
The Relationship of Glutathione-S-Transferase and Multi-Drug Resistance-Related Protein 1 in Nitric Oxide (NO) Transport and Storage


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. General Biology of Nitric Oxide
2. The Nitric Oxide Synthase Family of Enzymes
3. Physiological Roles of NO
3.1. NO and S-Nitrosylation
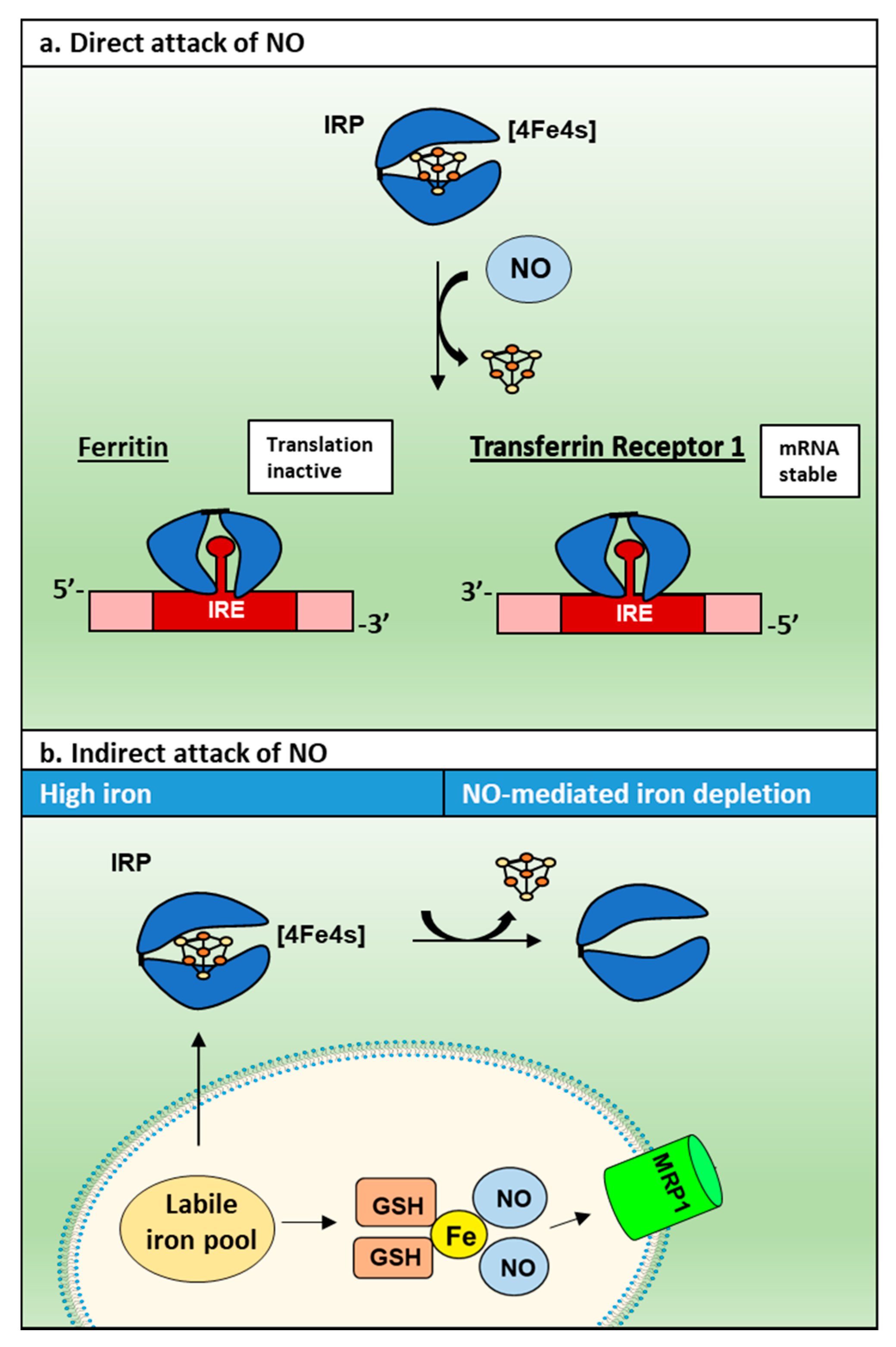
3.2. NO and Its Interaction with Cellular Iron
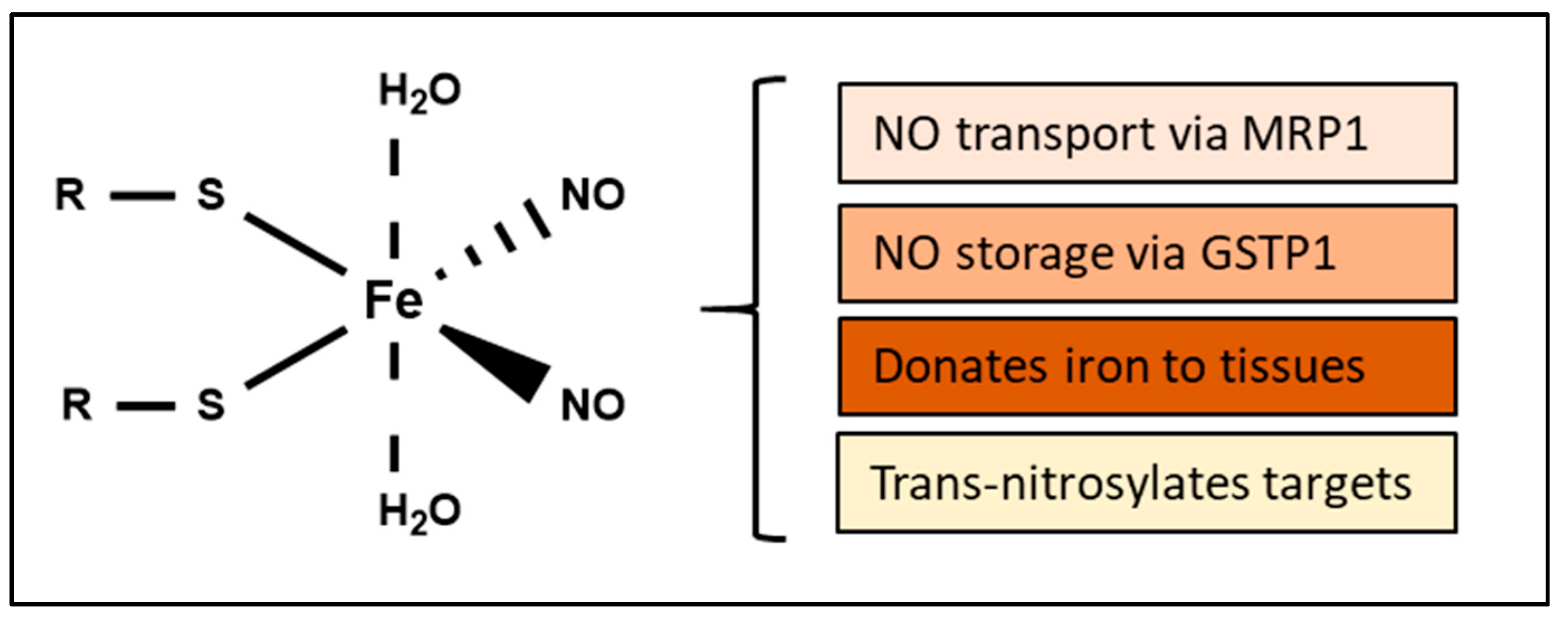
4. Biological Functions of DNICs
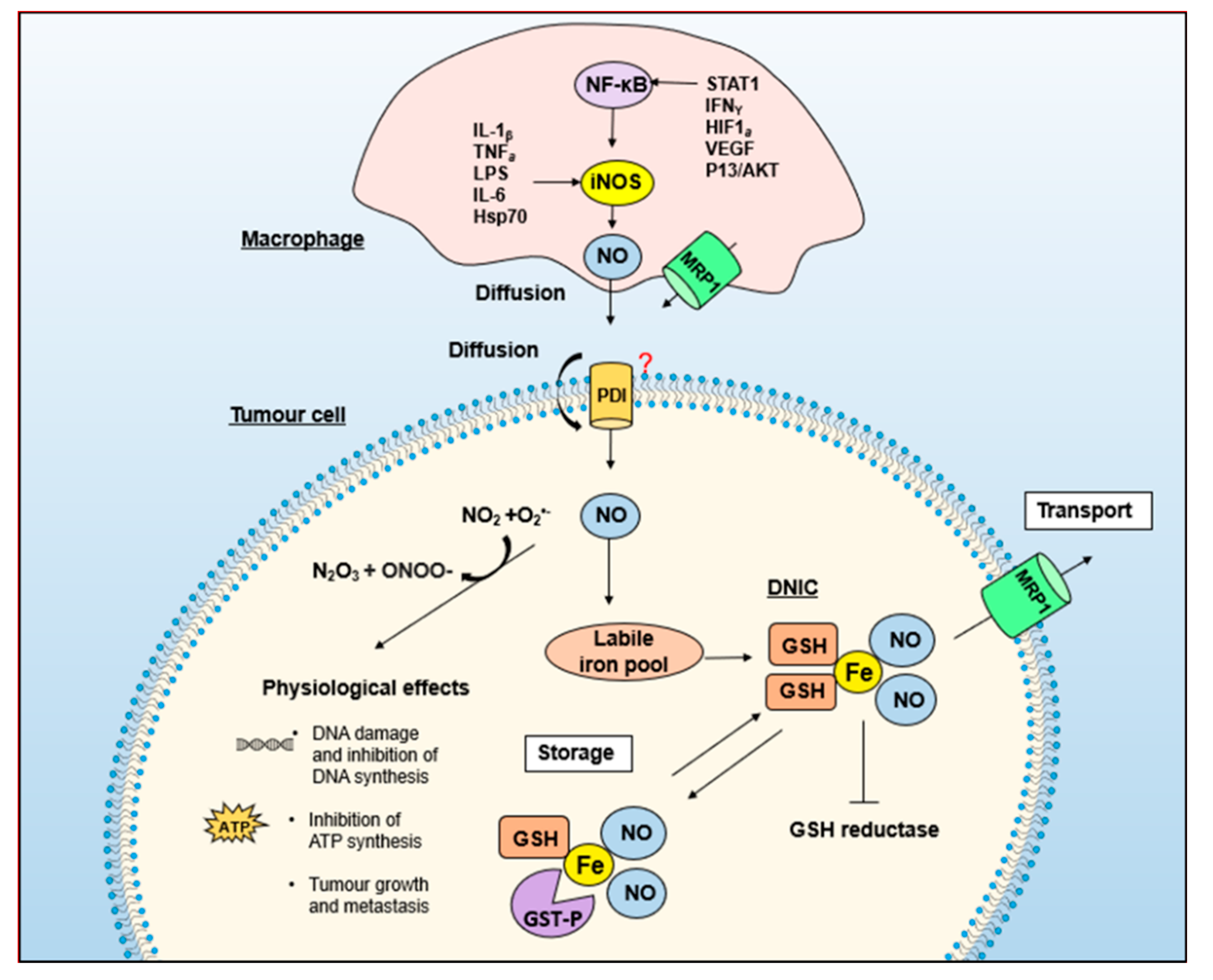
5. GSH and Energy Metabolism Are Essential for NO-Induced Iron Efflux from Cells
6. Multi-Drug Resistance-Related Protein 1 (MRP1) Mediates the Release of Iron and GSH from Cells as DNICs
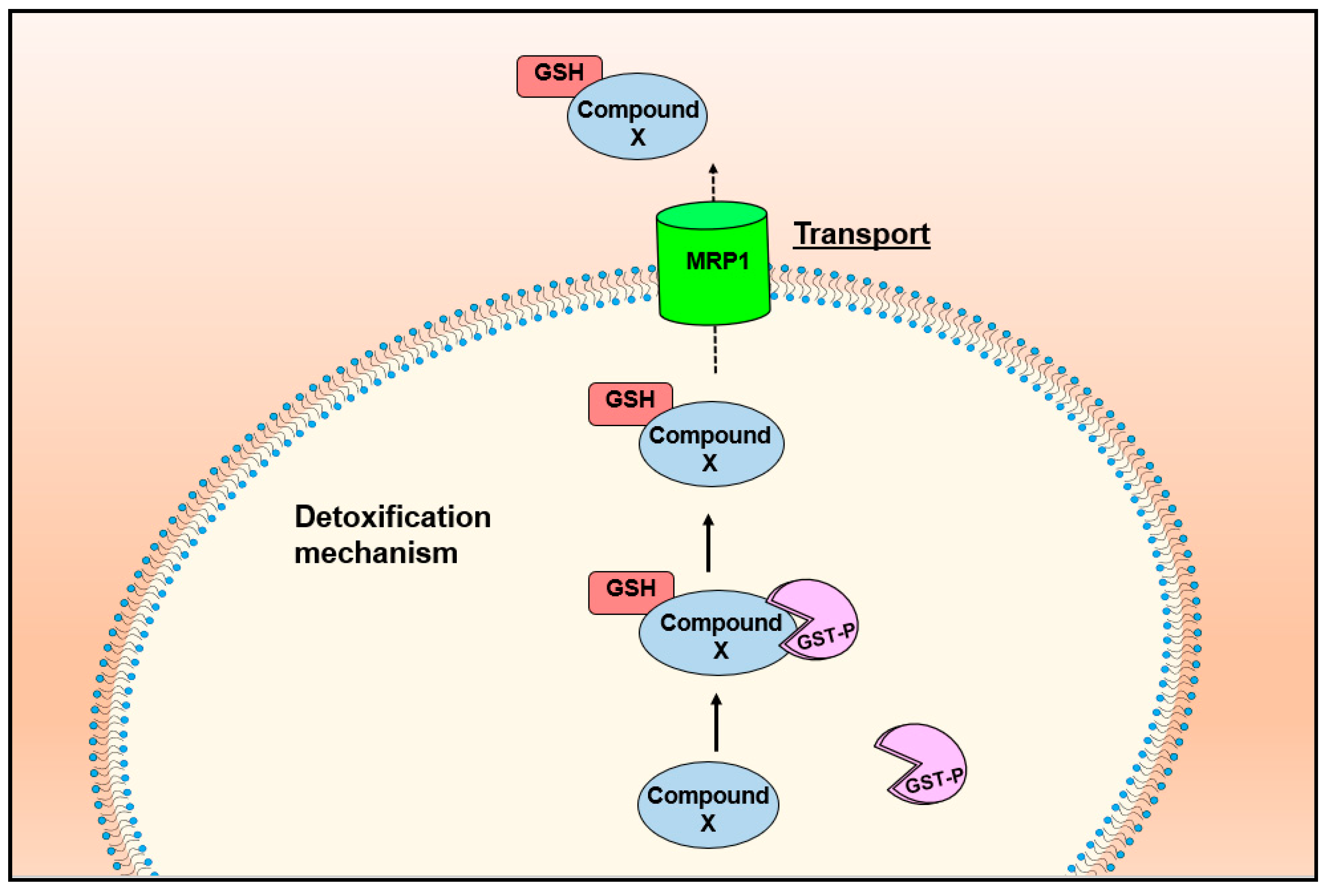
7. MRP1 Forms an Integrated Detoxification System with GSTs in Drug Resistance
8. The Potential Intermediary or Storage Role of DNICs by GST Enzymes
9. Implications of the MRP1–GST Interaction for Understanding NO Biology: DNICs as a Common Currency for the Storage and Transport of NO
10. Summary: DNIC Storage and Transport by GST and MRP1: NO and Beyond
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| AP1 | Activator protein 1 |
| ATF2 | Activating transcription factor 2 |
| BH4 | (6R-)5,6,7,8-tetrahydrobiopterin |
| CaM | Calmodulin |
| Cav-1 | Caveolin-1 |
| cGMP | Cyclic guanosine monophosphate |
| CN− | Cyanide |
| cNOS | Constitutive nitric oxide synthase |
| CNS | Central nervous system |
| CO | Carbon monoxide |
| cP450 | Cytochrome P450 |
| Cys | Cysteine |
| DMT1 | Divalent metal transporter 1 |
| DNIC | Dinitrosyl-dithiol-iron-complexes |
| EGF | Endothelial growth factor |
| eNOS | Endothelial nitric oxide synthase |
| EPR | Electron paramagnetic resonance |
| FAD | Flavin adenine dinucleotide |
| Fe | Iron |
| FMN | Flavin mononucleotide |
| GlcN | Glucosamine |
| GPXs | Glutathione peroxidase |
| GSH | Glutathione |
| GSNO | S-nitroglutathione |
| GST | Glutathione-S-Transferase |
| HSP | Heat shock protein |
| IL-1β | Interleukin-1β |
| iNOS | Inducible nitric oxide synthase |
| IRE | Iron regulatory element |
| IRP | Iron regulatory protein |
| LDL | Low-density lipoprotein |
| LPS | Lipopolysaccharide |
| MRP1 | Multi-drug resistance related protein 1 |
| NADPH | Nicotinamide-adenine-dinucleotide phosphate |
| NDRG1 | N-myc downstream regulated gene-1 |
| NF-κB | Nuclear factor-κB |
| NO | Nitric Oxide |
| NO− | Nitroxyl anion |
| NO+ | Nitrosonium cation |
| NO• | Nitric oxide radical |
| NOS | Nitric oxide synthase |
| PC-PLC | Phosphatidylcholine-specific phospholipase C |
| PDTC | Pyrrolidine dithiocarbamate |
| PPP | Pentose phosphate pathway |
| RNOS | Reactive nitric oxide species |
| ROS | Reactive oxygen species |
| Ser | Serine |
| sGC | Soluble guanylate cyclase |
| SOD | Superoxide dismutase |
| TCA | Tricarboxylic acid |
| Tf | Transcription factor |
| TfR1 | Transferrin receptor 1 |
| TGF-β1 | Transforming growth factor beta 1 |
| TNF-α | Tumor necrosis factor alpha |
| Tyr | Tyrosine |
| UTRs | Untranslated regions |
References
- McCleverty, J.A. Chemistry of Nitric Oxide Relevant to Biology. Chem. Rev. 2004, 104, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Rosselli, M.; Keller, R.; Dubey, R. Role of Nitric Oxide in the Biology, Physiology and Pathophysiology of Reproduction. Hum. Reprod. Update 1998, 4, 3–24. [Google Scholar] [CrossRef]
- Rajfer, J.; Aronson, W.J.; Bush, P.A.; Dorey, F.J.; Ignarro, L.J. Nitric Oxide as a Mediator of Relaxation of the Corpus Cavernosum in Response to Nonadrenergic, Noncholinergic Neurotransmission. N. Engl. J. Med. 1992, 326, 90–94. [Google Scholar] [CrossRef]
- Sanders, K.M.; Ward, S.M. Nitric Oxide as a Mediator of Nonadrenergic Noncholinergic Neurotransmission. Am. J. Physiol. Gastrointest. Liver Physiol. 1992, 262, G379–G392. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Li, Y.; Moss, D.; Parkinson, C.; Rogers, M.V.; Moncada, S. Resistance to Leishmania Major Infection Correlates with the Induction of Nitric Oxide Synthase in Murine Macrophages. Eur. J. Immunol. 1991, 21, 3009–3014. [Google Scholar] [CrossRef]
- Nappi, A.J.; Vass, E.; Frey, F.; Carton, Y. Nitric Oxide Involvement in Drosophila Immunity. Nitric Oxide 2000, 4, 423–430. [Google Scholar] [CrossRef]
- Pavanelli, W.R.; Gutierrez, F.R.S.; da Silva, J.J.N.; Costa, I.C.; de Menezes, M.C.N.D.; de Abreu Oliveira Msc, F.J.; Itano, E.N.; Watanabe, M.A.E. The Effects of Nitric Oxide on the Immune Response during Giardiasis. Braz. J. Infect. Dis. 2010, 14, 606–612. [Google Scholar] [CrossRef]
- Rees, D.D.; Palmer, R.M.; Moncada, S. Role of Endothelium-Derived Nitric Oxide in the Regulation of Blood Pressure. Proc. Natl. Acad. Sci. USA 1989, 86, 3375–3378. [Google Scholar] [CrossRef] [PubMed]
- Tatemoto, K.; Takayama, K.; Zou, M.-X.; Kumaki, I.; Zhang, W.; Kumano, K.; Fujimiya, M. The Novel Peptide Apelin Lowers Blood Pressure via a Nitric Oxide-Dependent Mechanism. Regul. Pept. 2001, 99, 87–92. [Google Scholar] [CrossRef]
- Lala, P.K.; Chakraborty, C. Role of Nitric Oxide in Carcinogenesis and Tumour Progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Ohshima, H.; Bartsch, H. Chronic Infections and Inflammatory Processes as Cancer Risk Factors: Possible Role of Nitric Oxide in Carcinogenesis. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 1994, 305, 253–264. [Google Scholar] [CrossRef]
- Albina, J.E.; Reichner, J.S. Role of Nitric Oxide in Mediation of Macrophage Cytotoxicity and Apoptosis. Cancer Metastasis Rev. 1998, 17, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z. Iron Depletion: Possible Cause of Tumor Cell Cytotoxicity Induced by Activated Macrophages. Biochem. Biophys. Res. Commun. 1984, 123, 716–723. [Google Scholar] [CrossRef]
- Richardson, D.; Ponka, P. The Molecular Mechanisms of the Metabolism and Transport of Iron in Normal and Neoplastic Cells. Biochim. Biophys. Acta 1997, 1331, 1–40. [Google Scholar] [CrossRef]
- Toledo, J.C.; Bosworth, C.A.; Hennon, S.W.; Mahtani, H.A.; Bergonia, H.A.; Lancaster, J.R. Nitric Oxide-Induced Conversion of Cellular Chelatable Iron into Macromolecule-Bound Paramagnetic Dinitrosyliron Complexes. J. Biol. Chem. 2008, 283, 28926–28933. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Li, H.; Poulos, T.L. Structure–Function Studies on Nitric Oxide Synthases. J. Inorg. Biochem. 2005, 99, 293–305. [Google Scholar] [CrossRef]
- Kröncke, K.-D.; Fehsel, K.; Suschek, C.; Kolb-Bachofen, V. Inducible Nitric Oxide Synthase-Derived Nitric Oxide in Gene Regulation, Cell Death and Cell Survival. Int. Immunopharmacol. 2001, 1, 1407–1420. [Google Scholar] [CrossRef]
- Kuchan, M.J.; Frangos, J.A. Role of Calcium and Calmodulin in Flow-Induced Nitric Oxide Production in Endothelial Cells. Am. J. Physiol. Cell Physiol. 1994, 266, C628–C636. [Google Scholar] [CrossRef] [PubMed]
- Schuh, K.; Uldrijan, S.; Telkamp, M.; Röthlein, N.; Neyses, L. The Plasmamembrane Calmodulin–Dependent Calcium Pump : A Major Regulator of Nitric Oxide Synthase, I. J. Cell Biol. 2001, 155, 201–206. [Google Scholar] [CrossRef]
- Deguchi, T.; Yoshioka, M. l-arginine Identified as an Endogenous Activator for Soluble Guanylate Cyclase from Neuroblastoma Cells. J. Biol. Chem. 1982, 257, 10147–10151. [Google Scholar] [CrossRef]
