
Suppression of NOX2-Derived Reactive Oxygen Species (ROS) Reduces Epithelial-to-MesEnchymal Transition Through Blocking SiO2-Regulated JNK Activation

 and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Crystalline Silica
2.2. Establishment of a Mouse Model of Silicosis
2.3. Single-Cell RNA Library Construction and Sequencing
2.4. Cell Cultures and Treatments
2.5. Cell Scratch Assay
2.6. Transwell Assay
2.7. Histological Staining
2.8. Immunofluorescence Staining
2.9. Western Blot Analysis
2.10. Wound Healing Assay
2.11. EdU and Ki67 Proliferation Assays
2.12. Statistical Analysis
3. Results
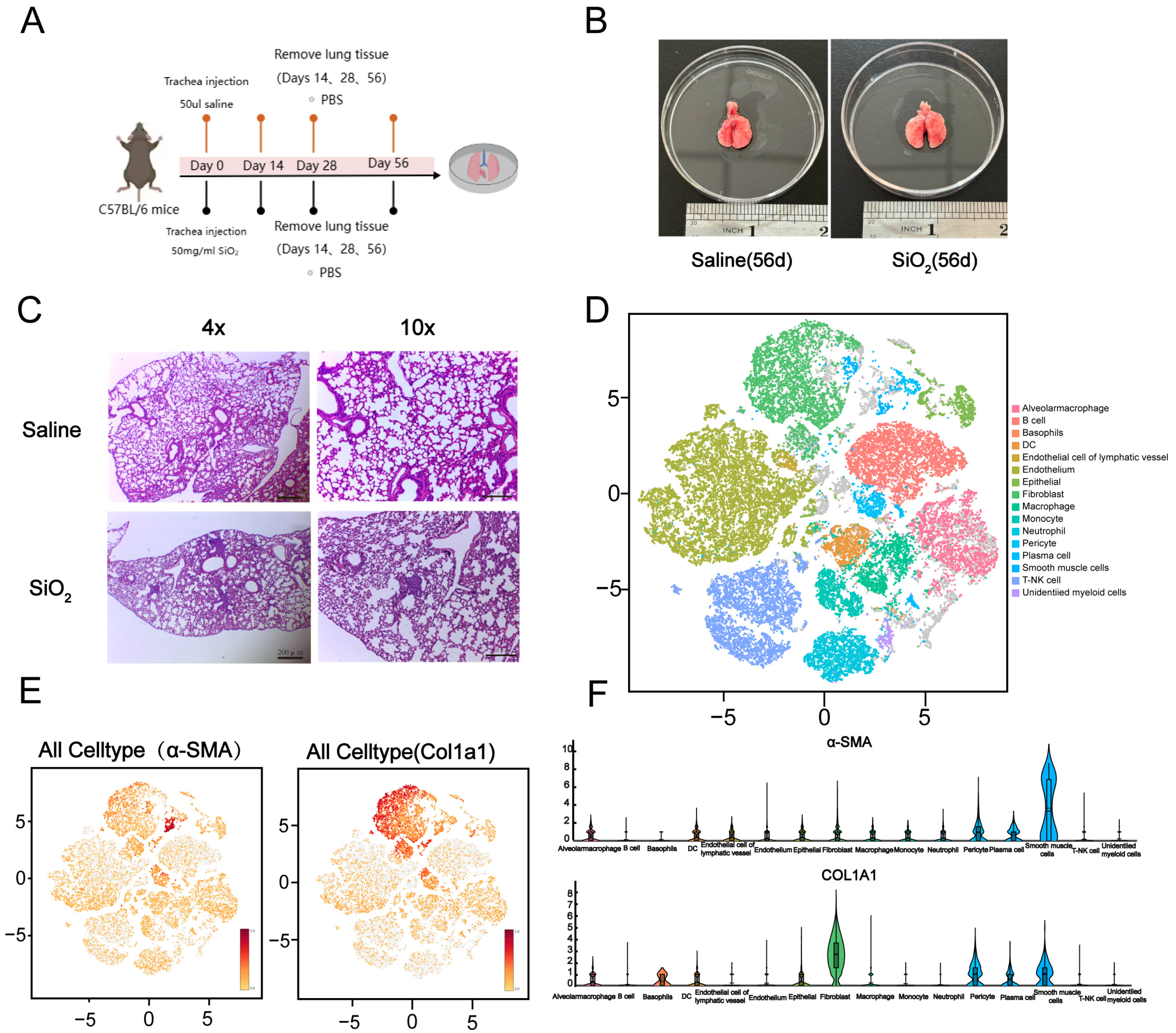
3.1. Generation of the Silicosis Mouse Model and Single-Cell Sequencing Classification of Mouse Lung
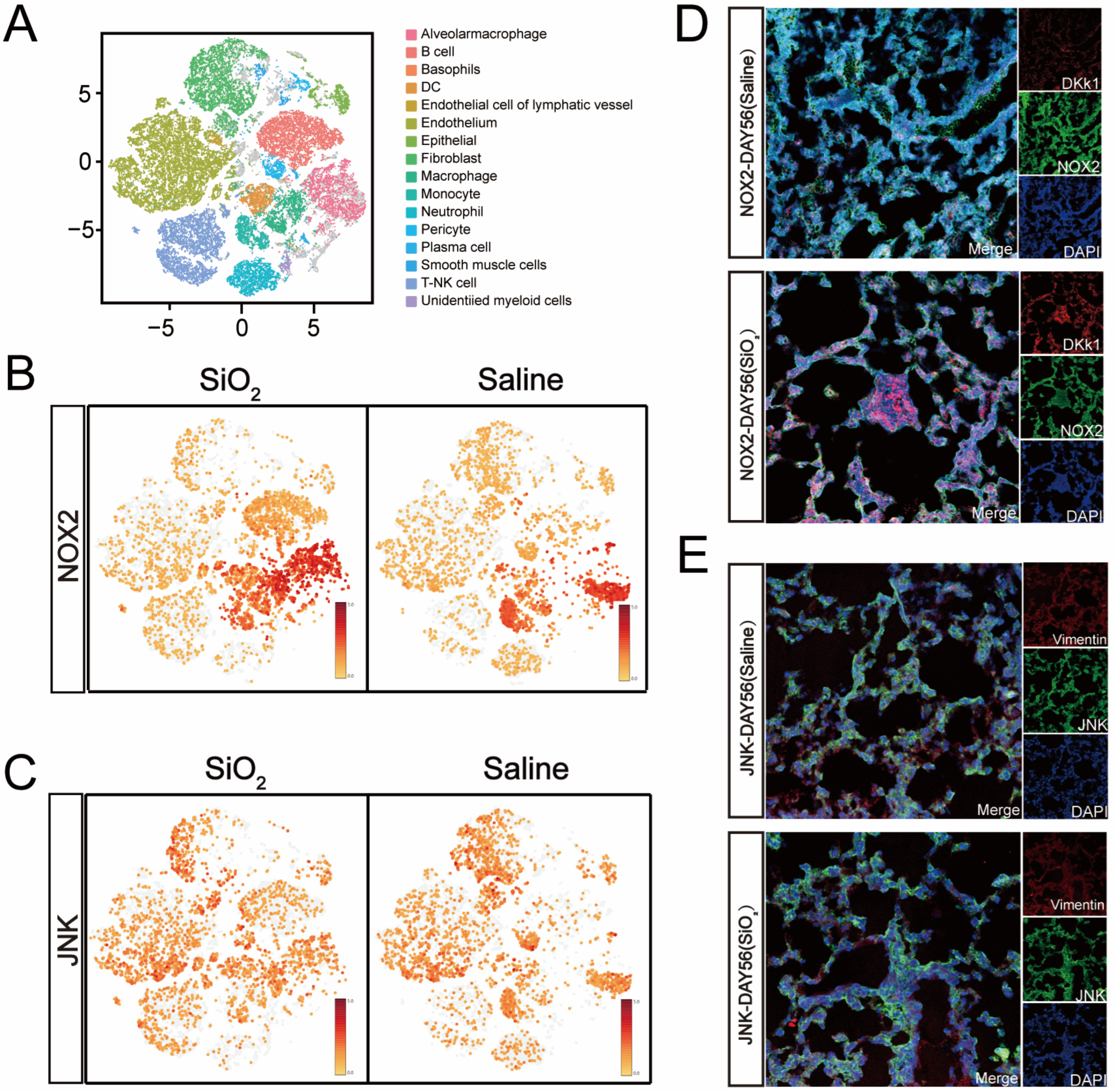
3.2. Increased NOX2/JNK Expression and Epithelial-to-Mesenchymal Transition in the Small Airways of the Silicosis Model
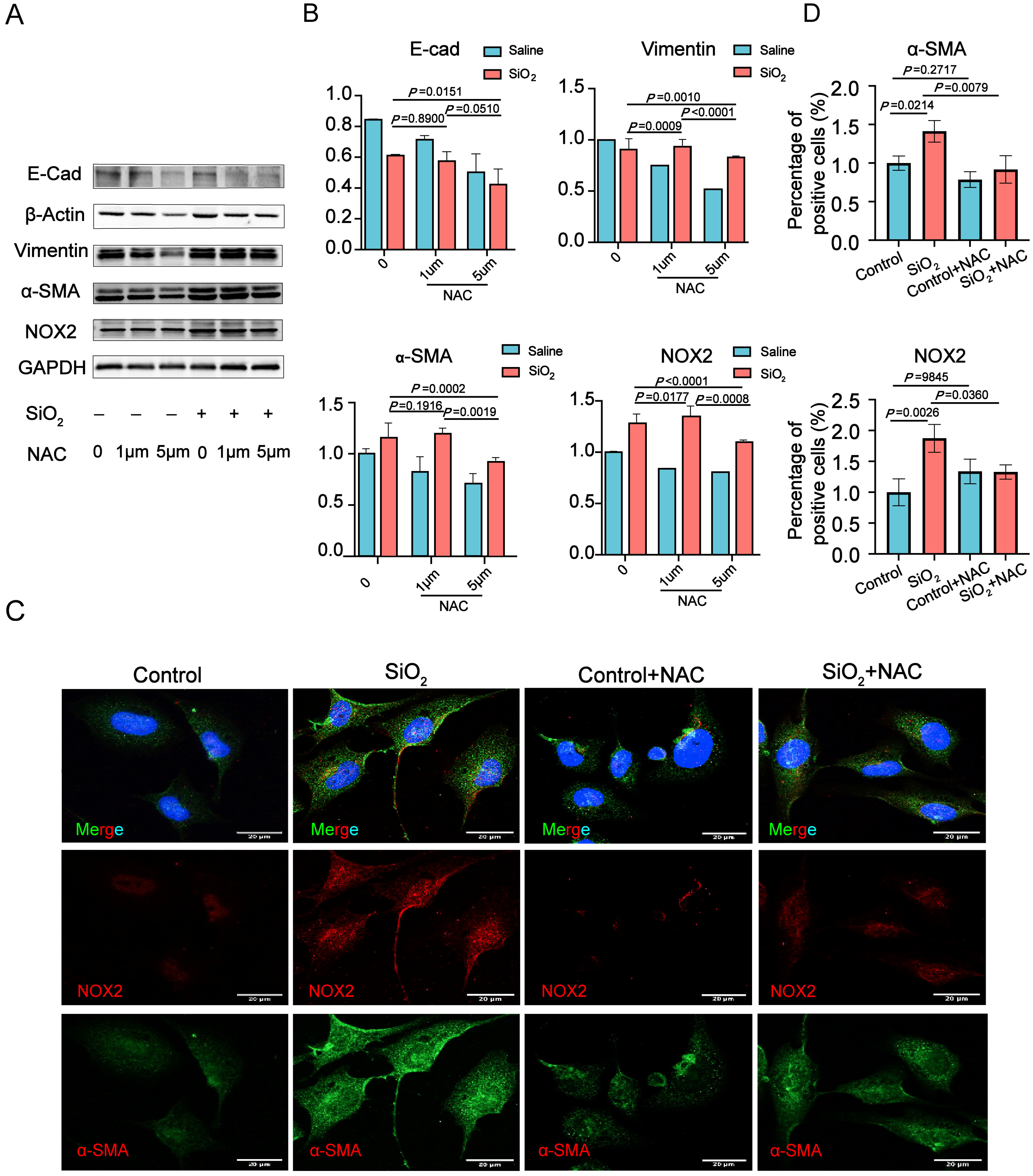
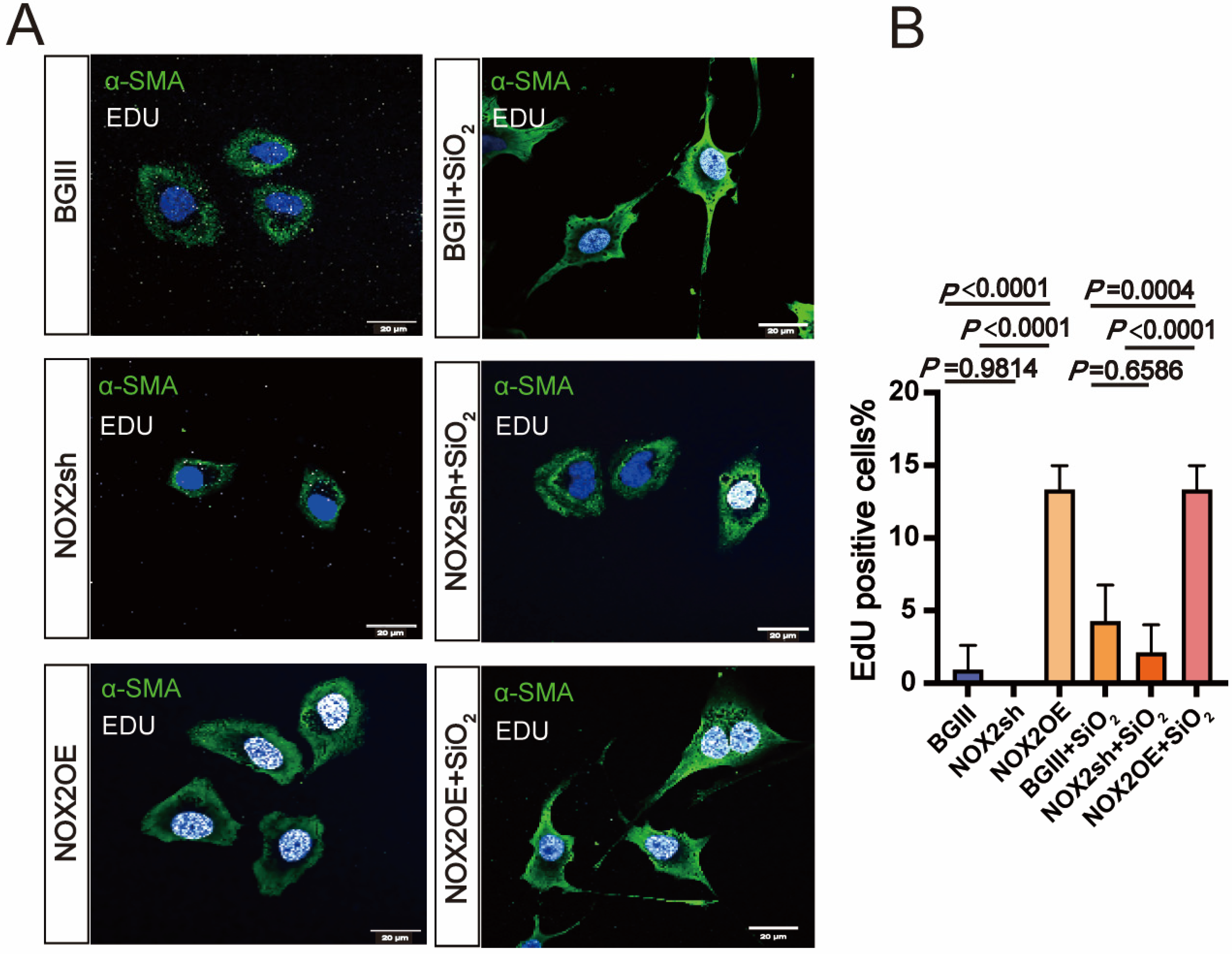
3.3. NOX2-Derived ROS Is Essential for the Epithelial-to-Mesenchymal Transition (EMT) in Human Lung Epithelial Cells Induced by SiO2
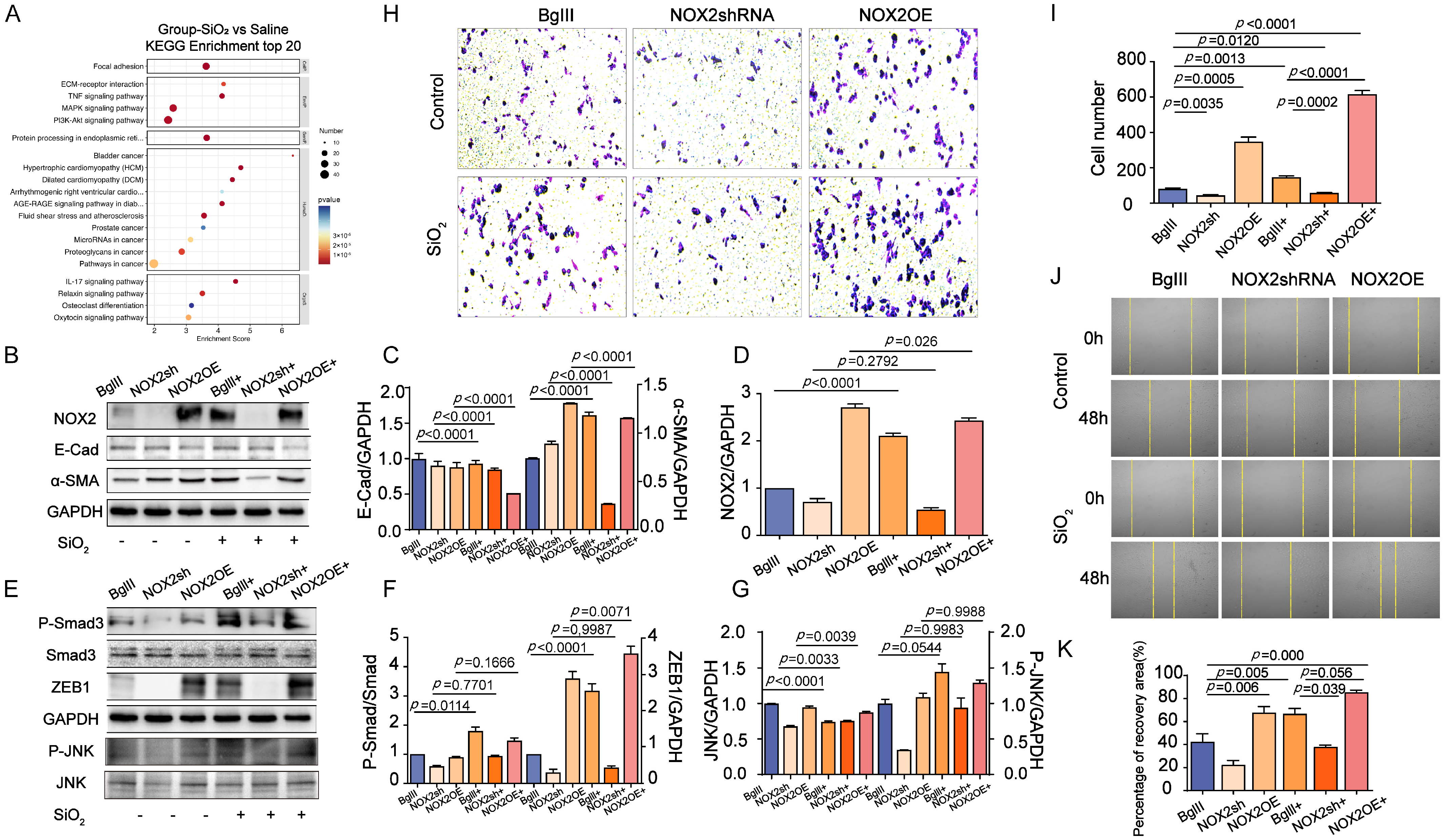
3.4. The Role of NOX2-Mediated JNK Signaling in Promoting EMT Transition in the Lung
3.5. Inhibition of JNK/C-Jun Signaling by SP600125 Effectively Reversed the Proliferation, Migration, and Invasion Capacities Induced by SiO2 in Human Epithelial Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vanka, K.S.; Shukla, S.; Gomez, H.M.; James, C.; Palanisami, T.; Williams, K.; Chambers, D.C.; Britton, W.J.; Ilic, D.; Hansbro, P.M.; et al. Understanding the pathogenesis of occupational coal and silica dust-associated lung disease. Eur. Respir. Rev. 2022, 31, 210250. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Shi, H.; Zhang, D.; Wang, C.; Zhao, F.; Li, L.; Xu, Z.; Jiang, J.; Li, J. Nebulized inhalation of LPAE-HDAC10 inhibits acetylation-mediated ROS/NF-κB pathway for silicosis treatment. J. Control. Release 2023, 364, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Abulikemu, A.; Zhao, X.; Xu, H.; Li, Y.; Ma, R.; Yao, Q.; Wang, J.; Sun, Z.; Li, Y.; Guo, C. Silica nanoparticles aggravated the metabolic associated fatty liver disease through disturbed amino acid and lipid metabolisms-mediated oxidative stress. Redox Biol. 2023, 59, 102569. