1. Introduction
The molecular crosstalk between the intestinal epithelium and luminal bacteria is a pivotal event in bacterial infections of the gastrointestinal tract. Many
Enterobacteriaceae show adhering and invading properties that reflect their virulent phenotype in intestinal infections. Changes of the composition of the gut microbiota with high abundance of
Enterobacteriaceae can be seen both in chronic intestinal inflammation, e.g., intestinal bowel disease (IBD) and in colitis models [
1,
2]. Thus, this bacterial family and especially
Escherichia coli (
E. coli) is gaining high interest for current research approaches in human and veterinary medicine. Even species from the
Enterobacteriaceae family that used to be considered as commensal bacteria in the gut, e.g., the uropathogenic
E. coli (UPEC), were shown to have a potential pathogenic impact on the intestinal epithelium.
Focal leaks in the colonic epithelial barrier were described in vitro on HT-29/B6 cells induced by the β-barrel pore-forming toxin (PFT) α-hemolysin (HlyA) of UPEC [
3]. Further studies could depict increased transepithelial permeability for macromolecules and intestinal inflammation due to the HlyA
+ E. coli 536 infection in an in vivo mouse model [
4]. Moreover, HlyA-producing bacteria were shown to be present in a higher abundance in active ulcerative colitis within the human colon mucosa [
4]. These results are in line with different studies, which associate intestinal colonization by
E. coli of the phylogenetic group B2 and D with the pathogenesis of ulcerative colitis [
5]. Whereas low numbers of HlyA-producing bacteria can also be found in healthy controls, data on the abundance of UPEC in intestinal diseases other than IBD are sparse. The regulation of epithelial barrier function during the infection with HlyA
+ E.coli by changes in tight junction (TJ) protein distribution was shown recently in porcine colon specimens [
6]. How the TJ proteins occludin or members of the claudin family are regulated by HlyA via intracellular signaling is still unknown. Moreover, the initial induction and further pathogenesis of epithelial
focal leaks by HlyA is not well-described. However, it was shown that
E. coli HlyA directly initiate intestinal barrier dysfunction, contributing on the one hand to diarrhea driven by a
leak flux mechanism and on the other hand to the
leaky gut phenomenon, allowing the entry of luminal antigens into the submucosa, which induce inflammation and subsequently cytokine-accelerated epithelial barrier disruption [
4]. These effects were assigned to calcium (Ca
2+) signaling in the epithelial cells [
7], since the HlyA pores in the host cell membrane mediate Ca
2+ influx through the pores [
8,
9].
Analysis on cell signaling in infected bladder cells with HlyA-harboring UPEC or HlyA alone lead to dephosphorylation of AKT/Protein kinase B (PKB), a key regulator of inflammatory responses, host cell survival, proliferation and metabolism [
9]. HlyA, but not other pore-forming toxins, induces a degradation of scaffolding and signaling proteins like paxillin and PAK-1 [
10]. Infection with HlyA-harboring UPEC leads to exfoliation of bladder epithelial cells. Whether this is due to the pathomechanism of the bacterium or part of the host defense mechanism to purge the infected cells so far remains unclear.
Since HlyA was shown to contribute to epithelial damage in intestinal diseases as IBD (i.e., ulcerative colitis) and other inflammatory diseases (i.e., cystitis), the higher toxinogenic potential of the HlyA+ E. coli could also depend on the invasion of bacteria in the epithelial layer. Therefore, it is of importance to identify host cell signals that are dysregulated by either toxins or the bacterial contact, mediating the epithelial damage and subsequently disease. In particular, epithelial cell polarity regulation, i.e., phosphoinositide (phosphatidylinositol phosphate (PIP)) signaling, triggered by the bacteria could in fact be a first step in the pathogenesis of the infection, inducing damage of the intestinal mucosa.
The Phosphatase and Tensin homolog deleted on chromosome ten (PTEN) is a phosphatase playing a role in regulation of cell polarity and tumor suppression. PTEN was described to localize to the plasma membrane, the cytosol as well as the nucleus. It dephosphorylates phosphatidylinositol-3,4,5-trisphosphate (PIP3), thereby suppressing AKT/PKB activation. In the process of epithelial morphogenesis and polarization, PTEN localizes in the apical membrane, enriching this compartment in phosphatidylinositol-4,5-bisphosphate (PIP2). PIP2 sums up for about 1% of the lipids in the plasma membrane. Nevertheless, as a source of three important second messengers, it is a key substrate in multiple membrane functions, e.g., endocytosis, exocytosis, enzyme activation and actin cytoskeleton organization, to name but a few [
11].
Besides the PIP3-dephosphorylation, PTEN has a plethora of functions. For instance, it also regulates Ca
2+-release from the endoplasmatic reticulum and mitochondria-associated membranes and modulates the cell cycle. Loss of PTEN function increases the amount of PIP3 in the plasma membrane and is associated with cell polarity changes, cell growth, cell migration and prolonged cell survival. In experimental studies, with inhibition of the PTEN pathway or deregulation of PTEN binding partners in intestinal epithelial cells, it was proposed that PTEN plays a role in colorectal cancer progression [
12]. In many types of cancer, PTEN expression was found to be altered [
13]. In colorectal cancers, PTEN alterations are associated with microsatellite instability, BRAF mutations and advanced metastases [
14]. Loss of PTEN gene function leads to disordered polarization of intestinal cells [
12]. Furthermore, PTEN loss in Caco-2 cells leads to higher Cdc42 levels and increased cell motility and invasion properties with upregulated expression of matrix metalloproteinases (MMP2, MMP9) and urokinase plasminogen activator (uPA) [
12]. Interestingly, disruption of PTEN in the intestinal epithelium of IL-10
−/− mice speeds up the onset and increases the severity of colitis [
15]. Reduced PTEN activity is known to increase cell migration in cancer cell lines [
12]. Interestingly, cell protrusions from the epithelial layer, leading to cell shedding, have also been described after HlyA infection in both bladder and intestinal epithelium [
7,
16,
17]. Single cell separation or collective cell migration like in metastasis could be associated to polarity changes and PTEN regulation and could be linked to pore-forming toxins like HlyA, as one aspect of intestinal pathology.
In order to identify pathomechanisms that might also serve as targets for future treatment options, we investigated the influence of HlyA from
E. coli 536 on cell polarity and epithelial barrier function of human colon carcinoma Caco-2 cells, with respect to the molecular targets and its subcellular distribution during infection. Since it was shown in other bacterial infection models that cell polarity and PIP signaling were affected during the first steps of infection [
18], we hypothesized that HlyA
+ E. coli influences cell polarity in a similar way. In order to assess the abundance of PIP2, we used a green fluorescent protein (GFP)-bound biosensor that targets PIP2 by a Pleckstrin Homology (PH) domain. The PH domain is part of the membrane targeting and activation of phospholipase Cδ (PLCδ) domain connected to a homology binding domain of AKT (GFP-PLCδ-PH construct). Moreover, we aimed at characterizing the concurrent effect on epithelial barrier function in the first hours of infection of Caco-2 monolayers and the cellular impairment during the course of the HlyA
+ E. coli infection.
3. Discussion
In inflammatory or inflammation-prone conditions, HlyA was described as potentiator of the
leaky gut phenomenon [
4,
7]. HlyA
+ UPEC strains can also be found in the large intestine of animals or humans, even in healthy conditions [
4]. In the phase of active inflammation in ulcerative colitis, they can be found in high numbers and in mouse models an infection with HlyA-producing
E. coli 536 causes intestinal inflammation [
4]. Whether or not they can play a role in human intestinal inflammation even without genetic predisposition remains unclear due to lack of study data. This can be explained partly because UPEC are not routinely assessed as, e.g., in infectious diarrhea, and partly because the nomenclature provokes confusion on different HlyA types, as the HlyA from UPEC is genetically different from the HlyA of enterohemorrhagic
E. coli (EHEC). Although strong effects on epithelial cell shedding or barrier dysfunction of UPEC HlyA on polarized intestinal cells were known for many years, the mode of virulence of UPEC in the intestine remained unclear, especially on the level of cell signaling. In this study, we could provide new findings on the pathomechanisms of UPEC HlyA on the intestinal epithelium.
