci. Rep. 2016, 6, 32251. [Google Scholar] [CrossRef]
- Voltz, E.M.; Koopman, J.S.; Ward, M.J.; Brown, A.L.; Frost, S.D.W. Simple epidemiological dynamics explain phylogenetic clustering of HIV from patients with recent infection. PLos Comput. Biol. 2012, 8, e1002552. [Google Scholar]
- Volz, E.M.; Ionides, E.; Romero-Severson, E.O.; Brandt, M.G.; Mokotoff, E.; Koopman, J.S. HIV-1 Transmission during Early Infection in Men Who Have Sex with Men: A Phylodynamic Analysis. PLoS Med. 2013, 10, e1001568. [Google Scholar] [CrossRef]
- Ratmann, O.; van Sighem, A.; Bezemer, D.; Gavryushkina, A.; Jurriaans, S.; Wensing, A.; de Wolf, F.; Reiss, P.; Fraser, C. Sources of HIV infection among men having sex with men and implications for prevention. Sci. Transl. Med. 2016, 8, 320ra2. [Google Scholar] [CrossRef]
- Audelin, A.M.; Cowan, S.A.; Obel, N.; Nielsen, C.; Jorgensen, L.B.; Gerstoft, J. Phylogenetics of the Danish HIV Epidemic: The Role of Very Late Presenters in Sustaining the Epidemic. J. Acq. Immun. Def. Synd. 2013, 62, 102–108. [Google Scholar] [CrossRef]
- Montaner, J.S.G.; Lima, V.D.; Barrios, R.; Yip, B.; Wood, E.; Kerr, T.; Shannon, K.; Harrigan, P.R.; Hogg, R.S.; Daly, P.; et al. Association of highly active antiretroviral therapy coverage, population viral load, and yearly new HIV diagnoses in British Columbia, Canada: A population-based study. Lancet 2010, 376, 532–539. [Google Scholar] [CrossRef]
- Brown, A.E.; Gifford, R.J.; Clewley, J.P.; Kucherer, C.; Masquelier, B.; Porter, K.; Balotta, C.; Back NK, T.; Jorgensen, L.B.; de Mendoza, C.; et al. Phylogenetic Reconstruction of Transmission Events from Individuals with Acute HIV Infection: Toward More-Rigorous Epidemiological Definitions. J. Infect. Dis. 2009, 199, 427–431. [Google Scholar] [CrossRef]
- Coburn, B.J.; Blower, S. A Major HIV Risk Factor for Young Men Who Have Sex With Men Is Sex with Older Partners. J. Acq. Immun. Def. Syndr. 2010, 54, 113–114. [Google Scholar] [CrossRef]
- Hurt, C.B.; Matthews, D.D.; Calabria, M.S.; Green, K.A.; Adimora, A.A.; Golin, C.E.; Hightow-Weidman, L.B. Sex With Older Partners Is Associated With Primary HIV Infection Among Men Who Have Sex with Men in North Carolina. J. Acq. Immun. Def. Syndr. 2010, 54, 185–190. [Google Scholar] [CrossRef]
- Nikolopoulos, G.K.; Pavlitina, E.; Muth, S.Q.; Schneider, J.; Psichogiou, M.; Williams, L.D.; Paraskevis, D.; Sypsa, V.; Magiorkinis, G.; Smyrnov, P.; et al. A network intervention that locates and intervenes with recently HIV-infected persons: The Transmission Reduction Intervention Project (TRIP). Sci. Rep. 2016, 6, 38100. [Google Scholar] [CrossRef]
- Vasylyeva, T.I.; Friedman, S.R.; Paraskevis, D.; Magiorkinis, G. Integrating molecular epidemiology and social network analysis to study infectious diseases: Towards a socio-molecular era for public health. Infect. Genet. Evol. 2016, 46, 248–255. [Google Scholar] [CrossRef]
- Kostaki, E.G.; Frampton, D.; Paraskevis, D.; Pantavou, K.; Ferns, B.; Raffle, J.; Grants, P.; Kozlakidis, Z.; Hadjikou, A.; Pavlitina, E.; et al. Near Full-length Genomic Sequencing and Molecular Analysis of HIV-Infected Individuals in a Network-based Intervention (TRIP) in Athens, Greece: Evidence that Transmissions Occur More Frequently from those with High HIV-RNA. Curr. HIV Res. 2018, 16, 345–353. [Google Scholar] [CrossRef]