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Chang, M.; Zhou, Q.; Zhang, L.; Wang, Y.; Guan, Y.; Li, H.; Zhao, Y.; Ding, C.; Hong, S.; et al. Activation of AMPK signalling by Metformin: Implication an important molecular mechanism for protecting against mice silicosis via inhibited endothelial cell-to-mesenchymal transition by regulating oxidative stress and apoptosis. Int. Immunopharmacol. 2023, 120, 110321. [Google Scholar] [CrossRef]
- Hegde, B.; Bodduluri, S.R.; Satpathy, S.R.; Alghsham, R.S.; Jala, V.R.; Uriarte, S.M.; Chung, D.H.; Lawrenz, M.B.; Haribabu, B. Inflammasome-Independent Leukotriene B4 Production Drives Crystalline Silica-Induced Sterile Inflammation. J. Immunol. 2018, 200, 3556–3567. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, Z.; Liu, J.; Song, M.; Zhang, T.; Chen, Y.; Hu, H.; Yang, P.; Li, B.; Song, X.; et al. Gefitinib and fostamatinib target EGFR and SYK to attenuate silicosis: A multi-omics study with drug exploration. Signal Transduct. Target. Ther. 2022, 7, 157. [Google Scholar] [CrossRef]
- Song, M.Y.; Wang, J.X.; Sun, Y.L.; Han, Z.F.; Zhou, Y.T.; Liu, Y.; Fan, T.H.; Li, Z.G.; Qi, X.M.; Luo, Y.; et al. Tetrandrine alleviates silicosis by inhibiting canonical and non-canonical NLRP3 inflammasome activation in lung macrophages. Acta Pharmacol. Sin. 2022, 43, 1274–1284. [Google Scholar] [CrossRef]
- Ni, S.; Li, D.; Wei, H.; Miao, K.S.; Zhuang, C. PPARγ Attenuates Interleukin-1β-Induced Cell Apoptosis by Inhibiting NOX2/ROS/p38MAPK Activation in Osteoarthritis Chondrocytes. Oxidative Med. Cell. Longev. 2021, 2021, 5551338. [Google Scholar] [CrossRef]
- Sul, O.J.; Ra, S.W. Quercetin Prevents LPS-Induced Oxidative Stress and Inflammation by Modulating NOX2/ROS/NF-kB in Lung Epithelial Cells. Molecules 2021, 26, 6949. [Google Scholar] [CrossRef]