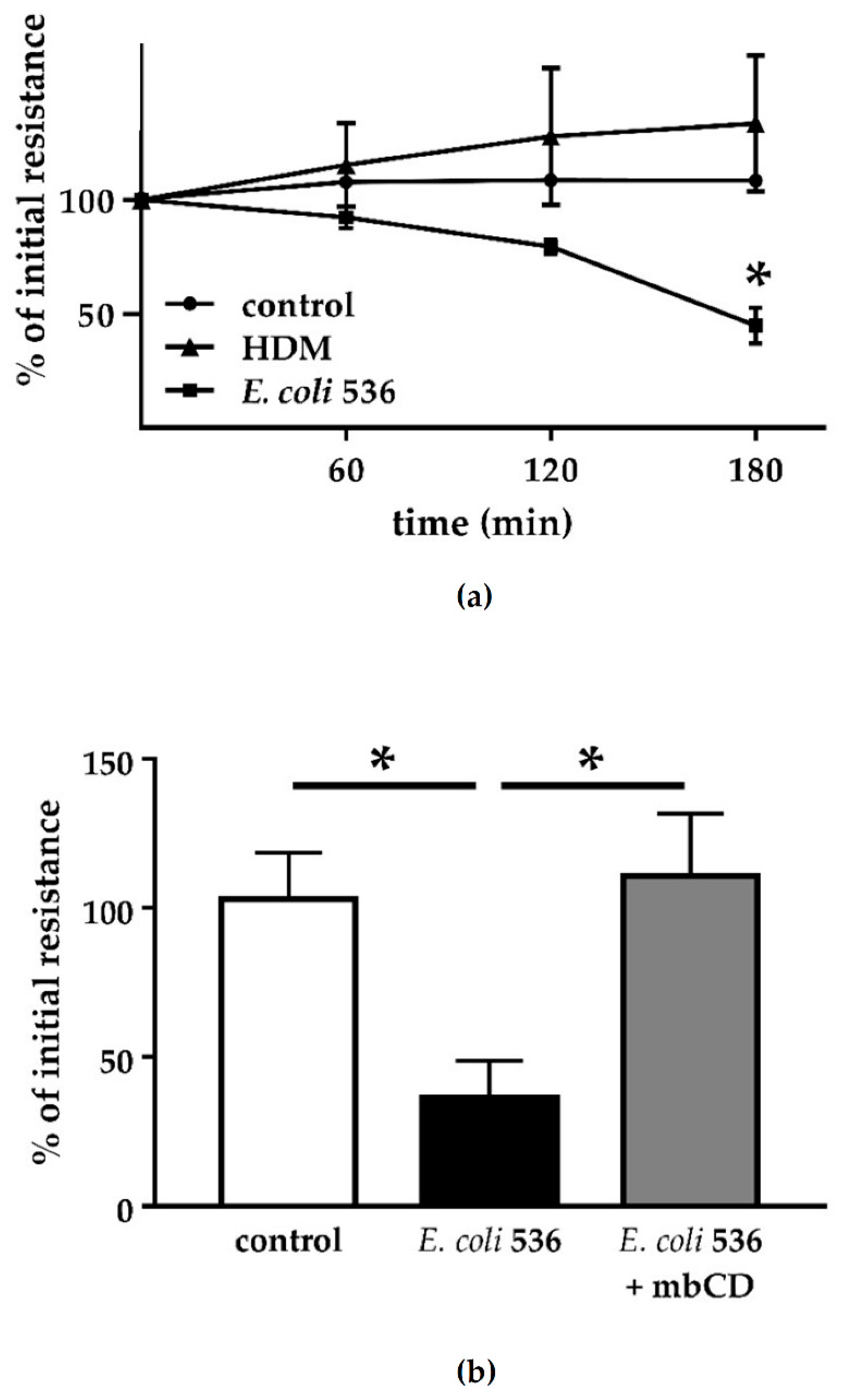
For bacterial adherence and invasion, a defined virulence factor of the UPEC pathovar are type-1 pili. The type-1 pilus adhesin FimH mediates bacterial invasion of human bladder epithelium [
20]. In our live cell imaging analysis, we could show bacterial invasion into intestinal epithelial cells with both pili expressing
E. coli 536 and the hemolysin-deficient mutant. Although invasion seems to be possible for UPEC strains even without HlyA, we could detect neither epithelial damage nor perturbation of the intestinal barrier after infection of Caco-2 cell monolayers. However, outer membrane proteins could have an effect on barrier function at later time points. Furthermore, incubation of sterile but HlyA-containing supernatant showed irreversible barrier-breaking effects on our cell monolayers, similar to infection with
E. coli 536 [
3,
7]. Thus, the early effects of UPEC on intestinal cells are likely due to its secreted toxin HlyA. Moreover, this may depend on the bacterial secretion of HlyA and the localization of the bacteria in proximity to the epithelial cells. The inhibition experiment with mbCD showed that the insertion of the heptameric HlyA pore proteins into the host cell membrane is responsible for the barrier defect. Similarly, it was shown for HlyA and further bacterial PFTs like aerolysin as well as other PFTs that the cellular disturbance depends on a proper insertion of the bacterial pores into the host cell membranes. When pore formation is disturbed, e.g., by zinc ions or other cations, the subsequent cellular pathologies are blocked [
6,
7,
21].