- Chou, W.-B.; Zeng, Y.-M.; Duan, S.-M.; Zhou, W.-H.; Gu, J.; Yang, G.-D. M2 Muscarinic Receptor of Spinal Cord Mediated Increase of NNOS Expression in Locus Coeruleus during Morphine Withdrawal. Acta Pharm. Sin. 2002, 23, 691–697. [Google Scholar]
- Catania, M.V.; Aronica, E.; Yankaya, B.; Troost, D. Increased Expression of Neuronal Nitric Oxide Synthase Spliced Variants in Reactive Astrocytes of Amyotrophic Lateral Sclerosis Human Spinal Cord. J. Neurosci. 2001, 21, RC148. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Chee, C.B.; Gaston, B.; Lilly, C.M.; Gerard, C.; Drazen, J.M.; Stamler, J.S. Constitutive and Inducible Nitric Oxide Synthase Gene Expression, Regulation, and Activity in Human Lung Epithelial Cells. Proc. Natl. Acad. Sci. USA 1994, 91, 10089–10093. [Google Scholar] [CrossRef] [PubMed]
- Shaul, P.W.; North, A.J.; Wu, L.C.; Wells, L.B.; Brannon, T.S.; Lau, K.S.; Michel, T.; Margraf, L.R.; Star, R.A. Endothelial Nitric Oxide Synthase Is Expressed in Cultured Human Bronchiolar Epithelium. J. Clin. Investig. 1994, 94, 2231–2236. [Google Scholar] [CrossRef] [PubMed]
- Cymeryng, C.B.; Lotito, S.P.; Colonna, C.; Finkielstein, C.; Pomeraniec, Y.; Grión, N.; Gadda, L.; Maloberti, P.; Podestá, E.J. Expression of Nitric Oxide Synthases in Rat Adrenal Zona Fasciculata Cells. Endocrinology 2002, 143, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric Oxide Synthase Isozymes. Characterization, Purification, Molecular Cloning, and Functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef]
- Lajoix, A.-D.; Reggio, H.; Chardès, T.; Péraldi-Roux, S.; Tribillac, F.; Roye, M.; Dietz, S.; Broca, C.; Manteghetti, M.; Ribes, G.; et al. A Neuronal Isoform of Nitric Oxide Synthase Expressed in Pancreatic β-Cells Controls Insulin Secretion. Diabetes 2001, 50, 1311–1323. [Google Scholar] [CrossRef]
- Brophy, C.M.; Knoepp, L.; Xin, J.; Pollock, J.S. Functional Expression of NOS 1 in Vascular Smooth Muscle. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H991–H997. [Google Scholar] [CrossRef]
- Boulanger, C.M.; Heymes, C.; Benessiano, J.; Geske, R.S.; Lévy, B.I.; Vanhoutte, P.M. Neuronal Nitric Oxide Synthase Is Expressed in Rat Vascular Smooth Muscle Cells. Circ. Res. 1998, 83, 1271–1278. [Google Scholar] [CrossRef]
- Macnaul, K.L.; Hutchinson, N.I. Differential Expression of INOS and CNOS MRNA in Human Vascular Smooth Muscle Cells and Endothelial Cells under Normal and Inflammatory Conditions. Biochem. Biophys. Res. Commun. 1993, 196, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Böhme, G.A.; Bon, C.; Lemaire, M.; Reibaud, M.; Piot, O.; Stutzmann, J.M.; Doble, A.; Blanchard, J.C. Altered Synaptic Plasticity and Memory Formation in Nitric Oxide Synthase Inhibitor-Treated Rats. Proc. Natl Acad Sci USA 1993, 90, 9191–9194. [Google Scholar] [CrossRef]
- Hölscher, C.; Rose, S.P. An Inhibitor of Nitric Oxide Synthesis Prevents Memory Formation in the Chick. Neurosci. Lett. 1992, 145, 165–167. [Google Scholar] [CrossRef]
- Togashi, H.; Sakuma, I.; Yoshioka, M.; Kobayashi, T.; Yasuda, H.; Kitabatake, A.; Saito, H.; Gross, S.S.; Levi, R. A Central Nervous System Action of Nitric Oxide in Blood Pressure Regulation. J. Pharm. Exp. 1992, 262, 343–347. [Google Scholar]
- El Karib, A.O.; Sheng, J.; Betz, A.L.; Malvin, R.L. The Central Effects of a Nitric Oxide Synthase Inhibitor (N Omega-Nitro-l-arginine) on Blood Pressure and Plasma Renin. Clin. Exp. Hypertens 1993, 15, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Jurzik, L.; Froh, M.; Straub, R.H.; Schölmerich, J.; Wiest, R. Up-Regulation of NNOS and Associated Increase in Nitrergic Vasodilation in Superior Mesenteric Arteries in Pre-Hepatic Portal Hypertension. J. Hepatol. 2005, 43, 258–265. [Google Scholar] [CrossRef]
- Schwarz, P.M.; Kleinert, H.; Förstermann, U. Potential Functional Significance of Brain-Type and Muscle-Type Nitric Oxide Synthase I Expressed in Adventitia and Media of Rat Aorta. Arter. Thromb. Vasc. Biol. 1999, 19, 2584–2590. [Google Scholar] [CrossRef] [PubMed]
- Vannini, F.; Kashfi, K.; Nath, N. The Dual Role of INOS in Cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef]
- Regenga, F.C.; Palandri, C.A.C.; Catelli, C.M.H.; Paula, D.A.; Biscegli, J.M.; Carlos, B.d.L.; Protásio, L.d. Influence of Hypoxia on Nitric Oxide Synthase Activity and Gene Expression in Children with Congenital Heart Disease. Circulation 2001, 103, 2272–2276. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Q.; Yuan, X.; Wang, T.; Luo, S.; Lei, H.; Xia, Y. Requirement of Heat Shock Protein 70 for Inducible Nitric Oxide Synthase Induction. Cell. Signal. 2013, 25, 1310–1317. [Google Scholar] [CrossRef]
- Ganster, R.W.; Taylor, B.S.; Shao, L.; Geller, D.A. Complex Regulation of Human Inducible Nitric Oxide Synthase Gene Transcription by Stat 1 and NF-ΚB. Proc. Natl. Acad. Sci. USA 2001, 98, 8638–8643. [Google Scholar] [CrossRef]
- Kleinert, H.; Wallerath, T.; Fritz, G.; Ihrig-Biedert, I.; Rodriguez-Pascual, F.; Geller, D.A.; Forstermann, U. Cytokine Induction of NO Synthase II in Human DLD-1 Cells: Roles of the JAK-STAT, AP-1 and NF-ΚB-Signaling Pathways. Br. J. Pharmacol. 1998, 125, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Morita, M.; Ishikawa, Y.; Rice, N.; Okamoto, S.; Kasahara, T.; Matsushima, K. Novel Mechanism of Glucocorticoid-Mediated Gene Repression. Nuclear Factor-Kappa B Is Target for Glucocorticoid-Mediated Interleukin 8 Gene Repression. J. Biol. Chem. 1994, 269, 13289–13295. [Google Scholar] [CrossRef]
- Tedeschi, E.; Menegazzi, M.; Margotto, D.; Suzuki, H.; Förstermann, U.; Kleinert, H. Anti-Inflammatory Actions of St. John’s Wort: Inhibition of Human Inducible Nitric-Oxide Synthase Expression by Down-Regulating Signal Transducer and Activator of Transcription-1α (STAT-1α) Activation. J. Pharm. Exp. 2003, 307, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, H.; Euchenhofer, C.; Ihrig-biedert, I.; Forstermann, U. Glucocorticoids Inhibit the Induction of Nitric Oxide Synthase II by Down-Regulating Cytokine-Induced Activity of Transcription Factor Nuclear Factor-KB. Mol. Pharm. 1995, 49, 15–21. [Google Scholar]
- Aktan, F. INOS-Mediated Nitric Oxide Production and Its Regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.W.; Cowan, J.A. Chemistry of Nitric Oxide with Protein-Bound Iron Sulfur Centers. Insights on Physiological Reactivity. J. Am. Chem. Soc. 1999, 121, 4093–4100. [Google Scholar] [CrossRef]
- Fehsel, K.; Jalowy, A.; Qi, S.; Burkart, V.; Hartmann, B.; Kolb, H. Islet Cell DNA Is a Target of Inflammatory Attack by Nitric Oxide. Diabetes 1993, 42, 496–500. [Google Scholar] [CrossRef]
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; Allen, J.S.; et al. DNA Deaminating Ability and Genotoxicity of Nitric Oxide and Its Progenitors. Science 1991, 254, 1001–1003. [Google Scholar] [CrossRef]
- Sase, K.; Michel, T. Expression of Constitutive Endothelial Nitric Oxide Synthase in Human Blood Platelets. Life Sci. 1995, 57, 2049–2055. [Google Scholar] [CrossRef]
- Gkaliagkousi, E.; Douma, S.; Zamboulis, C.; Ferro, A. Nitric Oxide Dysfunction in Vascular Endothelium and Platelets: Role in Essential Hypertension. J. Hypertens. 2009, 27, 2310–2320. [Google Scholar] [CrossRef]
- Balligand, J.-L.; Kobzik, L.; Han, X.; Kaye, D.M.; Belhassen, L.; O’Hara, D.S.; Kelly, R.A.; Smith, T.W.; Michel, T. Nitric Oxide-Dependent Parasympathetic Signaling Is Due to Activation of Constitutive Endothelial (Type III) Nitric Oxide Synthase in Cardiac Myocytes. J. Biol. Chem. 1995, 270, 14582–14586. [Google Scholar] [CrossRef] [PubMed]