- Bode, K.; Hauri-Hohl, M.; Jaquet, V.; Weyd, H. Unlocking the power of NOX2: A comprehensive review on its role in immune regulation. Redox Biol. 2023, 64, 102795. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jang, J.; Cha, S.-R.; Baek, H.; Lee, J.; Hong, S.-H.; Lee, H.-A.; Lee, T.-J.; Yang, S.-R. L-carnosine Attenuates Bleomycin-Induced Oxidative Stress via NFκB Pathway in the Pathogenesis of Pulmonary Fibrosis. Antioxidants 2022, 11, 2462. [Google Scholar] [CrossRef] [PubMed]
- Jarman, E.R.; Khambata, V.S.; Cope, C.; Jones, P.; Roger, J.; Ye, L.Y.; Duggan, N.; Head, D.; Pearce, A.; Press, N.J.; et al. An Inhibitor of NADPH Oxidase-4 Attenuates Established Pulmonary Fibrosis in a Rodent Disease Model. Am. J. Respir. Cell Mol. Biol. 2014, 50, 158–169. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Heather, L.C.; Luiken, J.J.F.P. CD36 as a gatekeeper of myocardial lipid metabolism and therapeutic target for metabolic disease. Physiol. Rev. 2024, 104, 727–764. [Google Scholar] [CrossRef]
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Rodari, M.M.; Cerf-Bensussan, N.; Parlato, M. Dysregulation of the immune response in TGF-β signalopathies. Front. Immunol. 2022, 13, 1066375. [Google Scholar] [CrossRef]
- Shao, M.; Wang, Y.; Dong, H.; Wang, L.; Zhang, X.; Han, X.; Sang, X.; Bao, Y.; Peng, M.; Cao, G. From liver fibrosis to hepatocarcinogenesis: Role of excessive liver H2O2 and targeting nanotherapeutics. Bioact. Mater. 2023, 23, 187–205. [Google Scholar] [CrossRef]
- Weng, M.S.; Chang, J.H.; Hung, W.Y.; Yang, Y.C.; Chien, M.H. The interplay of reactive oxygen species and the epidermal growth factor receptor in tumor progression and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 61. [Google Scholar] [CrossRef]
- Li, C.; Du, S.; Lu, Y.; Lu, X.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Blocking the 4-1BB Pathway Ameliorates Crystalline Silica-induced Lung Inflammation and Fibrosis in Mice. Theranostics 2016, 6, 2052–2067. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, M.; Wang, Z.; Cheng, Y.; Liu, H.; Zhou, Z.; Han, B.; Chen, B.; Yao, H.; Chao, J. Neogambogic acid prevents silica-induced fibrosis via inhibition of high-mobility group box 1 and MCP-1-induced protein 1. Toxicol. Appl. Pharmacol. 2016, 309, 129–140. [Google Scholar] [CrossRef] [PubMed]