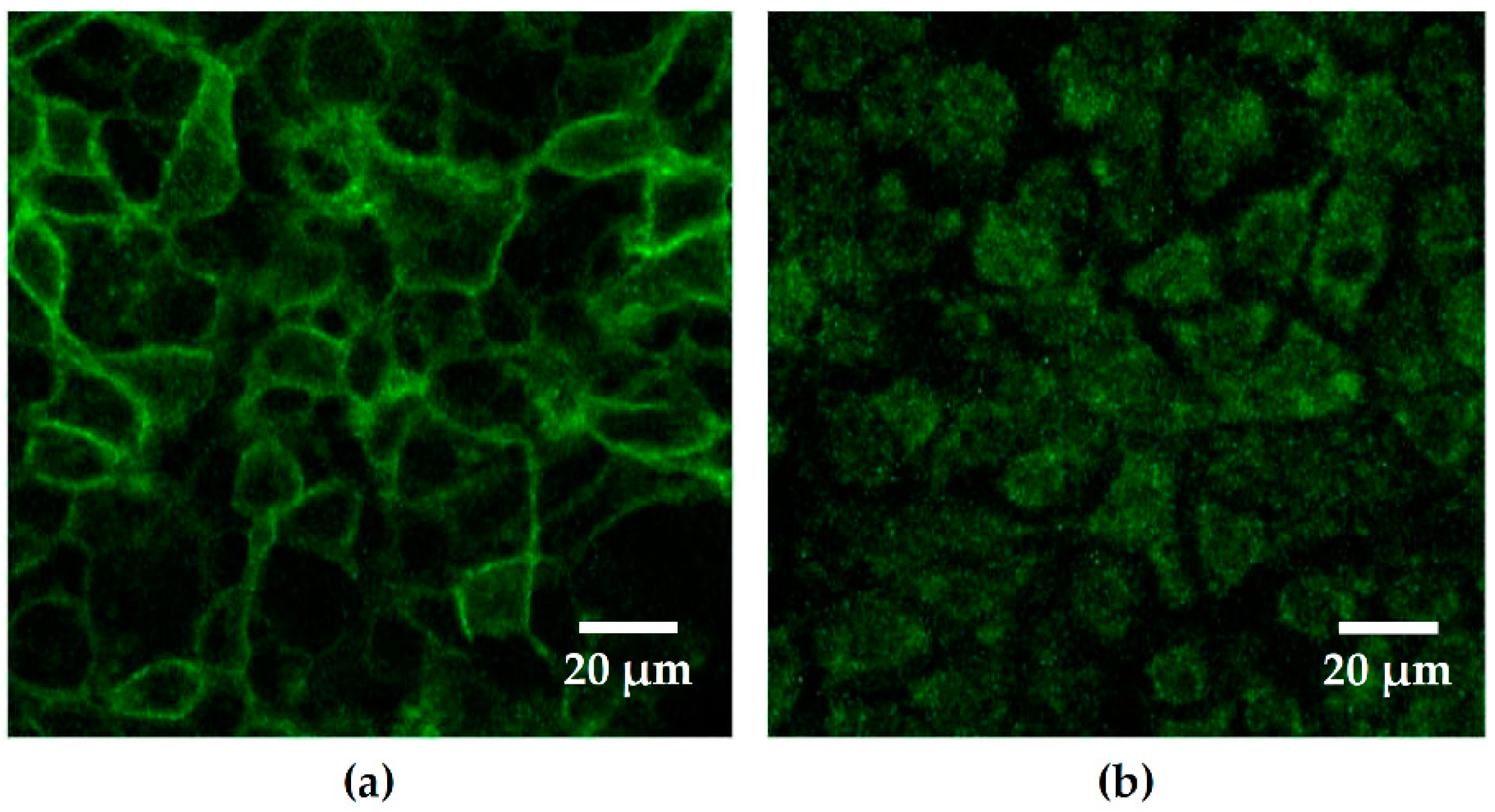
In previous studies on PIP distribution during bacterial infection on epithelial cells, both enteropathogenic
E. coli (EPEC) and
Pseudomonas aeruginosa caused transient focal PIP accumulations at the site of bacterial attachment [
22,
23]. In contrast to these observations, we did not see PIP accumulation in our infection model, not even after internalization of HlyA-negative HDM into Caco-2 cells. In our investigations, the PIP2 GFP-signal was eliminated from the membrane shortly after infection with
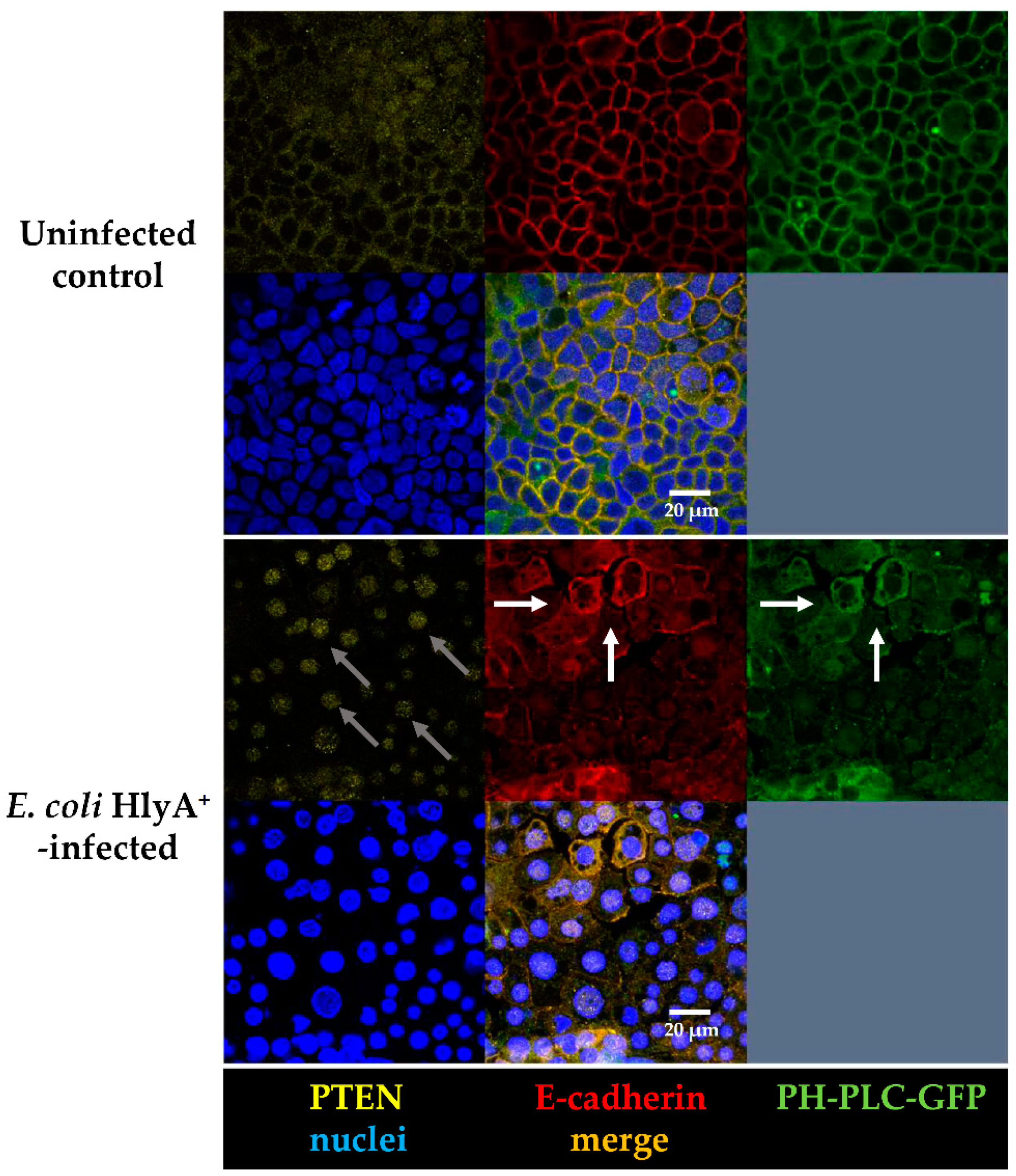
E. coli 536. Thus, a different mechanism linked to PIP2 elimination seems to be involved into UPEC pathogenesis. The PTEN is a dual-specificity phosphatase. It was first recognized as a tumor suppressor in the 1990s [
24], but nowadays, more and more of its wide range of biological functions are understood. The primary substrate of PTEN is PIP3, which is being hydrolyzed to PIP2 [
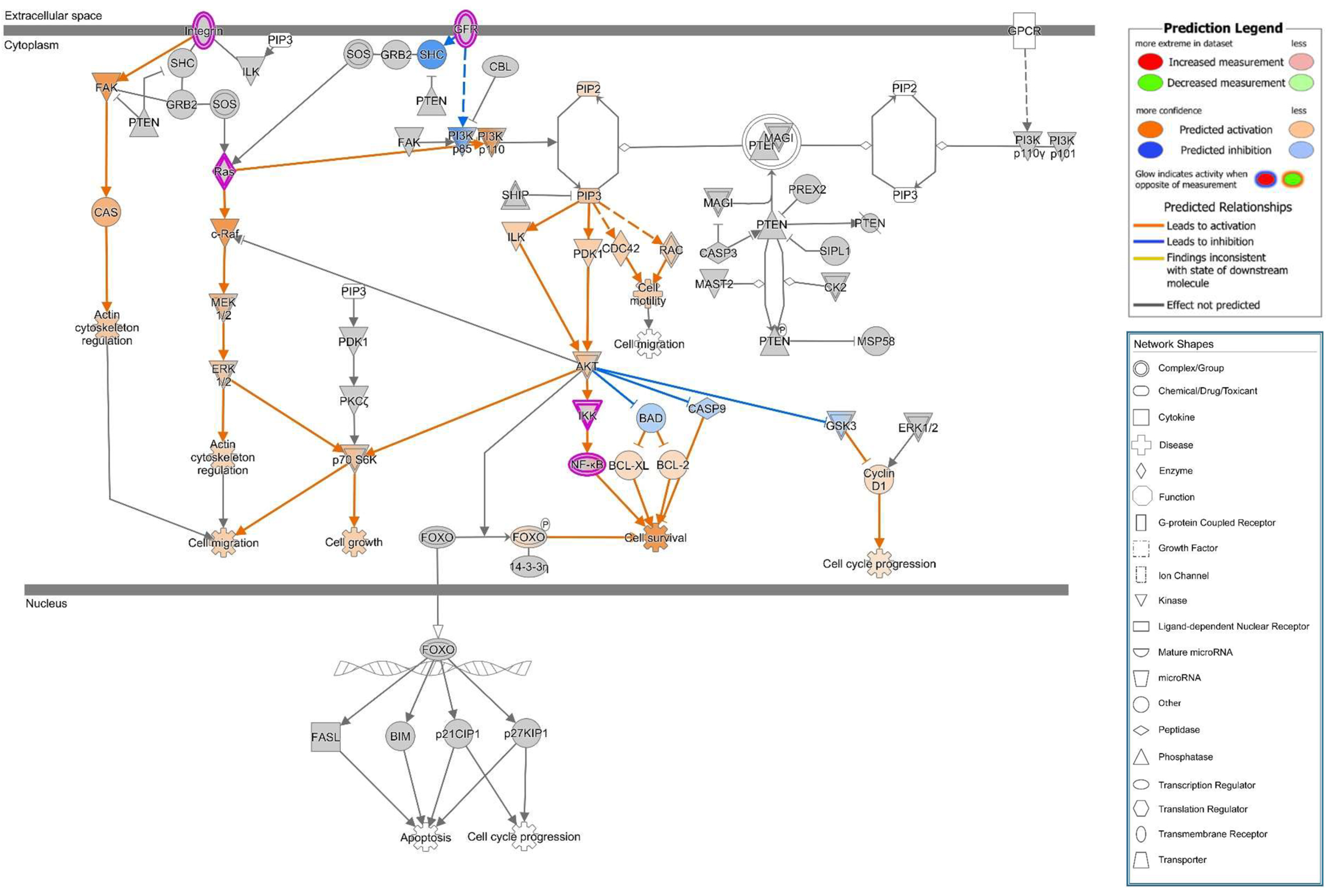
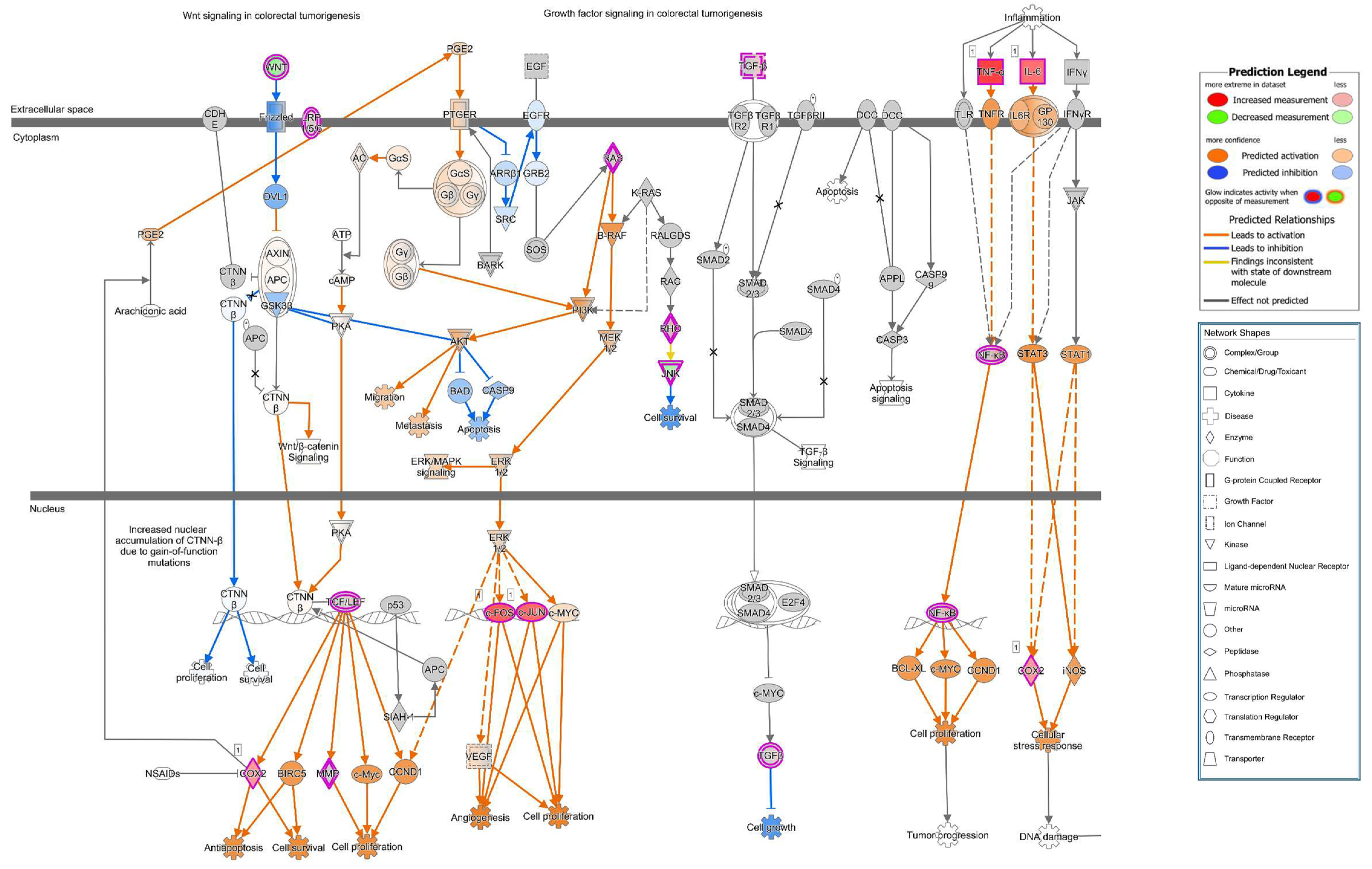
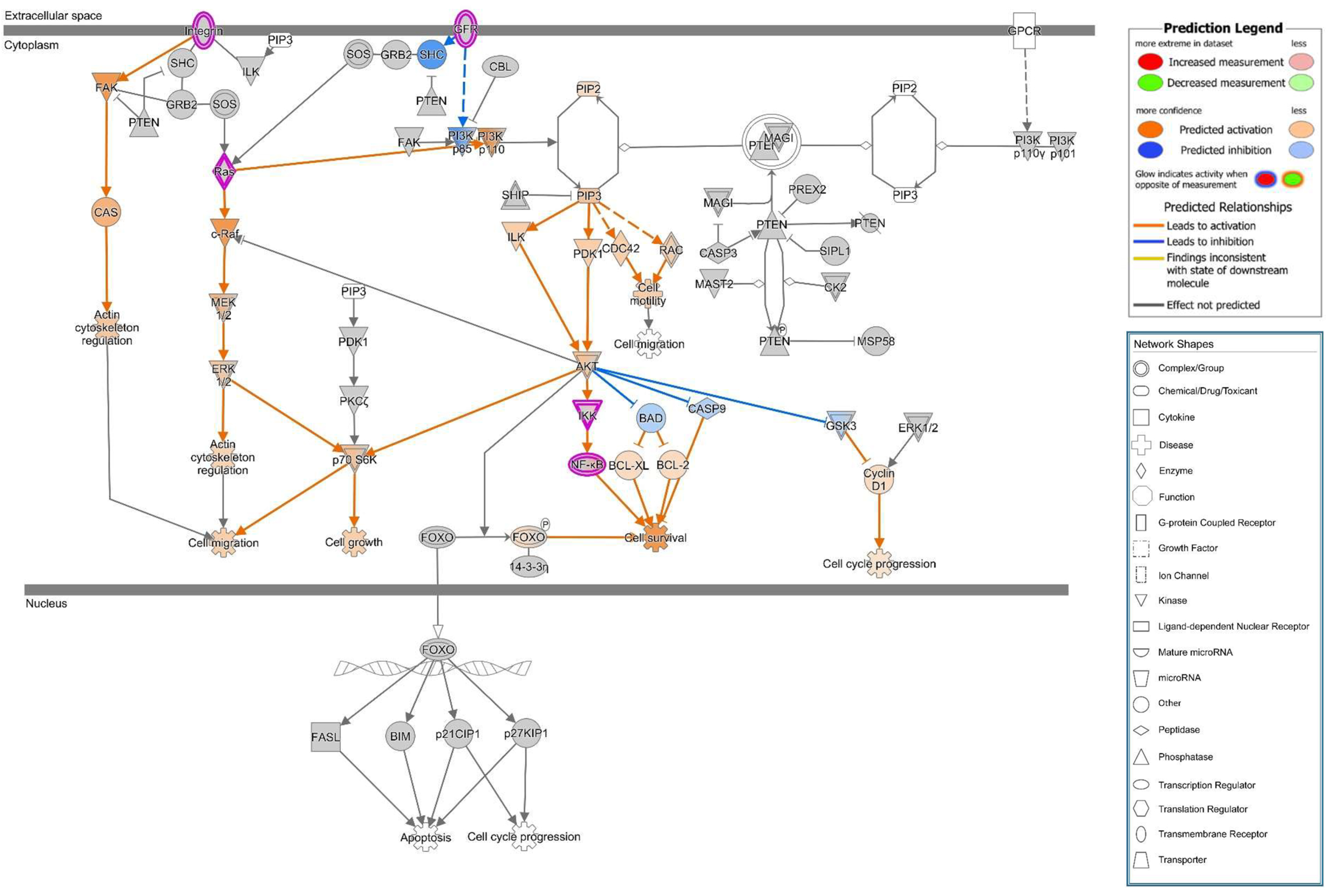
25]. PIP3 recruits AKT to the membrane and leads to its activation. Thus, PTEN acts as an AKT antagonist. In our study, we found PTEN signals to be reduced in the apical cell membrane micro-domain after HlyA-infection both in Caco-2 and in GFP-PH-PLC-transfected Caco-2 cells. Moreover, an increased PTEN signal was observed inside the nuclei. This PTEN regulation is in line with the signaling predictions from IPA with AKT activation and PTEN inhibition. Furthermore, this dysregulation can be associated to the predicted activation of the metastasis signaling pathway.