- Feron, O.; Belhassen, L.; Kobzik, L.; Smith, T.W.; Kelly, R.A.; Michel, T. Endothelial Nitric Oxide Synthase Targeting to Caveolae: Specific Interactions with Caveolin Isoforms in Cardiac Myocytes and Endothelial Cells. J. Biol. Chem. 1996, 271, 22810–22814. [Google Scholar] [CrossRef] [PubMed]
- Feron, O.; Dessy, C.; Opel, D.J.; Arstall, M.A.; Kelly, R.A.; Michel, T. Modulation of the Endothelial Nitric-Oxide Synthase-Caveolin Interaction in Cardiac Myocytes: Implications for the Automatic Regulation of Heart Rate. J. Biol. Chem. 1998, 273, 30249–30254. [Google Scholar] [CrossRef]
- Caviedes, A.; Varas-Godoy, M.; Lafourcade, C.; Sandoval, S.; Bravo-Alegria, J.; Kaehne, T.; Massmann, A.; Figueroa, J.P.; Nualart, F.; Wyneken, U. Endothelial Nitric Oxide Synthase Is Present in Dendritic Spines of Neurons in Primary Cultures. Front. Cell. Neurosci. 2017, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Doyle, C.A.; Slater, P. Localization of Neuronal and Endothelial Nitric Oxide Synthase Isoforms in Human Hippocampus. Neuroscience 1997, 76, 387–395. [Google Scholar] [CrossRef]
- Li, S.-T.; Pan, J.; Hua, X.-M.; Liu, H.; Shen, S.; Liu, J.-F.; Li, B.; Tao, B.-B.; Ge, X.-L.; Wang, X.-H.; et al. Endothelial Nitric Oxide Synthase Protects Neurons against Ischemic Injury through Regulation of Brain-Derived Neurotrophic Factor Expression. CNS Neurosci. Ther. 2014, 20, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Eis, A.L.W.; Brockman, D.E.; Pollock, J.S.; Myatt, L. Immunohistochemical Localization of Endothelial Nitric Oxide Synthase in Human Villous and Extravillous Trophoblast Populations and Expression during Syncytiotrophoblast Formation in Vitro. Placenta 1995, 16, 113–126. [Google Scholar] [CrossRef]
- Tracey, W.R.; Pollock, J.S.; Murad, F.; Nakane, M.; Forstermann, U. Identification of an Endothelial-like Type III NO Synthase in LLC-PK1 Kidney Epithelial Cells. Am. J. Physiol. Cell Physiol. 1994, 266, C22–C28. [Google Scholar] [CrossRef]
- Igarashi, J.; Michel, T. S1P and ENOS Regulation. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2008, 1781, 489–495. [Google Scholar] [CrossRef]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of Caveolae, Vascular Dysfunction, and Pulmonary Defects in Caveolin-1 Gene-Disrupted Mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef]
- García-Cardeña, G.; Fan, R.; Shah, V.; Sorrentino, R.; Cirino, G.; Papapetropoulos, A.; Sessa, W.C. Dynamic Activation of Endothelial Nitric Oxide Synthase by Hsp90. Nature 1998, 392, 821–824. [Google Scholar] [CrossRef]
- Harrison, R. Structure and Function of Xanthine Oxidoreductase: Where Are We Now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef]
- Zuckerbraun, B.S.; Shiva, S.; Ifedigbo, E.; Mathier, M.A.; Mollen, K.P.; Rao, J.; Bauer, P.M.; Choi, J.J.W.; Curtis, E.; Choi, A.M.K.; et al. Nitrite Potently Inhibits Hypoxic and Inflammatory Pulmonary Arterial Hypertension and Smooth Muscle Proliferation via Xanthine Oxidoreductase–Dependent Nitric Oxide Generation. Circulation 2010, 121, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Broillet, M.-C. S-Nitrosylation of Proteins. CMLS Cell. Mol. Life Sci. 1999, 55, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation in Health and Disease: A Current Perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef]
- Marshall, H.E.; Stamler, J.S. Inhibition of NF-ΚB by S-Nitrosylation. Biochemistry 2001, 40, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Leon, L.; Jeannin, J.-F.; Bettaieb, A. Post-Translational Modifications Induced by Nitric Oxide (NO): Implication in Cancer Cells Apoptosis. Nitric Oxide 2008, 19, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Sumbayev, V.V.; Budde, A.; Zhou, J.; Brüne, B. HIF-1 Alpha Protein as a Target for S-Nitrosation. FEBS Lett. 2003, 535, 106–112. [Google Scholar] [CrossRef]
- Yasinska, I.M.; Sumbayev, V.V. S-Nitrosation of Cys-800 of HIF-1alpha Protein Activates Its Interaction with P300 and Stimulates Its Transcriptional Activity. FEBS Lett. 2003, 549, 105–109. [Google Scholar] [CrossRef]
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-Nitrosylation of Mitochondrial Caspases. J. Cell Biol. 2001, 154, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Bosworth, C.A.; Toledo, J.C.; Zmijewski, J.W.; Li, Q.; Lancaster, J.R. Dinitrosyliron Complexes and the Mechanism(s) of Cellular Protein Nitrosothiol Formation from Nitric Oxide. Proc. Natl. Acad. Sci. USA 2009, 106, 4671–4676. [Google Scholar] [CrossRef] [PubMed]
- Hickok, J.R.; Vasudevan, D.; Thatcher, G.R.J.; Thomas, D.D. Is S-Nitrosocysteine a True Surrogate for Nitric Oxide? Antioxid. Redox Signal. 2012, 17, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Kong, X.L. Glutathione: A Key Component of the Cytoplasmic Labile Iron Pool. Biometals 2011, 24, 1179–1187. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox Environment of the Cell as Viewed through the Redox State of the Glutathione Disulfide/Glutathione Couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Watts, R.N.; Ponka, P.; Richardson, D.R. Effects of Nitrogen Monoxide and Carbon Monoxide on Molecular and Cellular Iron Metabolism: Mirror-Image Effector Molecules That Target Iron. Biochem. J. 2003, 369, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, H.; Darley-Usmar, V.; Freeman, B.A. Nitric Oxide Regulation of Tissue Free Radical Injury. Chem. Res. Toxicol. 1996, 9, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hanbauer, I.; Grisham, M.B.; Laval, F.; Nims, R.W.; Laval, J.; Cook, J.; Pacelli, R.; Liebmann, J.; Krishna, M.; et al. Chemical niology of nitric oxide: Regulation and protective and toxic mechanisms. In Current Topics in Cellular Regulation; Stadtman, E.R., Chock, P.B., Eds.; Academic Press: Cambridge, MA, USA, 1996; Volume 34, pp. 159–187. [Google Scholar]
- Giulivi, C.; Kato, K.; Cooper, C.E. Nitric Oxide Regulation of Mitochondrial Oxygen Consumption I: Cellular Physiology. Am. J. Physiol. Cell Physiol. 2006, 291, C1225–C1231. [Google Scholar] [CrossRef]
- Cooper, C.E.; Giulivi, C. Nitric Oxide Regulation of Mitochondrial Oxygen Consumption II: Molecular Mechanism and Tissue Physiology. Am. J. Physiol. Cell Physiol. 2007, 292, C1993–C2003. [Google Scholar] [CrossRef]
- Tranguch, S.; Steuerwald, N.; Huet-Hudson, Y.M. Nitric Oxide Synthase Production and Nitric Oxide Regulation of Preimplantation Embryo Development. Biol. Reprod. 2003, 68, 1538–1544. [Google Scholar] [CrossRef]
- Lee, M.; Arosio, P.; Cozzi, A.; Chasteen, N.D. Identification of the EPR-Active Iron-Nitrosyl Complexes in Mammalian Ferritins. Biochemistry 1994, 33, 3679–3687. [Google Scholar] [CrossRef] [PubMed]
- García-Pascual, Á.; Sancho, M.; Costa, G.; Triguero, D. Interstitial Cells of Cajal in the Urethra Are CGMP-Mediated Targets of Nitrergic Neurotransmission. Am. J. Physiol. Ren. Physiol. 2008, 295, F971–F983. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.H.W.; Lohmann, S.M.; Walter, U. The Nitric Oxide and CGMP Signal Transduction System: Regulation and Mechanism of Action. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1993, 1178, 153–175. [Google Scholar] [CrossRef]
- Denninger, J.W.; Marletta, M.A. Guanylate Cyclase and the ⋅NO/CGMP Signaling Pathway. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1411, 334–350. [Google Scholar] [CrossRef]