- DePianto, D.J.; Chandriani, S.; Abbas, A.R.; Jia, G.; N’Diaye, E.N.; Caplazi, P.; Kauder, S.E.; Biswas, S.; Karnik, S.K.; Ha, C.; et al. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 2015, 70, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, M.; Li, Z.; Hou, L.; Zhang, H.; Zhang, Z.; Hu, H.; Jiang, X.; Yang, J.; Zou, X.; et al. FcεRI deficiency alleviates silica-induced pulmonary inflammation and fibrosis. Ecotoxicol. Environ. Saf. 2022, 244, 114043. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Guo, H.; Cheng, Y.; Zhou, Z.; Zhang, W.; Han, B.; Luo, W.; Wang, J.; Xie, W.; Chao, J. circHECTD1 promotes the silica-induced pulmonary endothelial–mesenchymal transition via HECTD1. Cell Death Dis. 2018, 9, 396. [Google Scholar] [CrossRef]
- Keskinidou, C.; Vassiliou, A.; Dimopoulou, I.; Kotanidou, A.; Orfanos, S. Mechanistic Understanding of Lung Inflammation: Recent Advances and Emerging Techniques. J. Inflamm. Res. 2022, 15, 3501–3546. [Google Scholar] [CrossRef]
- Nocella, C.; D’Amico, A.; Cammisotto, V.; Bartimoccia, S.; Castellani, V.; Loffredo, L.; Marini, L.; Ferrara, G.; Testa, M.; Motta, G.; et al. Structure, Activation, and Regulation of NOX2: At the Crossroad between the Innate Immunity and Oxidative Stress-Mediated Pathologies. Antioxidants 2023, 12, 429. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H Oxidase Mediates TGF-β1–Induced Activation of Kidney Myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102. [Google Scholar] [CrossRef]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef]
- Sisto, M.; Ribatti, D.; Lisi, S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in TGFβ-Dependent EMT. Biomolecules 2021, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Fu, P.; Ma, L. Kidney fibrosis: From mechanisms to therapeutic medicines. Signal Transduct. Target. Ther. 2023, 8, 129. [Google Scholar] [CrossRef]