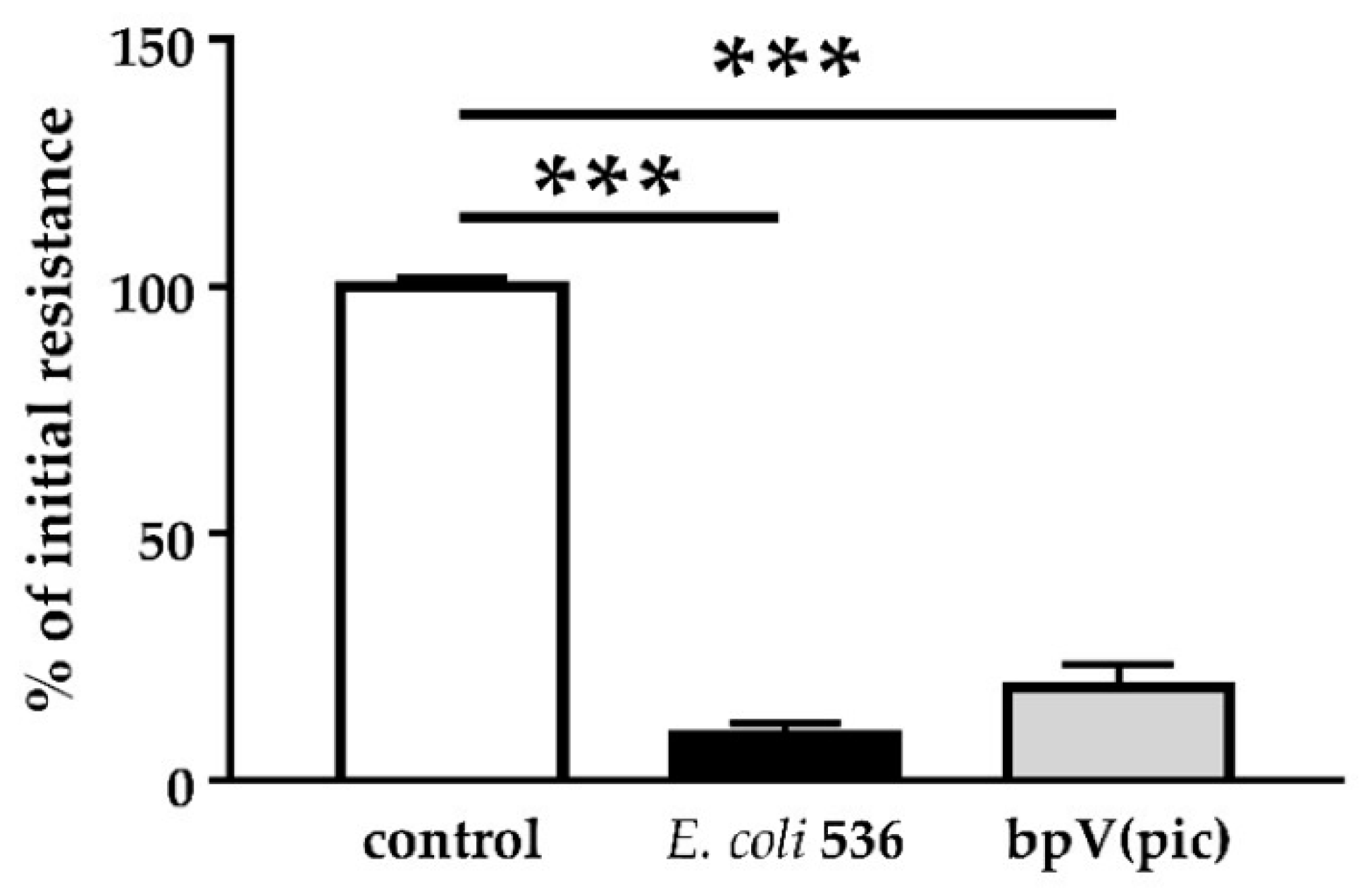
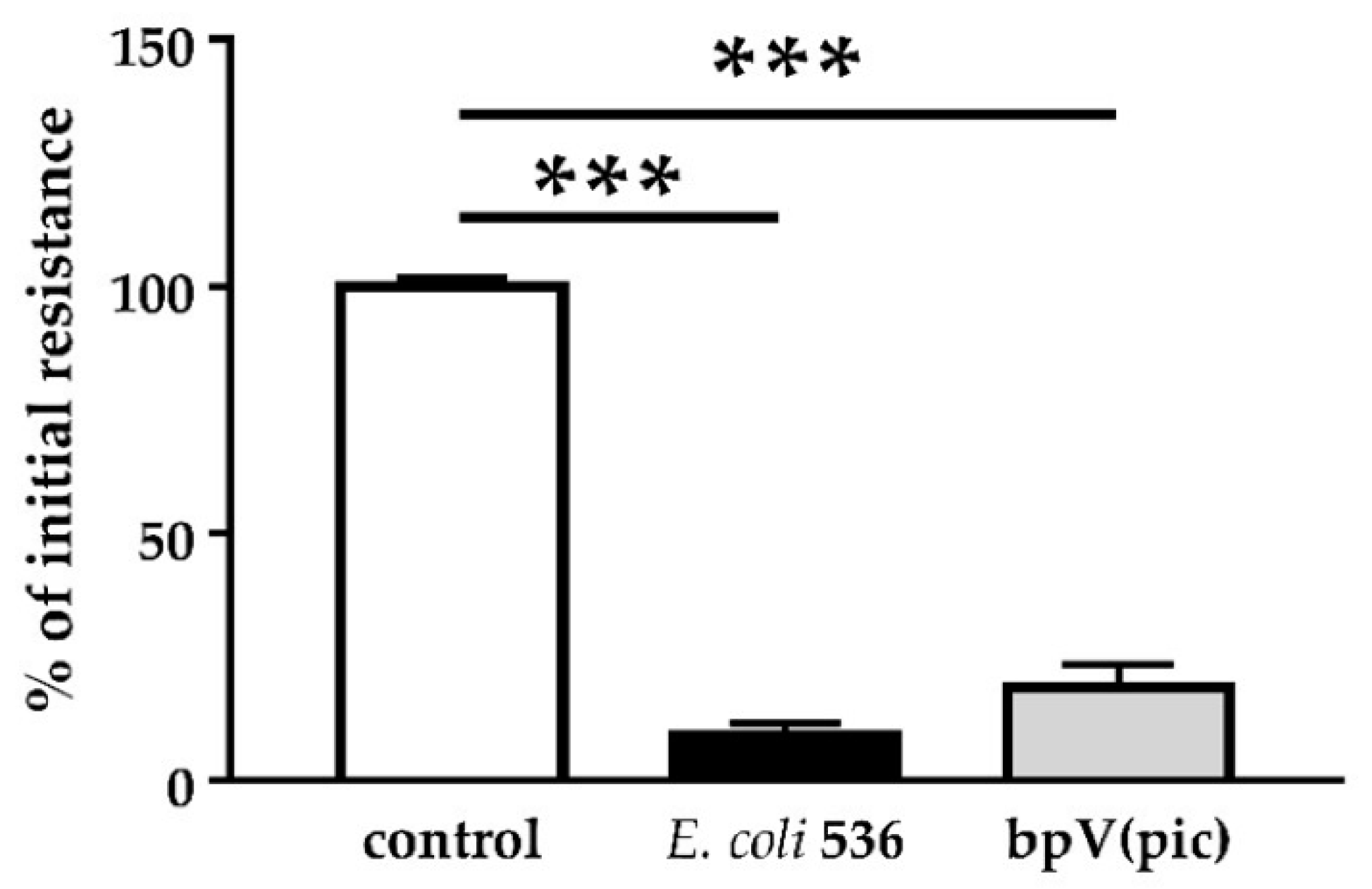
PTEN also plays an important role in cell metabolism. Other PTEN substrates belong to the Src family and take part in the focal adhesion complex. In this context, PTEN is known to be an inhibitor of cell migration [
26]. In fact, after PTEN inhibition with pbV(pic), we could observe increased numbers of cells that were detached from the monolayers. Infection with HlyA-secreting bacteria also leads to extensive detachment of cells from the tissue, morphologically similar to the bpV(pic)-induced cell exfoliation. In uroepithelial cells, this effect, called epithelial shedding, was previously described [
16]. Furthermore, Dhakal and colleagues found the PTEN substrate FAK from the focal adhesion complex to be reduced after HlyA-infection [
10], but to our knowledge, the present study is the first that links the HlyA effect to PTEN.