- Krumenacker, J.S.; Hanafy, K.A.; Murad, F. Regulation of Nitric Oxide and Soluble Guanylyl Cyclase. Brain Res. Bull. 2004, 62, 505–515. [Google Scholar] [CrossRef]
- Schlossmann, J.; Ammendola, A.; Ashman, K.; Zong, X.; Huber, A.; Neubauer, G.; Wang, G.-X.; Allescher, H.-D.; Korth, M.; Wilm, M.; et al. Regulation of Intracellular Calcium by a Signalling Complex of IRAG, IP 3 Receptor and CGMP Kinase Iβ. Nature 2000, 404, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Arshavsky, V.Y.; Lamb, T.D.; Pugh, E.N. G Proteins and Phototransduction. Annu. Rev. Physiol. 2002, 64, 153–187. [Google Scholar] [CrossRef] [PubMed]
- Mittal, B.; Tulsyan, S.; Kumar, S.; Mittal, R.D.; Agarwal, G. Cytochrome P450 in Cancer Susceptibility and Treatment. Adv. Clin. Chem. 2015, 71, 77–139. [Google Scholar] [PubMed]
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome P450 System. J. Adv. Pr. Oncol. 2013, 4, 263–268. [Google Scholar]
- Kim, Y.-M.; Bergonia, H.A.; Müller, C.; Pitt, B.R.; Watkins, W.D.; Lancaster, J.R. Loss and Degradation of Enzyme-Bound Heme Induced by Cellular Nitric Oxide Synthesis. J. Biol. Chem. 1995, 270, 5710–5713. [Google Scholar] [CrossRef]
- Rouault, T.A. The Role of Iron Regulatory Proteins in Mammalian Iron Homeostasis and Disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Kühn, L.C. Iron Regulatory Proteins and Their Role in Controlling Iron Metabolism. Metallomics 2015, 7, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.; Kühn, L.C. Noncoding 3’ Sequences of the Transferrin Receptor Gene Are Required for MRNA Regulation by Iron. EMBO J. 1987, 6, 1287–1293. [Google Scholar] [CrossRef]
- Müllner, E.W.; Kühn, L.C. A Stem-Loop in the 3’ Untranslated Region Mediates Iron-Dependent Regulation of Transferrin Receptor MRNA Stability in the Cytoplasm. Cell 1988, 53, 815–825. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Ferritin, Iron Homeostasis, and Oxidative Damage. Free Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef]
- Hubert, N.; Hentze, M.W. Previously Uncharacterized Isoforms of Divalent Metal Transporter (DMT)-1: Implications for Regulation and Cellular Function. Proc. Natl. Acad. Sci. USA 2002, 99, 12345–12350. [Google Scholar] [CrossRef]
- Gunshin, H.; Allerson, C.R.; Polycarpou-Schwarz, M.; Rofts, A.; Rogers, J.T.; Kishi, F.; Hentze, M.W.; Rouault, T.A.; Andrews, N.C.; Hediger, M.A. Iron-Dependent Regulation of the Divalent Metal Ion Transporter. FEBS Lett. 2001, 509, 309–316. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. IRP1 Regulates Erythropoiesis and Systemic Iron Homeostasis by Controlling HIF2α MRNA Translation. Blood 2013, 122, 1658–1668. [Google Scholar] [CrossRef]
- Yanatori, I.; Richardson, D.R.; Dhekne, H.S.; Toyokuni, S.; Kishi, F. CD63 Is Regulated by Iron via the IRE-IRP System and Is Important for Ferritin Secretion by Extracellular Vesicles. Blood 2021. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Weiss, G.; Hentze, M.W. Nitric Oxide and Oxidative Stress (H2O2) Control Mammalian Iron Metabolism by Different Pathways. Mol. Cell. Biol. 1996, 16, 3781–3788. [Google Scholar] [CrossRef]
- Richardson, D.; Neumannova, V.; Nagy, E.; Ponka, P. The Effect of Redox-Related Species of Nitrogen Monoxide on Transferrin and Iron Uptake and Cellular Proliferation of Erythroleukemia (K562) Cells. Blood 1995, 86, 3211–3219. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, S.L.; Watts, R.N.; Richardson, D.R. Nitrogen Monoxide Activates Iron Regulatory Protein 1 Rna-Binding Activity by Two Possible Mechanisms: Effect on the [4Fe-4S] Cluster and Iron Mobilization from Cells. Biochemistry 2000, 39, 2748–2758. [Google Scholar] [CrossRef]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Rouault, T.A. Mammalian Tissue Oxygen Levels Modulate Iron-Regulatory Protein Activities in Vivo. Science 2004, 306, 2087–2090. [Google Scholar] [CrossRef] [PubMed]
- Drapier, J.C.; Hirling, H.; Wietzerbin, J.; Kaldy, P.; Kühn, L.C. Biosynthesis of Nitric Oxide Activates Iron Regulatory Factor in Macrophages. EMBO J. 1993, 12, 3643–3649. [Google Scholar] [CrossRef]
- Weiss, G.; Goossen, B.; Doppler, W.; Fuchs, D.; Pantopoulos, K.; Werner-Felmayer, G.; Wachter, H.; Hentze, M.w. Translational Regulation via Iron-Responsive Elements by the Nitric Oxide/NO-Synthase Pathway. EMBO J. 1993, 12, 3651–3657. [Google Scholar] [CrossRef]
- Cairo, G.; Ronchi, R.; Recalcati, S.; Campanella, A.; Minotti, G. Nitric Oxide and Peroxynitrite Activate the Iron Regulatory Protein-1 of J774A.1 Macrophages by Direct Disassembly of the Fe-S Cluster of Cytoplasmic Aconitase. Biochemistry 2002, 41, 7435–7442. [Google Scholar] [CrossRef]
- Kim, S.; Ponka, P. Control of Transferrin Receptor Expression via Nitric Oxide-Mediated Modulation of Iron-Regulatory Protein 2. J. Biol Chem 1999, 274, 33035–33042. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Ponka, P. Effects of Interferon-γ and Lipopolysaccharide on Macrophage Iron Metabolism Are Mediated by Nitric Oxide-Induced Degradation of Iron Regulatory Protein 2. J. Biol. Chem. 2000, 275, 6220–6226. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.N.; Richardson, D.R. Examination of the Mechanism of Action of Nitrogen Monoxide on Iron Uptake from Transferrin. J. Lab. Clin. Med. 2000, 136, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Neumannova, V.; Ponka, P. Nitrogen Monoxide Decreases Iron Uptake from Transferrin but Does Not Mobilise Iron from Prelabelled Neoplastic Cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1995, 1266, 250–260. [Google Scholar] [CrossRef][Green Version]
- Vanin, A.F. Endothelium-Derived Relaxing Factor Is a Nitrosyl Iron Complex with Thiol Ligands. FEBS Lett. 1991, 289, 1–3. [Google Scholar] [CrossRef]
- Cleeter, M.W.J.; Cooper, J.M.; Darley-Usmar, V.M.; Moncada, S.; Schapira, A.H.V. Reversible Inhibition of Cytochrome c Oxidase, the Terminal Enzyme of the Mitochondrial Respiratory Chain, by Nitric Oxide: Implications for Neurodegenerative Diseases. FEBS Lett. 1994, 345, 50–54. [Google Scholar] [CrossRef]
- Woolum, J.C.; Tiezzi, E.; Commoner, B. Electron Spin Resonance of Iron-Nitric Oxide Complexes with Amino Acids, Peptides and Proteins. Biochim. Biophys. Acta (BBA) Protein Struct. 1968, 160, 311–320. [Google Scholar] [CrossRef]
- Vanin, A.F.; Bliumenfel'd, L.A.; Chetverikov, A.G. EPR Study of Non-Heme Iron Complexes in Cells and Tissues. Biofizika 1967, 12, 829–838. [Google Scholar] [PubMed]
- Landry, A.P.; Duan, X.; Huang, H.; Ding, H. Iron–Sulfur Proteins Are the Major Source of Protein-Bound Dinitrosyl Iron Complexes Formed in Escherichia Coli Cells under Nitric Oxide Stress. Free Radic. Biol. Med. 2011, 50, 1582–1590. [Google Scholar] [CrossRef]
- Hickok, J.R.; Sahni, S.; Mikhed, Y.; Bonini, M.G.; Thomas, D.D. Nitric Oxide Suppresses Tumor Cell Migration through N-Myc Downstream-Regulated Gene-1 (NDRG1) Expression: Role of Chelatable Iron. J. Biol. Chem. 2011, 286, 41413–41424. [Google Scholar] [CrossRef] [PubMed]
- Chekmarev, J.; Azad, M.G.; Richardson, D.R. The Oncogenic Signaling Disruptor, NDRG1: Molecular and Cellular Mechanisms of Activity. Cells 2021, 10, 2382. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.; Zhang, W.; Zhang, J.; Liu, X.; Shi, H.; Che, H.; Wang, W.; Li, F.; Yao, L. Human Differentiation-Related Gene NDRG1 Is a Myc Downstream-Regulated Gene That Is Repressed by Myc on the Core Promoter Region. Gene 2008, 417, 5–12. [Google Scholar] [CrossRef]