- Zeng, J.; Li, D.; Li, Z.; Zhang, J.; Zhao, X. Dendrobium officinale Attenuates Myocardial Fibrosis via Inhibiting EMT Signaling Pathway in HFD/STZ-Induced Diabetic Mice. Biol. Pharm. Bull. 2020, 43, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K. NADPH oxidase-derived reactive oxygen species: Dosis facit venenum. Exp. Physiol. 2019, 104, 447–452. [Google Scholar] [CrossRef]
- Yu, P.; Li, J.; Jiang, J.; Zhao, Z.; Hui, Z.; Zhang, J.; Zheng, Y.; Ling, D.; Wang, L.; Jiang, L.H.; et al. A dual role of transient receptor potential melastatin 2 channel in cytotoxicity induced by silica nanoparticles. Sci. Rep. 2015, 5, 18171. [Google Scholar] [CrossRef]
- Wu, Z.; Hou, Q.; Chen, T.; Jiang, X.; Wang, L.; Xu, J.; Wang, L. ROS-reactive PMS/PC drug delivery system improves new bone formation under diabetic conditions by promoting angiogenesis-osteogenesis coupling via down-regulating NOX2-ROS signalling axis. Biomaterials 2022, 291, 121900. [Google Scholar] [CrossRef]
- Grauers Wiktorin, H.; Aydin, E.; Hellstrand, K.; Martner, A.; Ghose, J. NOX2-Derived Reactive Oxygen Species in Cancer. Oxidative Med. Cell. Longev. 2020, 2020, 7095902. [Google Scholar] [CrossRef]
- Liu, M.C.; Blumenthal, M.N.; Walton-Bowen, K. Zileuton Provided Clinically Relevant Reductions in the Need for Rescue Medication and Oral Corticosteroids Compared to Placebo in Moderate and Severe Asthmatics. Chest 2005, 128, 245S–596S. [Google Scholar] [CrossRef]
- Carnesecchi, S.; Pache, J.-C.; Barazzone-Argiroffo, C. NOX enzymes: Potential target for the treatment of acute lung injury. Cell. Mol. Life Sci. 2012, 69, 2373–2385. [Google Scholar] [CrossRef]
- Zhang, W.X.; Kubes, P. NOX2 is the best defense a good offense. Blood 2022, 139, 2851–2853. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L. Mechanisms and consequences of oxidative stress in lung disease: Therapeutic implications for an aging populace. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L642–L653. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, S.; Deffert, C.; Pagano, A.; Garrido-Urbani, S.; Métrailler-Ruchonnet, I.; Schäppi, M.; Donati, Y.; Matthay, M.A.; Krause, K.-H.; Barazzone Argiroffo, C. NADPH Oxidase-1 Plays a Crucial Role in Hyperoxia-induced Acute Lung Injury in Mice. Am. J. Respir. Crit. Care Med. 2009, 180, 972–981. [Google Scholar] [CrossRef]