PTEN as a tumor suppressor belongs to the most frequently mutated genes in human cancers [
13,
24,
26]. Loss of PTEN causes disruption in chromosomal structure and numerical chromosome instability and leads to cell cycle progression [
24]. As reported, PTEN deficient intestinal cells have increased capacity to induce tumors [
12]. Moreover, in our experiments on Caco-2 cells, PTEN localization was changed into the nuclei, and cell proliferation was increased after HlyA
+ E. coli infection, pointing to changes in cell cycle regulation. Furthermore, after both HlyA
+ E. coli infection and PTEN inhibition with bpV(pic), the cell shedding from the epithelium was promoted. In a parallel experiment, PTEN inhibition by bpV(pic) decreased epithelial resistance to the same extend as HlyA did. Thus, PTEN inhibition could mimic the barrier defect of HlyA. Nevertheless, whether the increase in proliferation was the result of a PTEN-driven effect or due to an activated restoration mechanism of the epithelium, is up to future investigations.
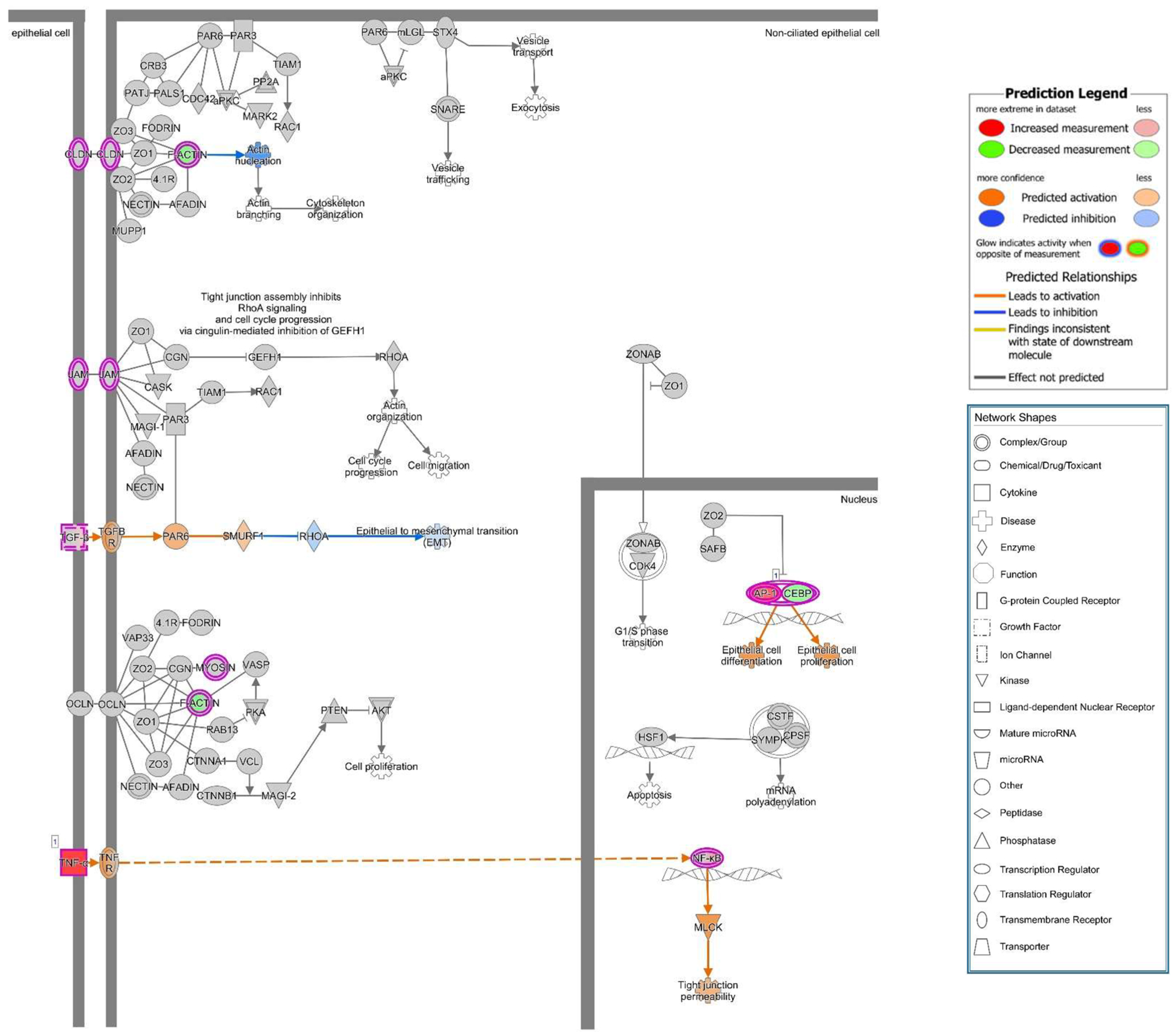
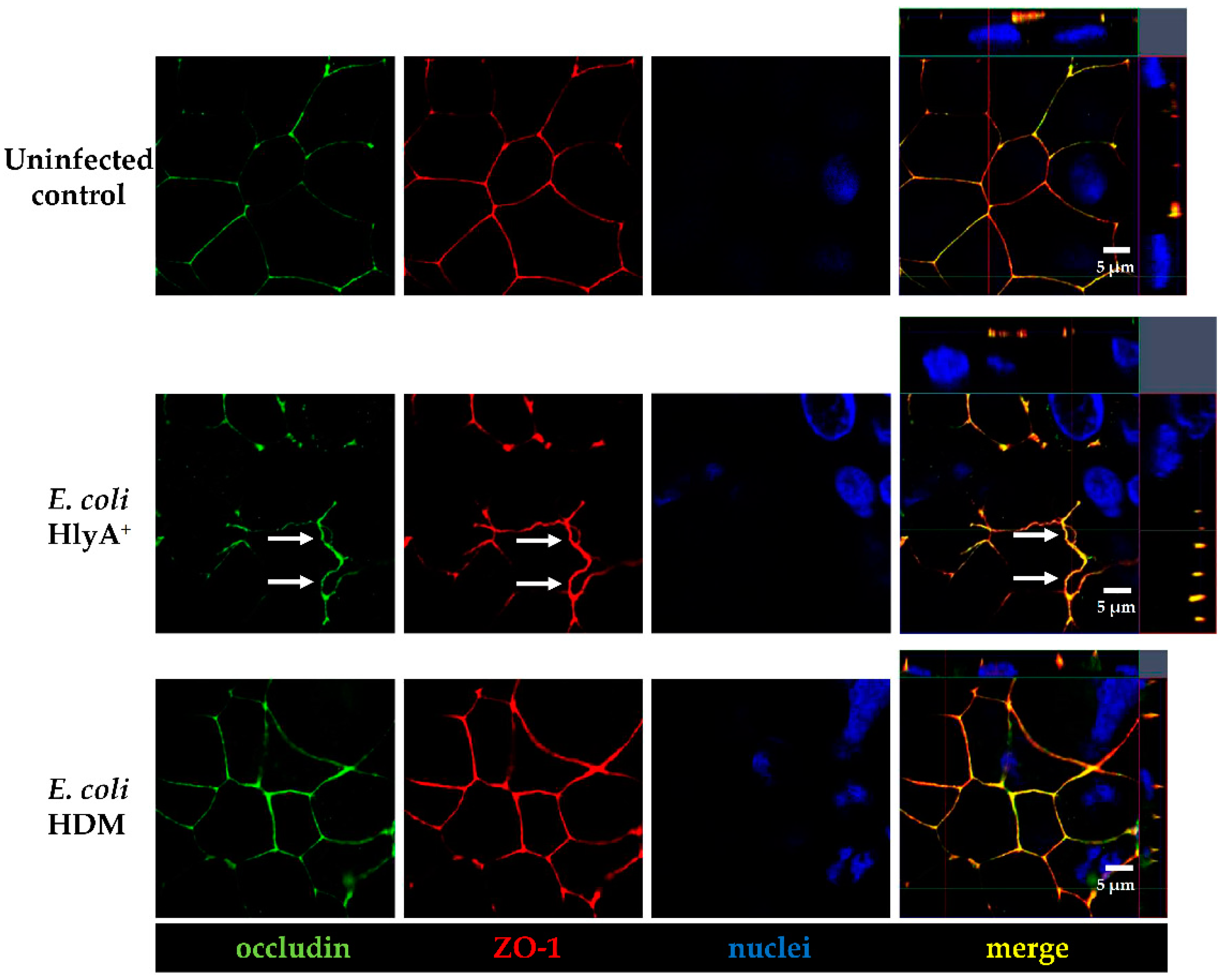
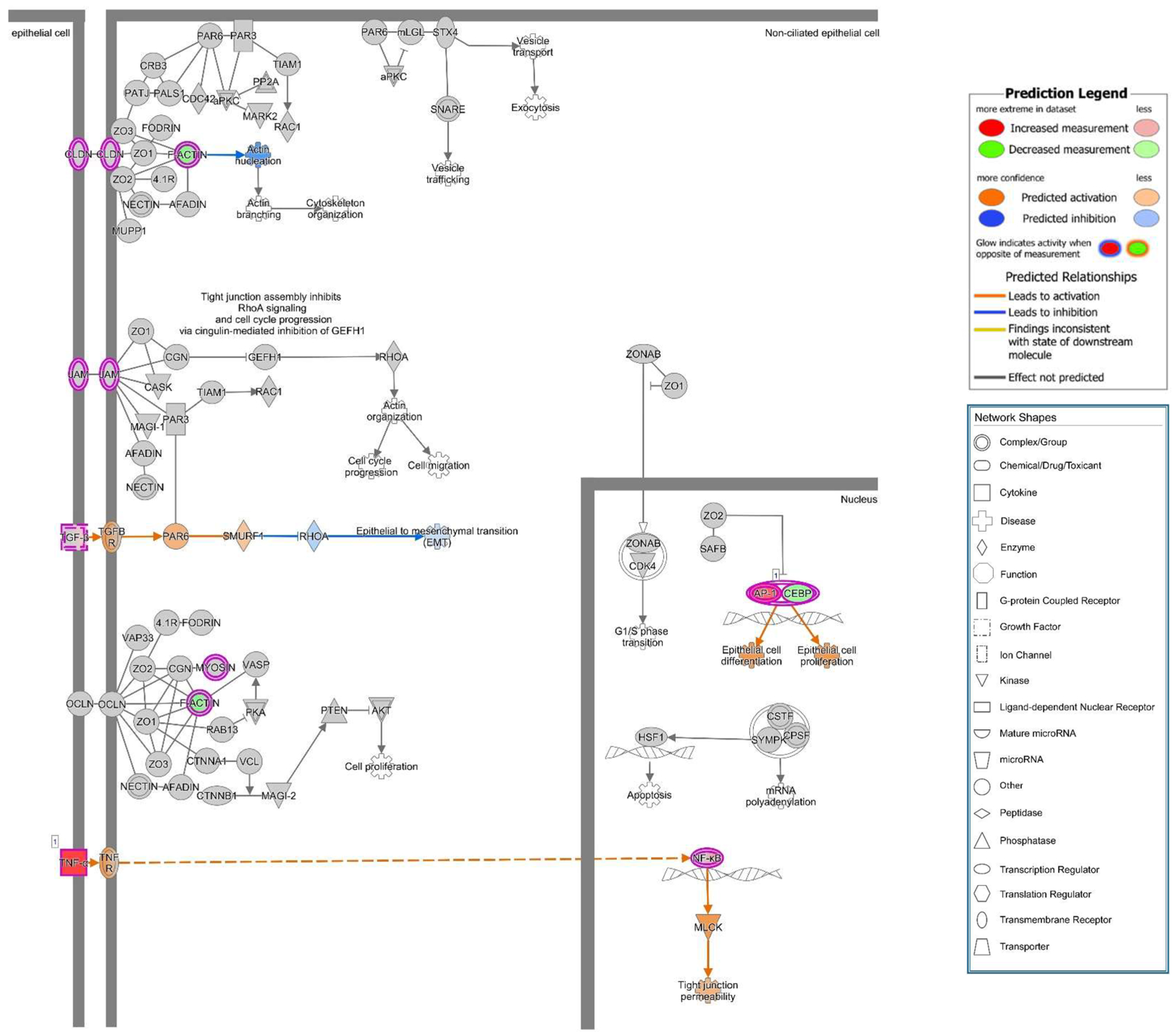
A previous study showed that upon suppression of PTEN, an up-regulation of ARF4 leads to altered expression of TJ proteins occludin and claudin-4 and increased epithelial permeability [
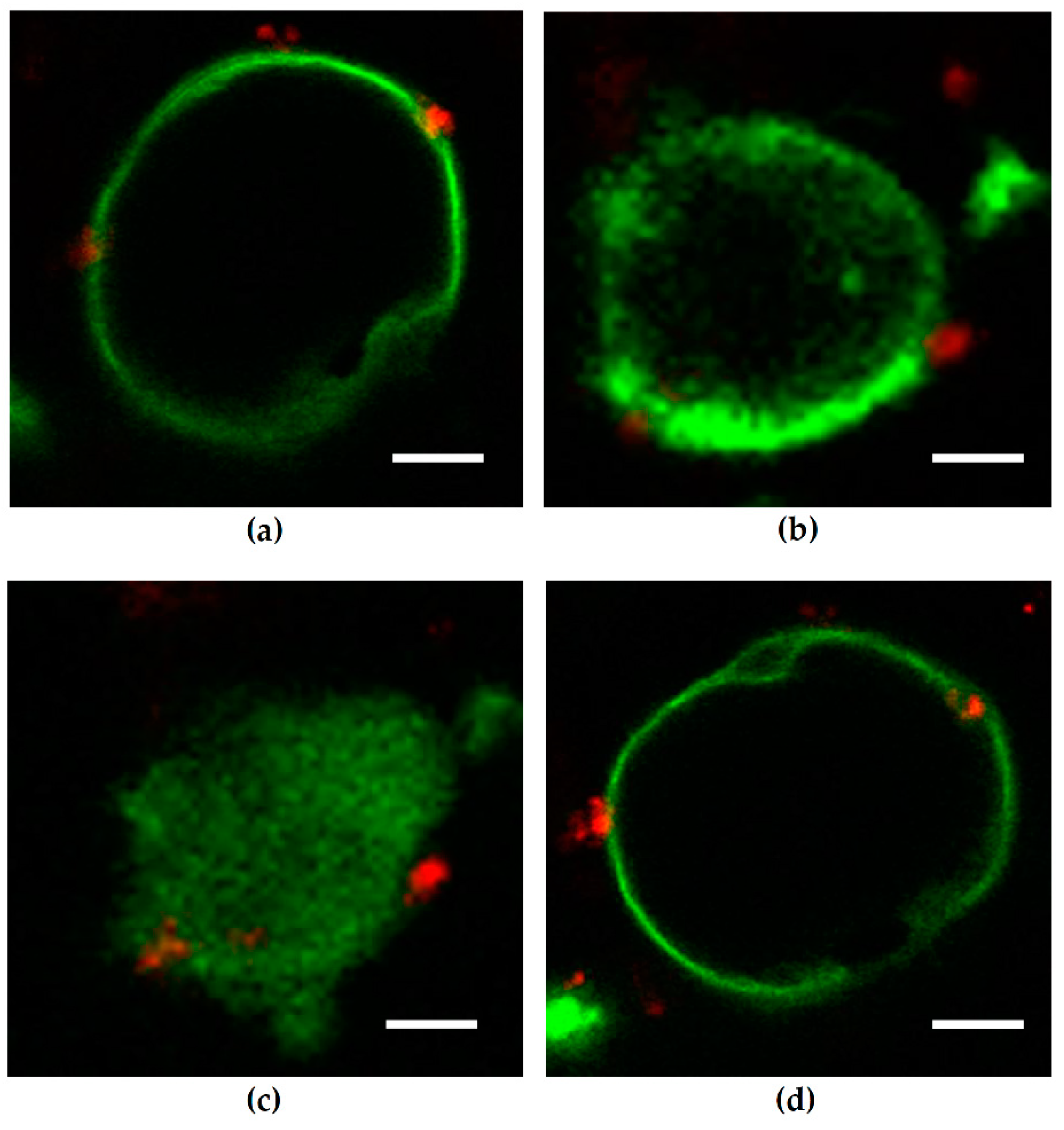
27]. Unfortunately, we found no association of ARF4 to the infection in our RNA-Seq data. However, in our study, we observed a change of distribution of the AJ protein E-cadherin from cell–cell borders towards the cytosol after infection of Caco-2 cells with HlyA-positive
E. coli 536. Under control conditions, PTEN and E-cadherin co-localize at the cell–cell border, and this interaction is a critical regulator of epithelial morphogenesis [
28]. We could detect both PTEN and E-cadherin reduction from the apical cell-cell contacts after HlyA
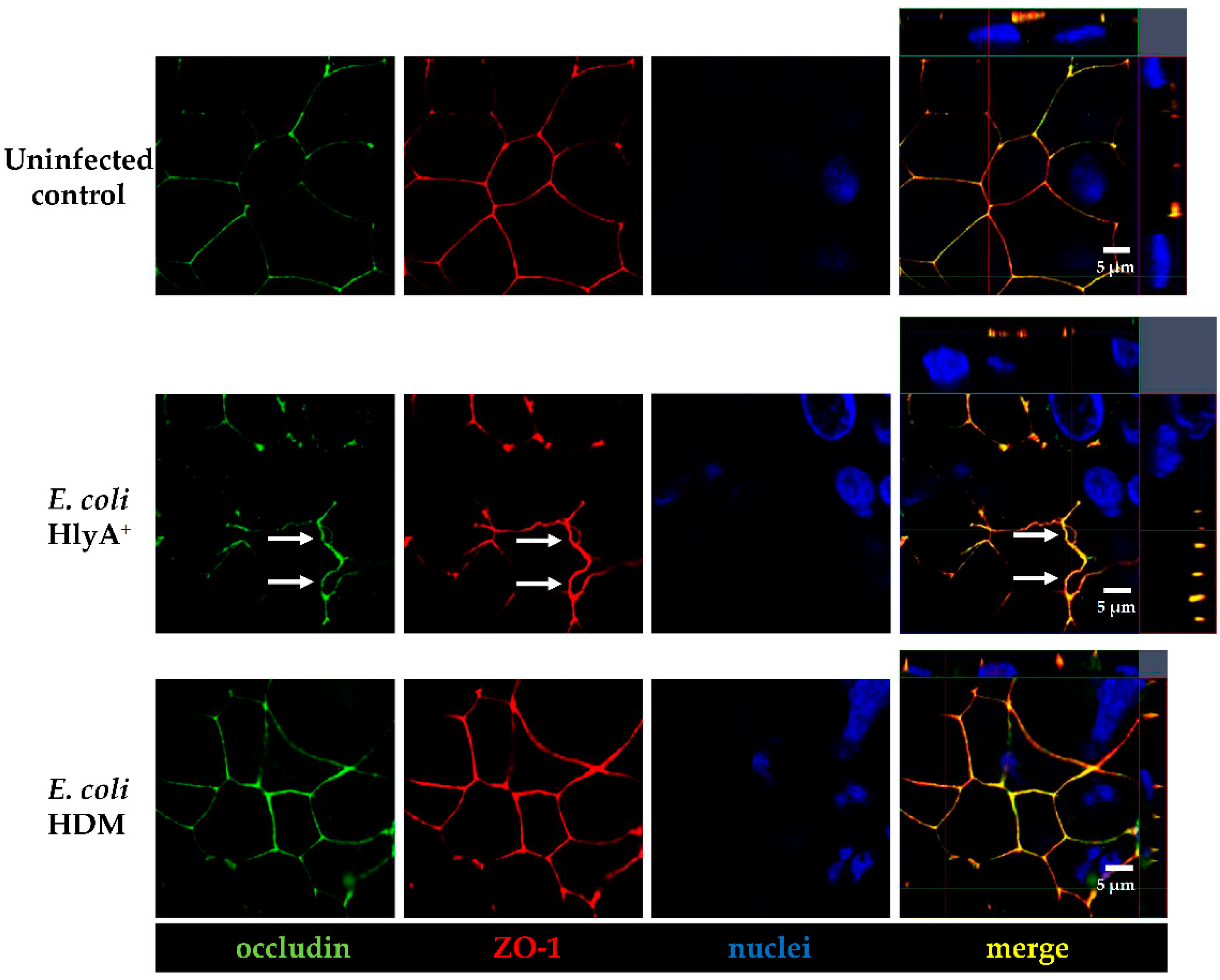
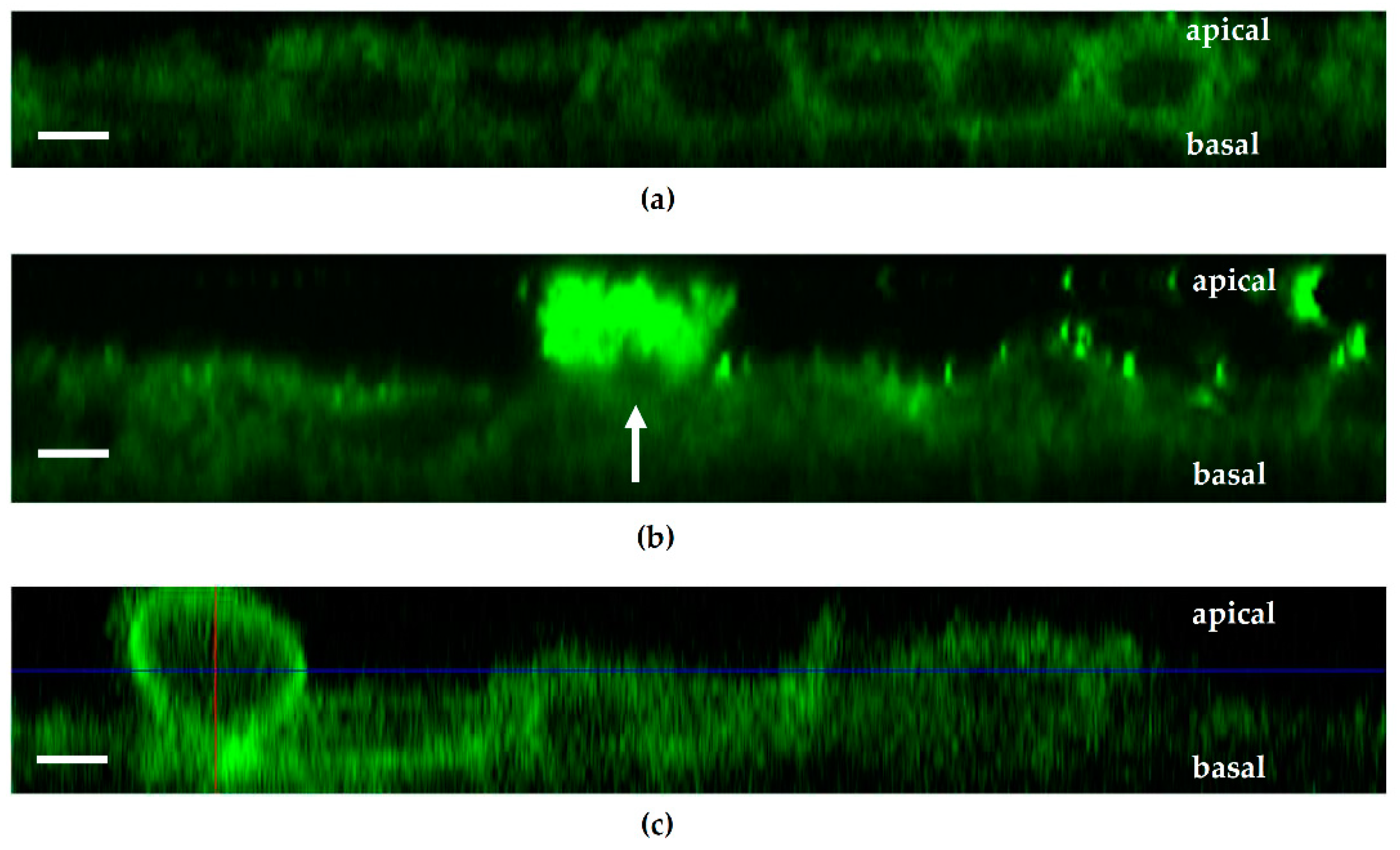
+ E. coli infection and measured a pronounced decrease in TER both after HlyA
+ E. coli infection and after PTEN inhibition with bpV(pic). Since it was shown that changes in PTEN abundance in proximity to the apical membrane affect both AJ and TJ signaling, our results could be explained by toxin-induced disorganization of the barrier-forming junctional complexes. For the latter, we found evidence during the first events of TJ protein redistribution of occludin and ZO-1 in the bicellular TJ during cell detachment induced by HlyA within 3 h. Similar to this, we previously observed a subcellular redistribution of the TJ proteins occludin, claudin-4 and claudin-5 as well as a disorganization of E-cadherin and F-actin in colonic mucosae from pigs infected with
E. coli 536 at 5 h after infection [
6]. The first evidence for the initial detachment of the TJ protein occludin represents the start of the barrier dysfunction and also indicates the beginning of the cell detachment, which might be the initiation point of
focal leaks and could end up in larger epithelial lesions, characteristic for the HlyA effect to induce
focal leaks [
3,
4]. This HlyA-induced barrier dysfunction by