- Tiffon, C. Histone Deacetylase Inhibition Restores Expression of Hypoxia-Inducible Protein NDRG1 in Pancreatic Cancer. Pancreas 2018, 47, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Salnikow, K.; Kluz, T.; Costa, M.; Piquemal, D.; Demidenko, Z.N.; Xie, K.; Blagosklonny, M.V. The Regulation of Hypoxic Genes by Calcium Involves C-Jun/AP-1, Which Cooperates with Hypoxia-Inducible Factor 1 in Response to Hypoxia. Mol. Cell Biol 2002, 22, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Salnikow, K.; Costa, M. Cap43, a Novel Gene Specifically Induced by Ni2+ Compounds. Cancer Res. 1998, 58, 2182–2189. [Google Scholar]
- Lane, D.J.R.; Saletta, F.; Rahmanto, Y.S.; Kovacevic, Z.; Richardson, D.R. N-Myc Downstream Regulated 1 (NDRG1) Is Regulated by Eukaryotic Initiation Factor 3a (EIF3a) during Cellular Stress Caused by Iron Depletion. PLoS ONE 2013, 8, e57273. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, D.; Zheng, Y.; Zhao, Q.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. Targeting the Metastasis Suppressor, NDRG1, Using Novel Iron Chelators: Regulation of Stress Fiber-Mediated Tumor Cell Migration via Modulation of the ROCK1/PMLC2 Signaling Pathway. Mol. Pharm. 2013, 83, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T.V.; Richardson, D.R. Iron Chelators with High Antiproliferative Activity Up-Regulate the Expression of a Growth Inhibitory and Metastasis Suppressor Gene: A Link between Iron Metabolism and Proliferation. Blood 2004, 104, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, D.; Yue, F.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. The Iron Chelators Dp44mT and DFO Inhibit TGF-β-Induced Epithelial-Mesenchymal Transition via up-Regulation of N-Myc Downstream-Regulated Gene 1 (NDRG1). J. Biol. Chem. 2012, 287, 17016–17028. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Iiizumi-Gairani, M.; Okuda, H.; Kobayashi, A.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Xing, F.; Fukuda, K.; Modur, V.; et al. KAI1 Gene Is Engaged in NDRG1 Gene-Mediated Metastasis Suppression through the ATF3-NFκB Complex in Human Prostate Cancer. J. Biol. Chem. 2011, 286, 18949–18959. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, F.; Izumi, H.; Kawahara, A.; Murakami, Y.; Kinoshita, H.; Kage, M.; Nishio, K.; Kohno, K.; Kuwano, M.; Ono, M. N-Myc Downstream Regulated Gene 1/Cap43 Suppresses Tumor Growth and Angiogenesis of Pancreatic Cancer through Attenuation of Inhibitor of ΚB Kinase β Expression. Cancer Res. 2009, 69, 4983–4991. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Pai, S.K.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; Takano, Y.; Saito, K.; Commes, T.; Piquemal, D.; et al. PTEN Up-Regulates the Tumor Metastasis Suppressor Gene Drg-1 in Prostate and Breast Cancer. Cancer Res. 2004, 64, 7655–7660. [Google Scholar] [CrossRef]
- Liu, W.; Xing, F.; Iiizumi-Gairani, M.; Okuda, H.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Hirota, S.; Kobayashi, A.; Mo, Y.-Y.; et al. N-Myc Downstream Regulated Gene 1 Modulates Wnt-β-Catenin Signalling and Pleiotropically Suppresses Metastasis. EMBO Mol. Med. 2012, 4, 93–108. [Google Scholar] [CrossRef]
- Kovacevic, Z.; Chikhani, S.; Lui, G.Y.L.; Sivagurunathan, S.; Richardson, D.R. The Iron-Regulated Metastasis Suppressor NDRG1 Targets NEDD4L, PTEN, and SMAD4 and Inhibits the PI3K and Ras Signaling Pathways. Antioxid. Redox Signal. 2013, 18, 874–887. [Google Scholar] [CrossRef]
- Lok, H.C.; Sahni, S.; Jansson, P.J.; Kovacevic, Z.; Hawkins, C.L.; Richardson, D.R. A Nitric Oxide Storage and Transport System That Protects Activated Macrophages from Endogenous Nitric Oxide Cytotoxicity. J. Biol. Chem. 2016, 291, 27042–27061. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.N.; Richardson, D.R. Nitrogen Monoxide (NO) and Glucose: Unexpected Links between Energy Metabolism and NO-Mediated Iron Mobilization from Cells. J. Biol. Chem. 2001, 276, 4724–4732. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.N.; Hawkins, C.; Ponka, P.; Richardson, D.R. Nitrogen Monoxide (NO)-Mediated Iron Release from Cells Is Linked to NO-Induced Glutathione Efflux via Multidrug Resistance-Associated Protein 1. Proc. Natl. Acad. Sci. USA 2006, 103, 7670–7675. [Google Scholar] [CrossRef] [PubMed]
- Lok, H.C.; Sahni, S.; Richardson, V.; Kalinowski, D.S.; Kovacevic, Z.; Lane, D.J.R.; Richardson, D.R. Glutathione S-Transferase and MRP1 Form an Integrated System Involved in the Storage and Transport of Dinitrosyl–Dithiolato Iron Complexes in Cells. Free Radic. Biol. Med. 2014, 75, 14–29. [Google Scholar] [CrossRef]
- Pellat, C.; Henry, Y.; Drapier, J.-C. IFN-γ-Activated Macrophages: Detection by Electron Paramagnetic Resonance of Complexes between l-arginine-Derived Nitric Oxide and Non-Heme Iron Proteins. Biochem. Biophys. Res. Commun. 1990, 166, 119–125. [Google Scholar] [CrossRef]
- Sergent, O.; Griffon, B.; Morel, I.; Chevanne, M.; Dubos, M.-P.; Cillard, P.; Cillard, J. Effect of Nitric Oxide on Iron-Mediated Oxidative Stress in Primary Rat Hepatocyte Culture. Hepatology 1997, 25, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Bergonia, H.A.; Di Silvio, M.; Sweetland, M.A.; Billiar, T.R.; Simmons, R.L.; Lancaster, J.R., Jr. Nonheme Iron-Nitrosyl Complex Formation in Rat Hepatocytes: Detection by Electron Paramagnetic Resonance Spectroscopy. Arch. Biochem. Biophys. 1993, 302, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Yoshimura, T. The Physiological Activity and in Vivo Distribution of Dinitrosyl Dithiolato Iron Complex. Jpn. J. Pharmacol. 2000, 82, 95–101. [Google Scholar] [CrossRef]
- Pedersen, J.Z.; De Maria, F.; Turella, P.; Federici, G.; Mattei, M.; Fabrini, R.; Dawood, K.F.; Massimi, M.; Caccuri, A.M.; Ricci, G. Glutathione Transferases Sequester Toxic Dinitrosyl-Iron Complexes in Cells: A Portection Mechanism against Excess Nitric Oxide. J. Biol. Chem. 2007, 282, 6364–6371. [Google Scholar] [CrossRef]
- Vanin, A.F.; Malenkova, I.V.; Mordvintsev, O.I.; Miul'sh, A. Dinitrosyl Complexes of Iron with Thiol-Containing Ligands and Their Reverse Conversion into Nitrosothiols. Biokhimiia 1993, 58, 1094–1103. [Google Scholar]
- Тimoshin, A.А.; Lakomkin, V.L.; Аbramov, A.А.; Ruuge, E.K.; Kapel’ko, V.I.; Chazov, E.I.; Vanin, A.F. The Hypotensive Effect of the Nitric Monoxide Donor Oxacom at Different Routs of Its Administration to Experimental Animals. Eur. J. Pharmacol. 2015, 765, 525–532. [Google Scholar] [CrossRef]
- Lakomkin, V.L.; Vanin, A.F.; Timoshin, A.A.; Kapelko, V.I.; Chazov, E.I. Long-Lasting Hypotensive Action of Stable Preparations of Dinitrosyl-Iron Complexes with Thiol-Containing Ligands in Conscious Normotensive and Hypertensive Rats. Nitric Oxide 2007, 16, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Saisavoey, T.; Sangtanoo, P.; Reamtong, O.; Karnchanatat, A. Anti-Inflammatory Effects of Lychee (Litchi Chinensis Sonn.) Seed Peptide Hydrolysate on RAW 264.7 Macrophage Cells. Food Biotechnol. 2018, 32, 79–94. [Google Scholar] [CrossRef]
