
iT4SE-EP: Accurate Identification of Bacterial Type IV Secreted Effectors by Exploring Evolutionary Features from Two PSI-BLAST Profiles


Abstract
1. Introduction
2. Results and Discussions
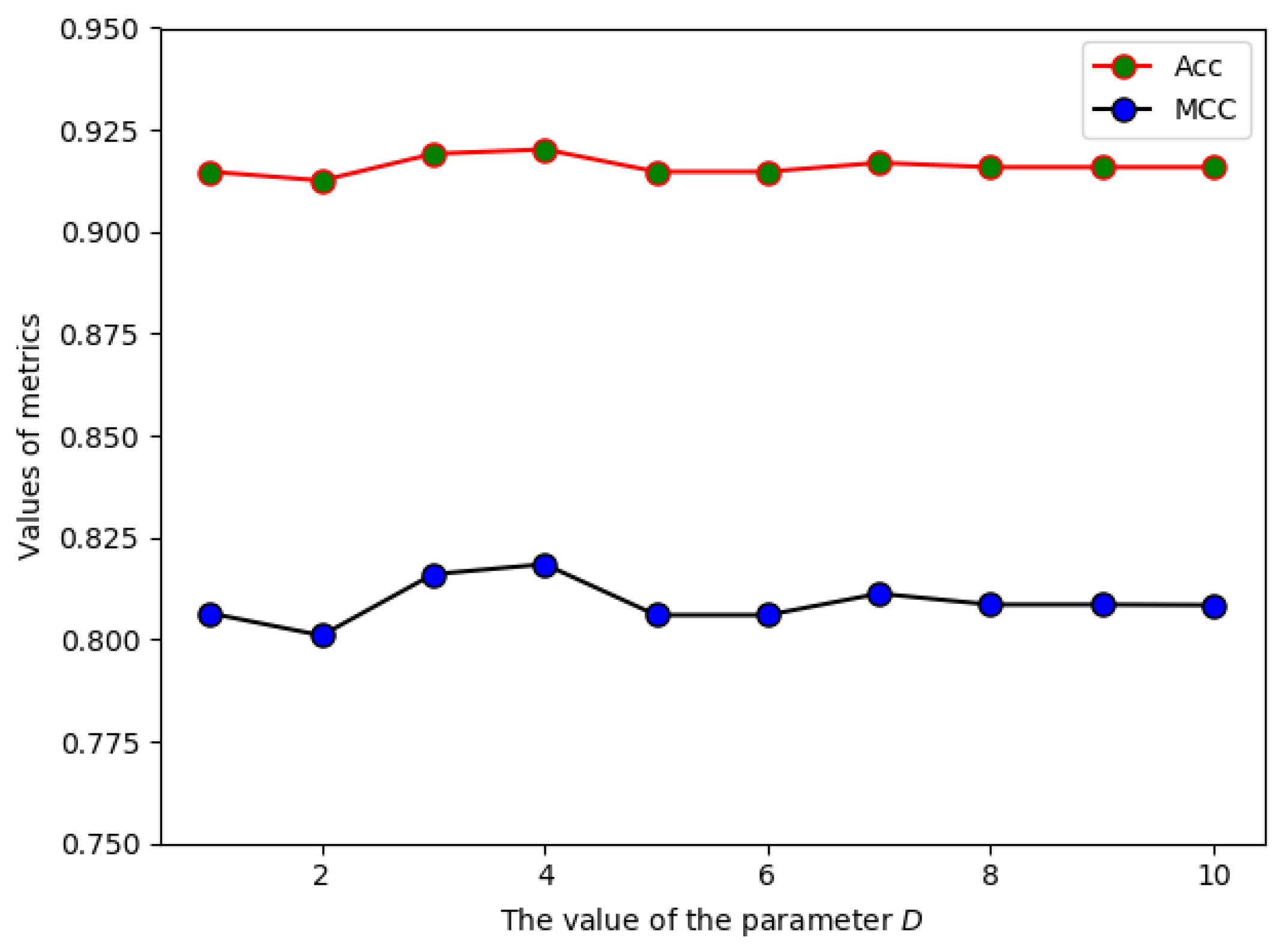