- Gautam, J.; Ku, J.-M.; Regmi, S.C.; Jeong, H.; Wang, Y.; Banskota, S.; Park, M.-H.; Nam, T.-g.; Jeong, B.-S.; Kim, J.-A. Dual Inhibition of NOX2 and Receptor Tyrosine Kinase by BJ-1301 Enhances Anticancer Therapy Efficacy via Suppression of Autocrine-Stimulatory Factors in Lung Cancer. Mol. Cancer Ther. 2017, 16, 2144–2156. [Google Scholar] [CrossRef]
- Hu, C.; Wu, Z.; Huang, Z.; Hao, X.; Wang, S.; Deng, J.; Yin, Y.; Tan, C. Nox2 impairs VEGF-A-induced angiogenesis in placenta via mitochondrial ROS-STAT3 pathway. Redox Biol. 2021, 45, 102051. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.-y.; Wang, Y.; Zhao, W.-x.; Li, F.-d.; Guo, P.-r.; Fan, Q.; Wu, X.-f. NOX2 inhibition stabilizes vulnerable plaques by enhancing macrophage efferocytosis via MertK/PI3K/AKT pathway. Redox Biol. 2023, 64, 102763. [Google Scholar] [CrossRef]
- Chen, C.; Yang, S.; Zhang, M.; Zhang, Z.; Hong, J.; Han, D.; Ma, J.; Zhang, S.B.; Okunieff, P.; Zhang, L. Triptolide mitigates radiation-induced pulmonary fibrosis via inhibition of axis of alveolar macrophages-NOXes-ROS-myofibroblasts. Cancer Biol. Ther. 2016, 17, 381–389. [Google Scholar] [CrossRef]
- Xiong, Y.; Cui, X.; Zhou, Y.; Chai, G.; Jiang, X.; Ge, G.; Wang, Y.; Sun, H.; Che, H.; Nie, Y.; et al. Dehydrocostus lactone inhibits BLM-induced pulmonary fibrosis and inflammation in mice via the JNK and p38 MAPK-mediated NF-κB signaling pathways. Int. Immunopharmacol. 2021, 98, 107780. [Google Scholar] [CrossRef]
- Ahmad, R. Induction of ROS-mediated cell death and activation of the JNK pathway by a sulfonamide derivative. Int. J. Mol. Med. 2019, 44, 1552–1562. [Google Scholar] [CrossRef]
- Li, X.; Ye, C.; Mulati, M.; Sun, L.; Qian, F. Ellipticine blocks synergistic effects of IL-17A and TNF-α in epithelial cells and alleviates severe acute pancreatitis-associated acute lung injury. Biochem. Pharmacol. 2020, 177, 113992. [Google Scholar] [CrossRef]