- Lin, Z.-S.; Lo, F.-C.; Li, C.-H.; Chen, C.-H.; Huang, W.-N.; Hsu, I.-J.; Lee, J.-F.; Horng, J.-C.; Liaw, W.-F. Peptide-Bound Dinitrosyliron Complexes (DNICs) and Neutral/Reduced-Form Roussin’s Red Esters (RREs/RRREs): Understanding Nitrosylation of [Fe–S] Clusters Leading to the Formation of DNICs and RREs Using a de Novo Design Strategy. Inorg. Chem. 2011, 50, 10417–10431. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.-J.; Wang, C.-C.; Chang, C.-M. Synthesis of Dinitrosyl Iron Complexes (DNICs) with Intramolecular Hydrogen Bonding. J. Organomet. Chem. 2008, 693, 3582–3586. [Google Scholar] [CrossRef]
- Yang, J.; Duan, X.; Landry, A.P.; Ding, H. Oxygen Is Required for the l-cysteine-Mediated Decomposition of Protein-Bound Dinitrosyl–Iron Complexes. Free Radic. Biol. Med. 2010, 49, 268–274. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Farrotti, A.; Stella, L.; Ketterman, A.J.; Pedersen, J.Z.; Allocati, N.; Lau, P.C.K.; Grosse, S.; Eltis, L.D.; et al. The Impact of Nitric Oxide Toxicity on the Evolution of the Glutathione Transferase Superfamily: A Proposal for an Evolutionart Driving Force. J. Biol. Chem. 2013, 288, 24936–24947. [Google Scholar] [CrossRef]
- Ballatori, N.; Hammond, C.L.; Cunningham, J.B.; Krance, S.M.; Marchan, R. Molecular Mechanisms of Reduced Glutathione Transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A Families of Membrane Proteins. Toxicol. Appl. Pharmacol. 2005, 204, 238–255. [Google Scholar] [CrossRef]
- Mokh, V.P.; Poltorakov, А.P.; Serezhenkov, V.А.; Vanin, A.F. On the Nature of a Compound Formed from Dinitrosyl-Iron Complexes with Cysteine and Responsible for a Long-Lasting Vasorelaxation. Nitric Oxide 2010, 22, 266–274. [Google Scholar] [CrossRef]
- Vanin, A.F.; Poltorakov, A.P.; Mikoyan, V.D.; Kubrina, L.N.; Burbaev, D.S. Polynuclear Water-Soluble Dinitrosyl Iron Complexes with Cysteine or Glutathione Ligands: Electron Paramagnetic Resonance and Optical Studies. Nitric Oxide 2010, 23, 136–149. [Google Scholar] [CrossRef]
- Pereira, J.C.M.; Iretskii, A.V.; Han, R.-M.; Ford, P.C. Dinitrosyl Iron Complexes with Cysteine. Kinetics Studies of the Formation and Reactions of DNICs in Aqueous Solution. J. Am. Chem. Soc. 2015, 137, 328–336. [Google Scholar] [CrossRef]
- Borodulin, R.R.; Kubrina, L.N.; Shvydkiy, V.O.; Lakomkin, V.L.; Vanin, A.F. A Simple Protocol for the Synthesis of Dinitrosyl Iron Complexes with Glutathione: EPR, Optical, Chromatographic and Biological Characterization of Reaction Products. Nitric Oxide 2013, 35, 110–115. [Google Scholar] [CrossRef]
- Roussin, F.Z. Research on Double Iron Nitrosulphides (New Class of Salts). Ann. Chem. Phy. 1858, 52, 285. [Google Scholar]
- Vanin, A.F. Dinitrosyl Iron Complexes with Thiol-Containing Ligands as a “Working Form” of Endogenous Nitric Oxide. Nitric Oxide 2016, 54, 15–29. [Google Scholar] [CrossRef]
- Keese, M.A.; Böse, M.; Mülsch, A.; Schirmer, R.H.; Becker, K. Dinitrosyl-Dithiol-Iron Complexes, Nitric Oxide (NO) Carriers in Vivo, as Potent Inhibitors of Human Glutathione Reductase and Glutathione-s-Transferase. Biochem. Pharmacol. 1997, 54, 1307–1313. [Google Scholar] [CrossRef]
- Ueno, T.; Suzuki, Y.; Fujii, S.; Vanin, A.F.; Yoshimura, T. In Vivo Nitric Oxide Transfer of a Physiological NO Carrier, Dinitrosyl Dithiolato Iron Complex, to Target Complex. Biochem. Pharmacol. 2002, 63, 485–493. [Google Scholar] [CrossRef]
- Becker, K.; Savvides, S.N.; Keese, M.; Schirmer, R.H.; Karplus, P.A. Enzyme Inactivation through Sulfhydryl Oxidation by Physiologic NO-Carriers. Nat. Struct. Biol. 1998, 5, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Ku, W.-C.; Feng, L.-T.; Tsai, M.-L.; Hsieh, C.-H.; Hsu, W.-H.; Liaw, W.-F.; Hung, C.-H.; Chen, Y.-J. Nitric Oxide Physiological Responses and Delivery Mechanisms Probed by Water-Soluble Roussin’s Red Ester and {Fe(NO)2}10 DNIC. J. Am. Chem. Soc. 2008, 130, 10929–10938. [Google Scholar] [CrossRef] [PubMed]
- Maria, F.D.; Pedersen, J.Z.; Caccuri, A.M.; Antonini, G.; Turella, P.; Stella, L.; Bello, M.L.; Federici, G.; Ricci, G. The Specific Interaction of Dinitrosyl-Diglutathionyl-Iron Complex, a Natural NO Carrier, with the Glutathione Transferase Superfamily: Suggestion for an Evolutionary Pressure in the Direction of the Storage of Nitric Oxide. J. Biol. Chem. 2003, 278, 42283–42293. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-C.; Lu, C.-Y.; Chen, Y.-L.; Lo, F.-C.; Wang, T.-Y.; Chen, Y.-J.; Yuan, S.-S.; Liaw, W.-F.; Wang, Y.-M. Water-Soluble Dinitrosyl Iron Complex (DNIC): A Nitric Oxide Vehicle Triggering Cancer Cell Death via Apoptosis. Inorg. Chem. 2016, 55, 9383–9392. [Google Scholar] [CrossRef] [PubMed]
- Rahmanto, Y.S.; Kalinowski, D.S.; Lane, D.J.R.; Lok, H.C.; Richardson, V.; Richardson, D.R. Nitrogen Monoxide (NO) Storage and Transport by Dinitrosyl-Dithiol-Iron Complexes: Long-Lived NO That Is Trafficked by Interacting Proteins. J. Biol. Chem. 2012, 287, 6960–6968. [Google Scholar] [CrossRef]
- Hickok, J.R.; Sahni, S.; Shen, H.; Arvind, A.; Antoniou, C.; Fung, L.W.M.; Thomas, D.D. Dinitrosyliron Complexes Are the Most Abundant Nitric Oxide-Derived Cellular Adduct: Biological Parameters of Assembly and Disappearance. Free Radic. Biol. Med. 2011, 51, 1558–1566. [Google Scholar] [CrossRef]
- Borodulin, R.R.; Kubrina, L.N.; Mikoyan, V.D.; Poltorakov, A.P.; Shvydkiy, V.О.; Burbaev, D.S.; Serezhenkov, V.A.; Yakhontova, E.R.; Vanin, A.F. Dinitrosyl Iron Complexes with Glutathione as NO and NO+ Donors. Nitric Oxide 2013, 29, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Galagan, M.E.; Oranovskaia, E.V.; Mordvintsev, P.I.; Medvedev, O.S.; Vanin, A.F. Hypotensive Effect of Dinitrosyl Iron Complexes in Experiments on Waking Animals. Biulleten Vsesoiuznogo Kardiol. Nauchnogo Tsentra AMN SSSR 1988, 11, 75–80. [Google Scholar] [PubMed]
- Mayer, B.; Kleschyov, A.L.; Stessel, H.; Russwurm, M.; Münzel, T.; Koesling, D.; Schmidt, K. Inactivation of Soluble Guanylate Cyclase by Stoichiometric S-Nitrosation. Mol. Pharm. 2009, 75, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Severina, I.S.; Bussygina, O.G.; Pyatakova, N.V.; Malenkova, I.V.; Vanin, A.F. Activation of Soluble Guanylate Cyclase by NO Donors—S-Nitrosothiols, and Dinitrosyl-Iron Complexes with Thiol-Containing Ligands. Nitric Oxide 2003, 8, 155–163. [Google Scholar] [CrossRef]
- Alencar, J.L.; Chalupsky, K.; Sarr, M.; Schini-Kerth, V.; Vanin, A.F.; Stoclet, J.-C.; Muller, B. Inhibition of Arterial Contraction by Dinitrosyl–Iron Complexes: Critical Role of the Thiol Ligand in Determining Rate of Nitric Oxide (NO) Release and Formation of Releasable NO Stores by S-Nitrosation. Biochem. Pharmacol. 2003, 66, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Vanin, A.F.; Mokh, V.P.; Serezhenkov, V.A.; Chazov, E.I. Vasorelaxing Activity of Stable Powder Preparations of Dinitrosyl Iron Complexes with Cysteine or Glutathione Ligands. Nitric Oxide 2007, 16, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Shumaev, K.B.; Kosmachevskaya, O.V.; Grachev, D.I.; A A Timoshin, A.A.; Topunov, A.F.; Lankin, V.Z.; Ruuge, E.K. Possible Mechanism of Antioxidant Action of Dinitrosyl Iron Complexes. Biomed. Khimiya 2021, 67, 162–168. [Google Scholar] [CrossRef]
- Shumaev, K.B.; Petrova, N.E.; Zabbarova, I.V.; Vanin, A.F.; Topunov, A.F.; Lankin, V.Z.; Ruuge, E.K. Interaction of Oxoferrylmyoglobin and Dinitrosyl-Iron Complexes. Biochemistry (Moscow) 2004, 69, 569–574. [Google Scholar] [CrossRef]
- Kleschyov, A.L.; Strand, S.; Schmitt, S.; Gottfried, D.; Skatchkov, M.; Sjakste, N.; Daiber, A.; Umansky, V.; Munzel, T. Dinitrosyl-Iron Triggers Apoptosis in Jurkat Cells despite Overexpression of Bcl-2. Free Radic. Biol. Med. 2006, 40, 1340–1348. [Google Scholar] [CrossRef]