- Smyrnov, P.; Williams, L.D.; Korobchuk, A.; Sazonova, Y.; Nikolopoulos, G.K.; Skaathun, B.; Morgan, E.; Schneider, J.; Vasylyeva, T.I.; Friedman, S.R. Risk network approaches to locating undiagnosed HIV cases in Odessa, Ukraine. J. Int. Aids Soc. 2018, 21, e25040. [Google Scholar] [CrossRef]
- Schueler, K.; Ferreira, M.; Nikolopoulos, G.; Skaathun, B.; Paraskevis, D.; Hatzakis, A.; Friedman, S.R.; Schneider, J.A. Pre-exposure Prophylaxis (PrEP) Awareness and Use Within High HIV Transmission Networks. AIDS Behav. 2019, 23, 1893–1903. [Google Scholar] [CrossRef]
- Vuylsteke, B.; Reyniers, T.; Lucet, C.; Nostlinger, C.; Deblonde, J.; Libois, A.; Sauvage, A.S.; Deprez, E.; Goffard, J.C.; Allard, S.D.; et al. High uptake of pre-exposure prophylaxis (PrEP) during early roll-out in Belgium: Results from surveillance reports. Sex. Health 2019, 16, 80–83. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Univariate Analysis | Multivariate Analysis | ||||||
|---|---|---|---|---|---|---|---|
| n | OR | p-Value | 95% Confidence Interval | OR | p-Value | 95% Confidence Interval | |
| Age (per year increase) | |||||||
| 927 | 0.991 | 0.181 | (0.979–1.004) | ||||
| Year of diagnosis | |||||||
| 2014 | 437 | 1.000 | - | Ref | |||
| 2016 | 490 | 1.020 | 0.893 | (0.767–1.357) | |||
| Gender | NS | ||||||
| Male | 717 | 1.000 | - | Ref | |||
| Female | 210 | 0.373 | <0.001 | (0.248–0.561) | |||
| Position in tree | |||||||
| Isolated | 479 | 1.000 | - | Ref | 1.000 | - | Ref |
| Clustered | 106 | 2.939 | <0.001 | (2.139–4.038) | 2.076 | <0.001 | (1.475–2.921) |
| Paired | 342 | 3.075 | <0.001 | (1.957–4.833) | 2.65 | <0.001 | (1.648–4.263) |
| Transmission risk | |||||||
| MSM | 413 | 1.000 | - | Ref | 1.000 | - | Ref |
| Heterosexual | 306 | 0.340 | <0.001 | (0.240–0.483) | 0.486 | <0.001 | (0.334–0.707) |
| Other | 13 | 0.127 | 0.048 | (0.016–0.982) | 0.224 | 0.164 | (0.027–1.839) |
| Unknown | 195 | 0.442 | <0.001 | (0.300–0.653) | 0.702 | 0.116 | (0.451–1.091) |
| Origin | NS | ||||||
| Belgian | 358 | 1.000 | - | Ref | |||
| African | 196 | 0.233 | <0.001 | (0.145–0.376) | |||
| European | 98 | 0.703 | 0.153 | (0.433–1.140) | |||
| Other | 66 | 0.580 | 0.071 | (0.321–1.048) | |||
| Unknown | 209 | 0.689 | 0.047 | (0.477–0.995) | |||
| Location of infection | |||||||
| Belgium | 312 | 1.000 | - | Ref | 1.000 | - | Ref |
| Africa | 73 | 0.062 | <0.001 | (0.019–0.203) | 0.140 | 0.001 | (0.042–0.470) |
| Europe | 32 | 0.763 | 0.488 | (0.356–1.638) | 0.939 | 0.878 | (0.421–2.092) |
| Other | 32 | 0.662 | 0.301 | (0.303–1.145) | 1.241 | 0.613 | (0.538–2.865) |
| Unknown | 478 | 0.456 | <0.001 | (0.335–0.621) | 0.62 | 0.008 | (0.436–0.881) |
| Subtype | NS | ||||||
| B | 455 | 1.000 | - | Ref | |||
| 02_AG | 108 | 0.732 | 0.191 | (0.458–1.168) | |||
| F | 79 | 1.220 | 0.430 | (0.744–2.001) | |||
| A | 78 | 0.436 | 0.008 | (0.237–0.803) | |||
| C | 66 | 0.315 | 0.002 | (0.152–0.653) | |||
| 01_AE | 33 | 0.997 | 0.993 | (0.471–2.109) | |||
| Other/undefined | 108 | 0.453 | 0.003 | (0.269–0.764) | |||
| Univariate Analysis | Multivariate Analysis | ||||||
|---|---|---|---|---|---|---|---|
| Risk Factors | n | OR | p-Value | 95% Confidence Interval | OR | p-Value | 95% Confidence Interval |
| Age (per year increase) | |||||||
| 554 | 0.958 | <0.001 | (0.958–0.974) | 0.928 | <0.001 | (0.904–0.953) | |