focal leak formation was shown to be independent of apoptosis induction or TJ protein expression changes [
3,
4]. A profound change of TJ protein expression was not expected in the short incubation time of
E. coli (4–5 h). In a previous study with orally
E. coli 536-infected germ-free mice, claudin-1, -2 and -3 expression in the colonic mucosa was not changed even 3 days after infection. Hence, TJ expression regulation seems to play a minor role [
4]. Thus, the TJ changes observed in the present study display the early events in cell loss towards
focal leak formation as well as single cell shedding. These pathomechanisms point to the
leak flux mechanism, the passive loss of water and solutes to the intestinal lumen, as type of diarrhea by HlyA-secreting
E. coli. Further progression of the TJ disruption or
focal leak induction contribute to the
leaky gut phenomenon [
4]. For future investigations, it will be worth to enlighten affected pathways contributing to the TJ and AJ detachment during the HlyA
+ E. coli infection. In this context, studying MLCK signaling in dependence of NF-κB and TNF signaling or the calcium pathway after influx of Ca
2+ ions through the HlyA pores would be as important as the investigation of the PI3K, AKT and PTEN pathway. However, Ca
2+ pathways also activate other pathways, including Ca
2+-dependent PKC isoforms, which were shown to rather strengthen barrier function in Caco-2 cells via the leukotriene pathway, and furthermore, the activation of prostaglandin signaling could lead to barrier dysfunction, both displaying further candidates in
E. coli 536 pathology [
29,
30]. Nevertheless, the mechanisms behind cell extrusion by HlyA were characterized here for the first time. It starts with the detachment of TJs and progresses to cell separation and
focal leak formation. First hints for signaling in this
focal leak pathomechanism via cell detachment were shown here. However, further in vitro and in vivo experimentation and confirmation of the HlyA-induced pathways are needed to decipher the complete pathological process and its presumed role in colorectal cancer metastasis signaling.
A recent clinical observation study found
E. coli HlyA and colibactin to be present in high abundance on colorectal cancer [
31]. This observation, together with our experimental association of HlyA to the
leaky gut with influx of noxious agents and to the pro-oncogenic PTEN inhibition as well as to the cell separation or even dissemination of colon carcinoma Caco-2 cells, could open new perspectives on the surveillance of these virulence factors in colorectal cancer and metastases. In future research approaches, a better understanding of the microbial influence on the progression of this disease could also lead to new treatment options. A recent study on serum levels of PTEN mRNA even suggested the use of PTEN as an efficient and independent diagnostic marker for colorectal cancer patients [
32].
For future directions in medical research, it could be implemented that the colonization of the intestine with certain E. coli harboring hemolysin types like UPEC HlyA or other virulence factors such as colibactin could apply as prognostic factor in IBD or colorectal carcinoma, when these E. coli types are present in or around inflammatory foci or lesions and/or overgrowing neoplastic tissue.