- Dinitrosyl Iron Complexes with Thiol-Containing Ligands and Apoptosis: Studies with HeLa Cell Cultures. Nitric Oxide 2011, 24, 151–159. [CrossRef]
- Interaction of Reactive Oxygen and Nitrogen Species with Albumin- and Methemoglobin-Bound Dinitrosyl-Iron Complexes. Nitric Oxide 2008, 18, 37–46. [CrossRef]
- Lok, H.C.; Rahmanto, Y.S.; Hawkins, C.L.; Kalinowski, D.S.; Morrow, C.S.; Townsend, A.J.; Ponka, P.; Richardson, D.R. Nitric Oxide Storage and Transport in Cells Are Mediated by Glutathione S-Transferase P1-1 and Multidrug Resistance Protein 1 via Dinitrosyl Iron Complexes. J. Biol. Chem. 2012, 287, 607–618. [Google Scholar] [CrossRef]
- Watts, R.N.; Richardson, D.R. The Mechanism of Nitrogen Monoxide (NO)-Mediated Iron Mobilization from Cells. Eur. J. Biochem. 2002, 269, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Zai, A.; Rudd, M.A.; Scribner, A.W.; Loscalzo, J. Cell-Surface Protein Disulfide Isomerase Catalyzes Transnitrosation and Regulates Intracellular Transfer of Nitric Oxide. J. Clin. Investig. 1999, 103, 393–399. [Google Scholar] [CrossRef]
- Krischel, V.; Bruch-Gerharz, D.; Suschek, C.; Kröncke, K.-D.; Ruzicka, T.; Kolb-Bachofen, V. Biphasic Effect of Exogenous Nitric Oxide on Proliferation and Differentiation in Skin Derived Keratinocytes but Not Fibroblasts. J. Investig. Dermatol. 1998, 111, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The Yin and Yang of Nitric Oxide in Cancer Progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef]
- Clancy, R.M.; Levartovsky, D.; Leszczynska-Piziak, J.; Yegudin, J.; Abramson, S.B. Nitric Oxide Reacts with Intracellular Glutathione and Activates the Hexose Monophosphate Shunt in Human Neutrophils: Evidence for S-Nitrosoglutathione as a Bioactive Intermediary. Proc. Natl. Acad. Sci. USA 1994, 91, 3680–3684. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Leslie, E.M.; Deeley, R.G.; Cole, S.P.C. ATPase Activity of Purified and Reconstituted Multidrug Resistance Protein MRP1 from Drug-Selected H69AR Cells. Biochim. Biophys. Acta (BBA) Biomembr. 1999, 1461, 69–82. [Google Scholar] [CrossRef][Green Version]
- Chang, X. A Molecular Understanding of ATP-Dependent Solute Transport by Multidrug Resistance-Associated Protein MRP1. Cancer Metastasis Rev. 2007, 26, 15–37. [Google Scholar] [CrossRef]
- Richardson, D. DNICs and intracellular iron: Nitrogen monoxide (NO)-mediated iron release from cells is linked to NO-mediated glutathione efflux via MRP1. In Radicals for Life: The Various Forms of Nitric Oxide; Elsevier Press: Cambridge, MA, USA, 2007; pp. 97–118. [Google Scholar]
- Turella, P.; Pedersen, J.Z.; Caccuri, A.M.; Maria, F.D.; Mastroberardino, P.; Bello, M.L.; Federici, G.; Ricci, G. Glutathione Transferase Superfamily Behaves like Storage Proteins for Dinitrosyl-Diglutathionyl-Iron Complex in Heterogeneous Systems. J. Biol. Chem. 2003, 278, 42294–42299. [Google Scholar] [CrossRef]
- Gilligan, D.M.; Panza, J.A.; Kilcoyne, C.M.; Waclawiw, M.A.; Casino, P.R.; Quyyumi, A. A Contribution of Endothelium-Derived Nitric Oxide to Exercise-Induced Vasodilation. Circulation 1994, 90, 2853–2858. [Google Scholar] [CrossRef] [PubMed]
- Meredith, I.T.; Currie, K.E.; Anderson, T.J.; Roddy, M.A.; Ganz, P.; Creager, M.A. Postischemic Vasodilation in Human Forearm Is Dependent on Endothelium-Derived Nitric Oxide. Am. J. Physiol. Heart Circ. Physiol. 1996, 270, H1435–H1440. [Google Scholar] [CrossRef] [PubMed]
- Drapier, J.C.; Hibbs, J.B. Murine Cytotoxic Activated Macrophages Inhibit Aconitase in Tumor Cells. Inhibition Involves the Iron-Sulfur Prosthetic Group and Is Reversible. J. Clin. Investig. 1986, 78, 790–797. [Google Scholar] [CrossRef]
- Drapier, J.-C.; Hibbs, J.B. Aconitases: A class of metalloproteins highly sensitive to nitric oxide synthesis. Methods Enzym. 1996, 269, 26–36. [Google Scholar]
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma Membrane Glutathione Transporters and Their Roles in Cell Physiology and Pathophysiology. Mol. Asp. Med. 2009, 30, 13–28. [Google Scholar] [CrossRef]
- Cole, S.P.C. Targeting Multidrug Resistance Protein 1 (MRP1, ABCC1): Past, Present, and Future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Hirrlinger, J.; König, J.; Keppler, D.; Lindenau, J.; Schulz, J.B.; Dringen, R. The Multidrug Resistance Protein MRP1 Mediates the Release of Glutathione Disulfide from Rat Astrocytes during Oxidative Stress. J. Neurochem. 2001, 76, 627–636. [Google Scholar] [CrossRef]
- Grant, C.E.; Valdimarsson, G.; Hipfner, D.R.; Almquist, K.C.; Cole, S.P.C.; Deeley, R.G. Overexpression of Multidrug Resistance-Associated Protein (MRP) Increases Resistance to Natural Product Drugs. Cancer Res. 1994, 54, 357–361. [Google Scholar]
- Depeille, P.; Cuq, P.; Passagne, I.; Evrard, A.; Vian, L. Combined Effects of GSTP1 and MRP1 in Melanoma Drug Resistance. Br. J. Cancer 2005, 93, 216–223. [Google Scholar] [CrossRef]
- Diah, S.K.; Smitherman, P.K.; Aldridge, J.; Volk, E.L.; Schneider, E.; Townsend, A.J.; Morrow, C.S. Resistance to Mitoxantrone in Multidrug-Resistant MCF7 Breast Cancer Cells: Evaluation of Mitoxantrone Transport and the Role of Multidrug Resistance Protein Family Proteins. Cancer Res. 2001, 61, 5461–5467. [Google Scholar]
- Haber, M.; Smith, J.; Bordow, S.B.; Flemming, C.; Cohn, S.L.; London, W.B.; Marshall, G.M.; Norris, M.D. Association of High-Level MRP1 Expression With Poor Clinical Outcome in a Large Prospective Study of Primary Neuroblastoma. JCO 2006, 24, 1546–1553. [Google Scholar] [CrossRef]
- Swerts, K.; Moerloose, B.D.; Dhooge, C.; Laureys, G.; Benoit, Y.; Philippé, J. Prognostic Significance of Multidrug Resistance-Related Proteins in Childhood Acute Lymphoblastic Leukaemia. Eur. J. Cancer 2006, 42, 295–309. [Google Scholar] [CrossRef]
- Benyahia, B.; Huguet, S.; Declèves, X.; Mokhtari, K.; Crinière, E.; Bernaudin, J.F.; Scherrmann, J.M.; Delattre, J.Y. Multidrug Resistance-Associated Protein MRP1 Expression in Human Gliomas: Chemosensitization to Vincristine and Etoposide by Indomethacin in Human Glioma Cell Lines Overexpressing MRP1. J. Neurooncol. 2004, 66, 65–70. [Google Scholar] [CrossRef]
- Dong, Q.; Zhou, C.; Ren, H.; Zhang, Z.; Cheng, F.; Xiong, Z.; Chen, C.; Yang, J.; Gao, J.; Zhang, Y.; et al. Lactate-Induced MRP1 Expression Contributes to Metabolism-Based Etoposide Resistance in Non-Small Cell Lung Cancer Cells. Cell Commun. Signal. 2020, 18, 167. [Google Scholar] [CrossRef]
- Fang, Z.; Chen, W.; Yuan, Z.; Liu, X.; Jiang, H. LncRNA-MALAT1 Contributes to the Cisplatin-Resistance of Lung Cancer by Upregulating MRP1 and MDR1 via STAT3 Activation. Biomed. Pharmacother. 2018, 101, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.S.; Peklak-Scott, C.; Bishwokarma, B.; Kute, T.E.; Smitherman, P.K.; Townsend, A.J. Multidrug Resistance Protein 1 (MRP1, ABCC1) Mediates Resistance to Mitoxantrone via Glutathione-Dependent Drug Efflux. Mol. Pharm. 2006, 69, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.S.; Smitherman, P.K.; Diah, S.K.; Schneider, E.; Townsend, A.J. Coordinated Action of Glutathione S-Transferases (GSTs) and Multidrug Resistance Protein 1 (MRP1) in Antineoplastic Drug Detoxification: Mechanism of GST A1-1 and MRP1- Associated Resistance to Chlorambucil in MCF7 Breast Carcinoma Cells. J. Biol. Chem. 1998, 273, 20114–20120. [Google Scholar] [CrossRef]