| Year of diagnosis | |||||||
| 2014 | 276 | 1.000 | - | Ref | |||
| 2016 | 278 | 0.899 | 0.551 | (0.635–1.274) | |||
| Infection Stage | NS | ||||||
| Early | 158 | 1.000 | - | Ref | |||
| Late | 396 | 4.287 | <0.001 | (2.659–6.913) | |||
| Gender | |||||||
| Male | 418 | 1.000 | - | Ref | 1.000 | - | Ref |
| Female | 136 | 13.639 | <0.001 | (8.504–21.876) | 2.529 | 0.013 | (1.214–5.266) |
| Position in tree | NS | ||||||
| Isolated | 273 | 1.000 | - | Ref | |||
| Clustered | 73 | 0.153 | <0.001 | (0.097–0.241) | |||
| Paired | 208 | 0.418 | 0.002 | (0.242–0.723) | |||
| Transmission risk | |||||||
| MSM | 280 | 1.000 | - | Ref | 1.000 | - | Ref |
| Heterosexual | 234 | 11.339 | <0.001 | (7.221–17.806) | 3.414 | 0.001 | (1.710–6.816) |
| Other | 10 | 18.083 | <0.001 | (4.452–73.457) | 2.885 | 0.267 | (0.445–18.710) |
| Unknown | 30 | 11.625 | <0.001 | (5.130–26.341) | 6.794 | 0.001 | (2.124–21.728) |
| Location of infection | |||||||
| Belgium | 245 | 1.000 | - | Ref | 1.000 | - | Ref |
| Africa | 70 | 104.963 | <0.001 | (38.861–283.506) | 17.405 | <0.001 | (5.551–54.574) |
| Europe | 14 | 0.621 | 0.652 | (0.078–4.936) | 0.854 | 0.894 | (0.083–8.755) |
| Other | 14 | 0.621 | 0.652 | (0.078–4.936) | 3.833 | 0.281 | (0.333–44.090) |
| Not reported | 211 | 7.556 | <0.001 | (4.664–12.241) | 4.799 | <0.001 | (2.554–9.017) |
| Subtype | |||||||
| B | 259 | 1.000 | - | Ref | 1.000 | - | Ref |
| 02_AG | 80 | 21.311 | <0.001 | (11.303–40.181) | 8.875 | <0.001 | (3.963–19.873) |
| F | 51 | 1.909 | 0.144 | (0.802–4.546) | 1.196 | 0.729 | (0.433–3.301) |
| A | 36 | 26.678 | <0.001 | (11.452–62.149) | 8.423 | <0.001 | (2.878–24.648) |
| C | 45 | 41.043 | <0.001 | (17.601–95.707) | 22.628 | <0.001 | (7.247–70.653) |
| 01_AE | 13 | 0.855 | 0.883 | (0.106–6.875) | 0.564 | 0.629 | (0.055–5.745) |
| Other/undefined | 70 | 22.387 | <0.001 | (11.550–43.392) | 6.581 | <0.001 | (2.753–15.728) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verhofstede, C.; Mortier, V.; Dauwe, K.; Callens, S.; Deblonde, J.; Dessilly, G.; Delforge, M.-L.; Fransen, K.; Sasse, A.; Stoffels, K.; et al. Exploring HIV-1 Transmission Dynamics by Combining Phylogenetic Analysis and Infection Timing. Viruses 2019, 11, 1096. https://doi.org/10.3390/v11121096
Verhofstede C, Mortier V, Dauwe K, Callens S, Deblonde J, Dessilly G, Delforge M-L, Fransen K, Sasse A, Stoffels K, et al. Exploring HIV-1 Transmission Dynamics by Combining Phylogenetic Analysis and Infection Timing. Viruses. 2019; 11(12):1096. https://doi.org/10.3390/v11121096
Chicago/Turabian StyleVerhofstede, Chris, Virginie Mortier, Kenny Dauwe, Steven Callens, Jessika Deblonde, Géraldine Dessilly, Marie-Luce Delforge, Katrien Fransen, André Sasse, Karolien Stoffels, and et al. 2019. "Exploring HIV-1 Transmission Dynamics by Combining Phylogenetic Analysis and Infection Timing" Viruses 11, no. 12: 1096. https://doi.org/10.3390/v11121096
APA StyleVerhofstede, C., Mortier, V., Dauwe, K., Callens, S., Deblonde, J., Dessilly, G., Delforge, M.-L., Fransen, K., Sasse, A., Stoffels, K., Van Beckhoven, D., Vanroye, F., Vaira, D., Vancutsem, E., & Van Laethem, K. (2019). Exploring HIV-1 Transmission Dynamics by Combining Phylogenetic Analysis and Infection Timing. Viruses, 11(12), 1096. https://doi.org/10.3390/v11121096