This could have implications for a treatment option, e.g., with probiotics like E. coli Nissle or antibiotics against these bacteria or combination of such strategies, in order to attenuate the course of the disease.
5. Materials and Methods
5.1. Bacterial Cultivation and Infection
Hemolysin-producing
E. coli 536 and the
E. coli 536 hemolysin-deficient mutant (HDM) (536ΔhlyI_ΔhlyII::cat) [
33] were cultured at 37 °C overnight in Luria–Bertani broth. One hour before infection, the media on Caco-2 cells was replaced by a media not containing antibodies. An aliquot was brought to log phase (2 colonies in 10 mL culture medium at 37 °C and 220 rpm for 3 h) and a standard infection dose of 50 µL containing 10
5 colony-forming units (CFU) was added to the apical compartment of epithelial cells, containing 500 µL of media, with a multiplicity of infection (MOI) of 100 to the epithelial cells. Cells were incubated with bacteria from 1 h up to 5 h.
5.2. Cell Culture
Human Caco-2 colon carcinoma cells [
34] were cultured in liquid medium MEM- with Gutamax
TM-1 (Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with 1% penicillin-streptomycin (P/S, Corning, Glendale, AZ, USA) and 15% fetal calf serum (FCS, Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) in 25 cm
2 culture flasks (Corning, Glendale, AZ, USA). The cells were maintained at 37 °C with 5% CO
2 and were fed every two-days. After 14 days, Caco-2 cells reached confluency; they were washed with phosphate-buffered saline (PBS, Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA), incubated for 30 to 60 min at 37 °C with trypsin-EDTA solution (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) and seeded on Millicell PCF filters (0.4 µm pore size, 12 mm Diameter, Merck Millipore ltd., Darmstadt, Germany). Cells were grown on filter supports for 10 to 14 days until forming a confluent monolayer before bacterial infection.
5.3. Transepithelial Resistance
The measurement of transepithelial electrical resistance (TER) of the colonic epithelial monolayers was performed as previously described [
35]. A fixed pair of electrodes connected to an Ohmmeter (EVOM, WPI, Sarasota, FL, USA) was used to measure the transepithelial electrical resistance of confluent monolayers on filters, at a stable temperature of 37 °C. Correction values for the resistance of the empty filter and the area were considered. Resistance measurements were performed before as well as after infection with
E. coli 536 and
E. coli HDM or after inhibition of PTEN with bpV(pic) (1µM, Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), PI3K with Ly294002 (50µM, Cell Signaling Technology, Danvers, MA, USA), PLC with U73122 (5µM, Millipore, Merck KGaA, Darmstadt, Germany), MLCK with ML-7 or H-7 (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), PKC with H7 or Calphostin C (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) or ERK 1/2 with U0126 (Cell Signaling Technology, Danvers, MA, USA) from 1 h up to 5 h after infection or inhibition.