- Ma, W.; Huang, Q.; Xiong, G.; Deng, L.; He, Y. The protective effect of Hederagenin on pulmonary fibrosis by regulating the Ras/JNK/NFAT4 axis in rats. Biosci. Biotechnol. Biochem. 2020, 84, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, C.D.; Wu, R.F.; Terada, L.S. ROS signaling and ER stress in cardiovascular disease. Mol. Asp. Med. 2018, 63, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xiang, Y.; Xu, X.; Fang, D.; Li, D.; Ni, F.; Zhu, X.; Chen, B.; Zhou, M. High Glucose-Induced ROS Production Stimulates Proliferation of Pancreatic Cancer via Inactivating the JNK Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 6917206. [Google Scholar] [CrossRef]
- Shen, C.; Jiang, Y.; Lin, J.; He, Y.; Liu, Y.; Fang, D. Purinergic receptor P2X7 activates NOX2/JNK signaling to participate in granulosa cell inflammation and apoptosis in polycystic ovary syndrome. J. Bioenerg. Biomembr. 2023, 55, 313–322. [Google Scholar] [CrossRef]
- Seo, S.Y.; Joo, S.H.; Lee, S.-O.; Yoon, G.; Cho, S.-S.; Choi, Y.H.; Park, J.W.; Shim, J.-H. Activation of p38 and JNK by ROS Contributes to Deoxybouvardin-Mediated Intrinsic Apoptosis in Oxaliplatin-Sensitive and -Resistant Colorectal Cancer Cells. Antioxidants 2024, 13, 866. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Az-Zahra, F.; Wongso, H.; Setyawati, L.U.; Novitasari, D.; Ikram, E.H.K. Molecular Mechanism of Natural Food Antioxidants to Regulate ROS in Treating Cancer: A Review. Antioxidants 2024, 13, 207. [Google Scholar] [CrossRef]
- Solinas, G.; Karin, M. JNK1 and IKKβ: Molecular links between obesity and metabolic dysfunction. FASEB J. 2010, 24, 2596–2611. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Cat # | Dilution Ratio | Company | ||
|---|---|---|---|---|---|
| WB | IHC | IF | |||
| α-SMA | Ab5694 | 1:2000 | - | 1:200 | Abcam, Cambridge, MA, USA |
| NOX2 | DF6520 | 1:1000 | - | 1:200 | Affinity, Changzhou, China |
| JNK | AF6318 | 1:1000 | - | - | Affinity, Cincinnati, OH, USA |
| P-JNK | AF3318 | 1:1000 | - | - | Affinity, Cincinnati, OH, USA |
| P-C-Jun | AF3095 | 1:1000 | - | - | Affinity, Cincinnati, OH, USA |
| E-Cad | Ab76055 | 1:1000 | - | - | Abcam, Cambridge, MA, USA |