- Allen, J.D.; Brinkhuis, R.F.; van Deemter, L.; Wijnholds, J.; Schinkel, A.H. Extensive Contribution of the Multidrug Transporters P-Glycoprotein and Mrp1 to Basal Drug Resistance. Cancer Res. 2000, 60, 5761–5766. [Google Scholar] [PubMed]
- Wormhoudt, L.W.; Commandeur, J.N.M.; Vermeulen, N.P.E. Genetic Polymorphisms of Human N-Acetyltransferase, Cytochrome P450, Glutathione-s-Transferase, and Epoxide Hydrolase Enzymes: Relevance to Xenobiotic Metabolism and Toxicity. Crit. Rev. Toxicol. 1999, 29, 59–124. [Google Scholar] [CrossRef]
- Harwaldt, P.; Rahlfs, S.; Becker, K. Glutathione S-Transferase of the Malarial Parasite Plasmodium Falciparum: Characterization of a Potential Drug Target. Biol. Chem. 2002, 383, 821–830. [Google Scholar] [CrossRef]
- Mannervik, B.; Board, P.G.; Hayes, J.D.; Listowsky, I.; Pearson, W.R. Nomenclature for mammalian soluble glutathione transferases. Methods Enzym. 2005, 401, 1–8. [Google Scholar]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, Function and Evolution of Glutathione Transferases: Implications for Classification of Non-Mammalian Members of an Ancient Enzyme Superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione Transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.S.; Diah, S.; Smitherman, P.K.; Schneider, E.; Townsend, A.J. Multidrug Resistance Protein and Glutathione S-Transferase P1-1 Act in Synergy to Confer Protection from 4-Nitroquinoline 1-Oxide Toxicity. Carcinogenesis 1998, 19, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Morrow, C.S.; Smitherman, P.K.; Townsend, A.J. Combined Expression of Multidrug Resistance Protein (MRP) and Glutathione S-Transferase P1-1 (GSTP1-1) in MCF7 Cells and High Level Resistance to the Cytotoxicities of Ethacrynic Acid but Not Oxazaphosphorines or Cisplatin. Biochem. Pharmacol. 1998, 56, 1013–1021. [Google Scholar] [CrossRef]
- Cesareo, E.; Parker, L.J.; Pedersen, J.Z.; Nuccetelli, M.; Mazzetti, A.P.; Pastore, A.; Federici, G.; Caccuri, A.M.; Ricci, G.; Adams, J.J.; et al. Nitrosylation of Human Glutathione Transferase P1-1 with Dinitrosyl Diglutathionyl Iron Complex in Vitro and in Vivo. J. Biol. Chem. 2005, 280, 42172–42180. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.L.; Nuccetelli, M.; Caccuri, A.M.; Stella, L.; Parker, M.W.; Rossjohn, J.; McKinstry, W.J.; Mozzi, A.F.; Federici, G.; Polizio, F.; et al. Human Glutathione Transferase P1-1 and Nitric Oxide Carriers: A New Role for an Old Enzyme. J. Biol. Chem. 2001, 276, 42138–42145. [Google Scholar] [CrossRef] [PubMed]
- Stella, L.; Pallottini, V.; Moreno, S.; Leoni, S.; De Maria, F.; Turella, P.; Federici, G.; Fabrini, R.; Dawood, K.F.; Bello, M.L.; et al. Electrostatic Association of Glutathione Transferase to the Nuclear Membrane: Evidence of an Enzyme Defence Barrier at the Nuclear Envelope. J. Biol. Chem. 2007, 282, 6372–6379. [Google Scholar] [CrossRef]
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct Mechanisms of Ferritin Delivery to Lysosomes in Iron-Depleted and Iron-Replete Cells. Mol. Cell. Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef]
- Lewandowska, H.; Męczyńska, S.; Sochanowicz, B.; Sadło, J.; Kruszewski, M. Crucial Role of Lysosomal Iron in the Formation of Dinitrosyl Iron Complexes in Vivo. J. Biol. Inorg. Chem. 2007, 12, 345–352. [Google Scholar] [CrossRef]
- Canto, P.; Canto-Cetina, T.; Juárez-Velázquez, R.; Rosas-Vargas, H.; Rangel-Villalobos, H.; Canizales-Quinteros, S.; Velázquez-Wong, A.C.; Villarreal-Molina, M.T.; Fernández, G.; Coral-Vázquez, R. Methylenetetrahydrofolate Reductase C677T and Glutathione S−transferase P1 A313G Are Associated with a Reduced Risk of Preeclampsia in Maya-Mestizo Women. Hypertens Res. 2008, 31, 1015–1019. [Google Scholar] [CrossRef]
- Olson, E.; Pravenec, M.; Landa, V.; Koh-Tan, H.H.C.; Dominiczak, A.F.; McBride, M.W.; Graham, D. Transgenic Overexpression of Glutathione S-Transferase μ-Type 1 Reduces Hypertension and Oxidative Stress in the Stroke-Prone Spontaneously Hypertensive Rat. J. Hypertens. 2019, 37, 985–996. [Google Scholar] [CrossRef]
- Tin, A.; Scharpf, R.; Estrella, M.M.; Yu, B.; Grove, M.L.; Chang, P.P.; Matsushita, K.; Köttgen, A.; Arking, D.E.; Boerwinkle, E.; et al. The Loss of GSTM1 Associates with Kidney Failure and Heart Failure. J. Am. Soc. Nephrol. 2017, 28, 3345–3352. [Google Scholar] [CrossRef]
- Ueno, T.; Suzuki, Y.; Fujii, S.; Vanin, A.F.; Yoshimura, T. Invivo Distribution and Behavior of Paramagnetic Dinitrosyl Dithiolato Iron Complex in the Abdomen of Mouse. Free Radic. Res. 1999, 31, 525–534. [Google Scholar] [CrossRef]
- Boese, M.; Mordvintcev, P.I.; Vanin, A.F.; Busse, R.; Mülsch, A. S-Nitrosation of Serum Albumin by Dinitrosyl-Iron Complex. J. Biol. Chem. 1995, 270, 29244–29249. [Google Scholar] [CrossRef] [PubMed]
- Vanin, A.F.; Malenkova, I.V.; Serezhenkov, V.A. Iron Catalyzes Both Decomposition and Synthesis Ofs-Nitrosothiols: Optical and Electron Paramagnetic Resonance Studies. Nitric Oxide 1997, 1, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Lippton, H.; Edwards, J.C.; Baricos, W.H.; Hyman, A.L.; Kadowitz, P.J.; Gruetter, C.A.J.L.; Edwards, J.C.; Baricos, A.H. Mechanism of Vascular Smooth Muscle Relaxation by Organic Nitrates, Nitrites, Nitroprusside and Nitric Oxide: Evidence for the Involvement of S-Nitrosothiols as Active Intermediates. J. Pharm. Exp. 1981, 218, 739–749. [Google Scholar]
- Ignarro, L.J.; Byrns, R.E.; Buga, G.M.; Wood, K.S.; Chaudhuri, G. Pharmacological Evidence That Endothelium-Derived Relaxing Factor Is Nitric Oxide: Use of Pyrogallol and Superoxide Dismutase to Study Endothelium-Dependent and Nitric Oxide-Elicited Vascular Smooth Muscle Relaxation. J. Pharm. Exp. 1988, 244, 181–189. [Google Scholar]
- Ignarro, L.J. Heme-Dependent Activation of Guanylate Cyclase by Nitric Oxide: A Novel Signal Transduction Mechanism. JVR 1991, 28, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zalcenstein, A.; Oren, M. Nitric Oxide Promotes P53 Nuclear Retention and Sensitizes Neuroblastoma Cells to Apoptosis by Ionizing Radiation. Cell Death Differ. 2003, 10, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z.; Rachlin, E.M. Nitric Oxide: A Cytotoxic Activated Macrophage Effector Molecule. Biochem. Biophys. Res. Commun. 1988, 157, 87–94. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russell, T.M.; Azad, M.G.; Richardson, D.R. The Relationship of Glutathione-S-Transferase and Multi-Drug Resistance-Related Protein 1 in Nitric Oxide (NO) Transport and Storage. Molecules 2021, 26, 5784. https://doi.org/10.3390/molecules26195784
Russell TM, Azad MG, Richardson DR. The Relationship of Glutathione-S-Transferase and Multi-Drug Resistance-Related Protein 1 in Nitric Oxide (NO) Transport and Storage. Molecules. 2021; 26(19):5784. https://doi.org/10.3390/molecules26195784
Chicago/Turabian StyleRussell, Tiffany M., Mahan Gholam Azad, and Des R. Richardson. 2021. "The Relationship of Glutathione-S-Transferase and Multi-Drug Resistance-Related Protein 1 in Nitric Oxide (NO) Transport and Storage" Molecules 26, no. 19: 5784. https://doi.org/10.3390/molecules26195784
APA StyleRussell, T. M., Azad, M. G., & Richardson, D. R. (2021). The Relationship of Glutathione-S-Transferase and Multi-Drug Resistance-Related Protein 1 in Nitric Oxide (NO) Transport and Storage. Molecules, 26(19), 5784. https://doi.org/10.3390/molecules26195784