5.4. Transfection
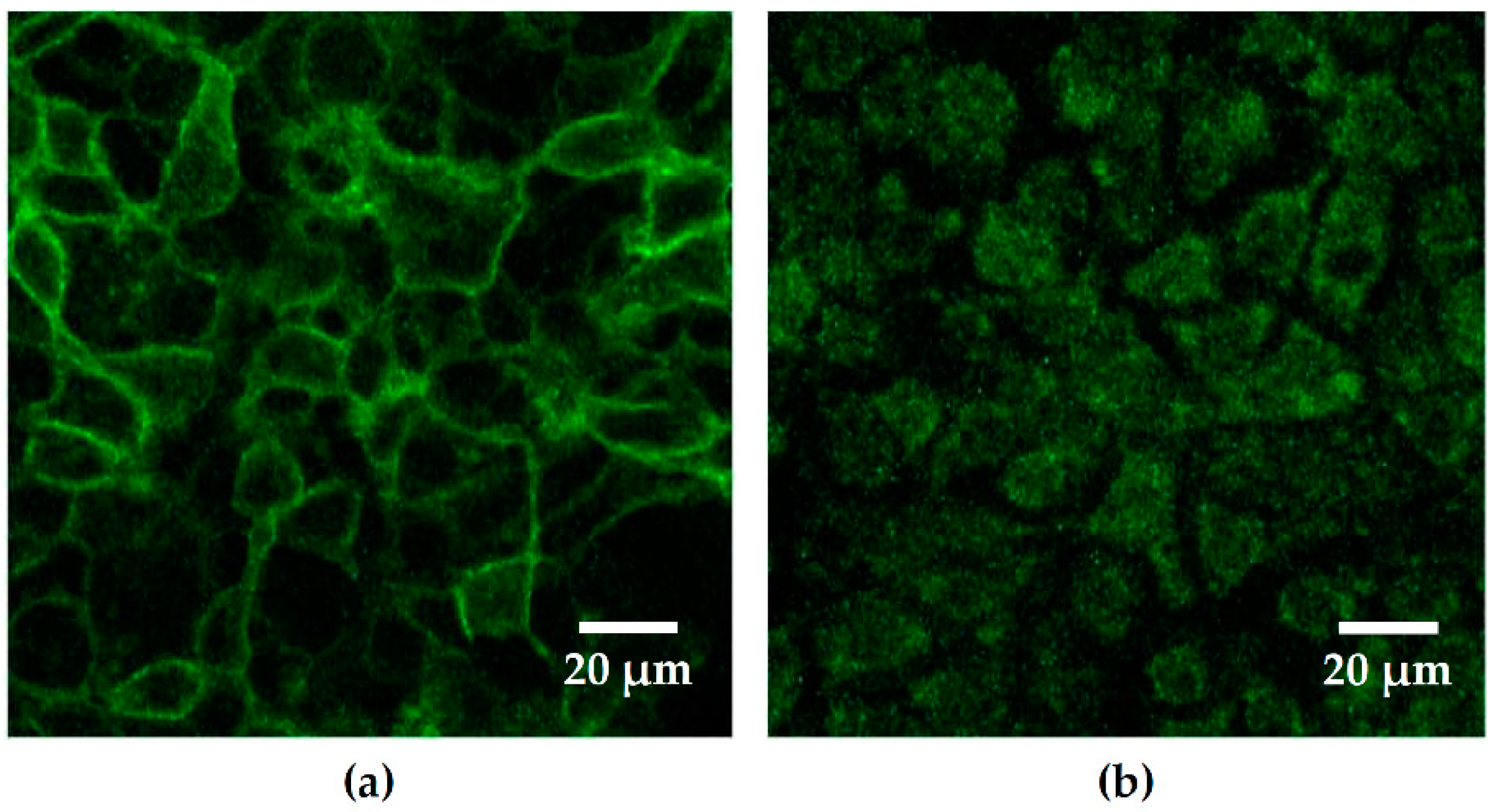
Caco-2 cells were transiently transfected with the DNA construct PH-PLCδ-eGFP used as a reporter for PIP2. The constructs were obtained from Prof. Michael Krauß (FMP, Leibniz Forschungsinstitut für molekulare Pharmakologie, Berlin, Germany). Caco-2 cells were grown for 14 days and split and grown again for three days. After three days, the cells were trypsinized with 1 mL trypsin-EDTA for 10 min at 37 °C. The cells were counted, and the cells suspension concentration was adapted to reach 6 × 104 cells/mL. A total of 750 000 cells were seeded in Lab-Tek II chambered coverglass well (0.7 cm2, Thermo Fisher Scientific Inc., Waltham, MA, USA), and cells were grown for 5 h in RPMI media + 10% FCS at 37 °C and 5% CO2. After 5 h, the DNA construct was added on the cells with media without FCS and Lipofectamin-2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA). After 18 h of growth, media without P/S was added to fill the wells completely and after 24 h the media was changed for RPMI + 10% FCS. Two days after lipofection, cells were checked for successful transfection with GFP localized at the apical membrane of cells via LSM. Seven days after transfection, cells were infected with bacteria, and TER was measured before and throughout the infection period. For stable transfection, the same protocol was applied; however, two days after transfection, cells were checked for successful transfection, and G418 (Geneticin, PAN biotech, Aidenbach, Germany) was added on the cells at a concentration of 1000 µg/mL to select viable clones. After the non-transfected cells died, the concentration of G418 was reduced to 600 µg/mL, and successful clones were selected and grown separately in 24 wells plates until confluency. They were then transferred to 12-well plate, 6-well plate and finally 25 cm2 flasks. The successfully transfected clone #29 was selected and used for further experiments, and this Caco-2 cell line with stable GFP-PLC-PH expression formed a confluent epithelial cells monolayer after appropriate culturing. These polarized monolayers were able to establish an apical-basolateral barrier as measured with high transepithelial electrical resistances (TER).
5.5. Live Cell Imaging
PH-PLCdelta-eGFP-transfected Caco-2 cells were cultured in Lab-Tek wells or in inverted filter supports (0.4 µm pore size, 12 mm Diameter, Merck Millipore ltd., Darmstadt, Germany). Seven days after confluency, images were taken using a water immersion objective (LSM780, Zeiss, Jena, Germany). Incubation chamber under the LSM objective was heated to 37 °C, and cells were kept under constant 5% CO2 atmosphere in ambient air throughout the live cell imaging experiment. E. coli 536 and HDM were stained with Hoechst 33342 solution (dilution 0.67 µg/mL, Cell Signaling Technology, Danvers, MA, USA) for visualization and added to the cell filters with standard infection dose.
5.6. Visualization of E. coli HlyA and Epithelial Integrity by Immunofluorescence Staining
Caco-2 cell monolayers were fixed with 2% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA). Then, cells were treated with 25 mM glycine (Carl Roth, Karlsruhe, Germany) in order to quench the protein linking up. TritonX-100 (Serva electrophoresis GmbH, Heidelberg, Germany) at 1% in phosphate-buffered saline with calcium and magnesium (PBS+, Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) was used for permeabilization for 2 h and subsequently stopped with blocking solution (10% goat serum, 1% bovine serum albumin, 0.8% TritonX-100 in PBS+) for 3 h. The primary antibody was added in blocking solution overnight. Staining with a secondary IgG antibody (Alexa Fluor, Thermo Fisher Scientific Inc., Waltham, MA, USA) was performed after several washing incubations with PBS+ (Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA). DAPI (4’-6-diamidino-2-phenylindole dihydrochloride, Roche Diagnostics, Mannheim, Germany) and DY-647P1-Phalloidin (1:200 dilution, Dyomics, Jena, Germany) were used for staining nuclei and actin cytoskeleton, respectively, at room temperature for 30 min. The following antibodies were used: Occludin (rabbit, 1:200 dilution, Invitrogen, Thermo Fisher Scientific Inc., Waltham, MA, USA, no. 711500), ZO-1 (mouse, 1:100 dilution, BD Biosciences, San Jose, CA, USA, no. 610966), E-Cadherin (mouse, 1:200 dilution, BD Biosciences, San Jose, CA, USA, no. 610182), PTEN (rabbit, 1:100 dilution, Cell Signaling Technology, Danvers, MA, USA, no. 9559S), anti-human Ki-67 Alexa Fluor 647 (mouse, 1:200 dilution, Biolegend, San Diego, CA, USA, no. 350509).
5.7. Epithelial Cell Counting after Bacterial Treatment for Cell Separation Assay
Caco-2 monolayers were infected with E. coli 536 or control infection with HDM for 5 h; then, bacterial cells were inactivated by addition of 100 mg/L gentamicin (Carl Roth GmbH, Karlsruhe, Germany), and the cell culture medium (DMEM) was transferred as supernatant on fresh 24-well coated with 0.002% poly-L-lysine (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), and the seeded Caco-2 cells were incubated for another 24 h in a cell culture incubator. The number of viable cells shedded from the infected Caco-2 monolayers was estimated the next day by microscopic counting of attached cells in a low-power field. Dead cells were counted microscopically by trypan blue staining and excluded from analysis.
5.8. Statistical Analysis
Data are expressed as mean values ± standard error of the mean (SEM). Statistical analysis was done with GraphPad Prism (GraphPad Software version 5, San Diego, CA, USA). Student’s t-test or 1-way ANOVA and t-test with Bonferroni correction was used for multiple comparisons. p < 0.05 was considered statistically significant.
5.9. Bioinformatic Pathway Analysis from RNA-Sequencing Expression Data
RNA was extracted from Caco-2 monolayers after 5 h incubation in an infection experiment with
E. coli 536 (3 cell monolayer filters each) using the mirVana RNA Isolation Kit (Ambion, Life Technologies, Carlsbad, CA, USA). The following methods were described previously [
36]. Briefly, library preparation of RNAs cDNA library and sequencing was performed by Illumina’s RNA-Seq prep kit following the manufacturer’s instructions. The purified DNA was quantified and diluted to 10 nM for cluster generation and sequencing on an Illumina NovaSeq6000 Sequencing System. The reads were mapped against the human genome GRCh37/hg19 using STAR Aligner v2.7.1a with double pass alignment [
37]. First mapping was obtained using Ensembl v73 coordinates as a framework. Second-pass mapping added splice sites that were found in the first run. RNA-Seq expression analysis Gene-read coverages were obtained using Bioconductor package Rsubread [
38]. The Bioconductor package DESeq2 was used to quantify the differential gene expression between two conditions in form of log2-fold changes with their corresponding
p-values [
39,
40].
p-values were corrected for multiple testing according to Benjamini Hochberg to match the false discovery rate, and a cut-off of
p < 0.05 (5% FDR) was chosen. Fastq files containing the raw reads from sequencing are available at Gene Expression Omnibus (GEO) archive under National Centre for Biotechnology Information (NCBI) website with GEO accession ID GSE169213.
Canonical pathway analysis was performed with the Ingenuity Upstream Regulator Analysis software (IPA, Qiagen Silicon Valley, Redwood City, CA, USA).

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}