| E-Cad | AF0131 | 1:2000 | - | - | Affinity, Changzhou, China |
| Vimentin | Ab92547 | 1:1000 | - | 1:200 | Abcam, Cambridge, MA, USA |
| Smad2/3 | #8685 | 1:1000 | - | - | Cell Signaling Technology (CST), Danvers, MA, USA |
| P-Smad2/3 | #8828 | 1:1000 | - | - | Cell Signaling Technology (CST), Danvers, MA, USA |
| Zeb1 | DF7414 | 1:1000 | - | - | Affinity, Cincinnati, OH, USA |
| anti-GAPDH | AF7021 | 1:2000 | - | - | Affinity, Changzhou, China |
| Goat anti-rabbit IgG (H + L)/HRP antibody | S0001 | 1:1000 | - | - | Affinity, Cincinnati, OH, USA |
| Goat anti-mouse IgG (H + L)/HRP antibody | S0002 | 1:1000 | - | - | Abcam, Cambridge, MA, USA |
| Anti-collagen 1 antibody | Ab34710 | 1:5000 | - | - | Abcam, Cambridge, MA, USA |
| 488-conjugated donkey anti-mouse | 711-545-150 | - | - | 1:200 | Jackson ImmunoResearch, West Grove, PA, USA |
| 647-conjugated donkey anti-rabbit | 711-605-152 | - | - | 1:200 | Jackson ImmunoResearch, West Grove, PA, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, G.; Gong, L.; Wang, K.; Sun, X.; Liu, Z.; Cai, Q. Suppression of NOX2-Derived Reactive Oxygen Species (ROS) Reduces Epithelial-to-MesEnchymal Transition Through Blocking SiO2-Regulated JNK Activation. Toxics 2025, 13, 365. https://doi.org/10.3390/toxics13050365
Xiang G, Gong L, Wang K, Sun X, Liu Z, Cai Q. Suppression of NOX2-Derived Reactive Oxygen Species (ROS) Reduces Epithelial-to-MesEnchymal Transition Through Blocking SiO2-Regulated JNK Activation. Toxics. 2025; 13(5):365. https://doi.org/10.3390/toxics13050365
Chicago/Turabian StyleXiang, Guanhan, Liang Gong, Kai Wang, Xiaobo Sun, Zhihong Liu, and Qian Cai. 2025. "Suppression of NOX2-Derived Reactive Oxygen Species (ROS) Reduces Epithelial-to-MesEnchymal Transition Through Blocking SiO2-Regulated JNK Activation" Toxics 13, no. 5: 365. https://doi.org/10.3390/toxics13050365
APA StyleXiang, G., Gong, L., Wang, K., Sun, X., Liu, Z., & Cai, Q. (2025). Suppression of NOX2-Derived Reactive Oxygen Species (ROS) Reduces Epithelial-to-MesEnchymal Transition Through Blocking SiO2-Regulated JNK Activation. Toxics, 13(5), 365. https://doi.org/10.3390/toxics13050365