2.1. The Effect of Parameter D on the Prediction Performance
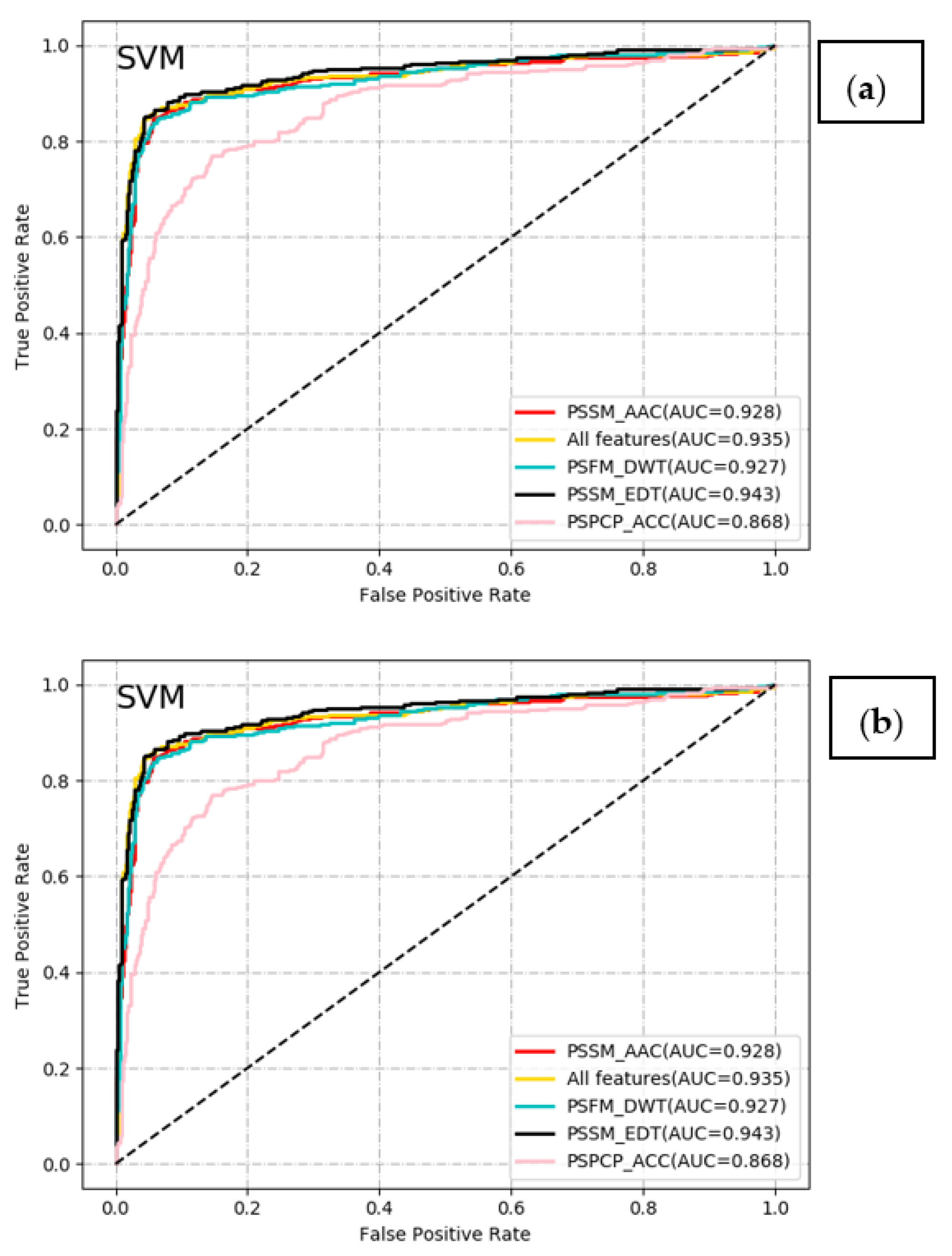
2.2. Comparative Analysis of Different Classifiers with Different Feature Encoding Schemes
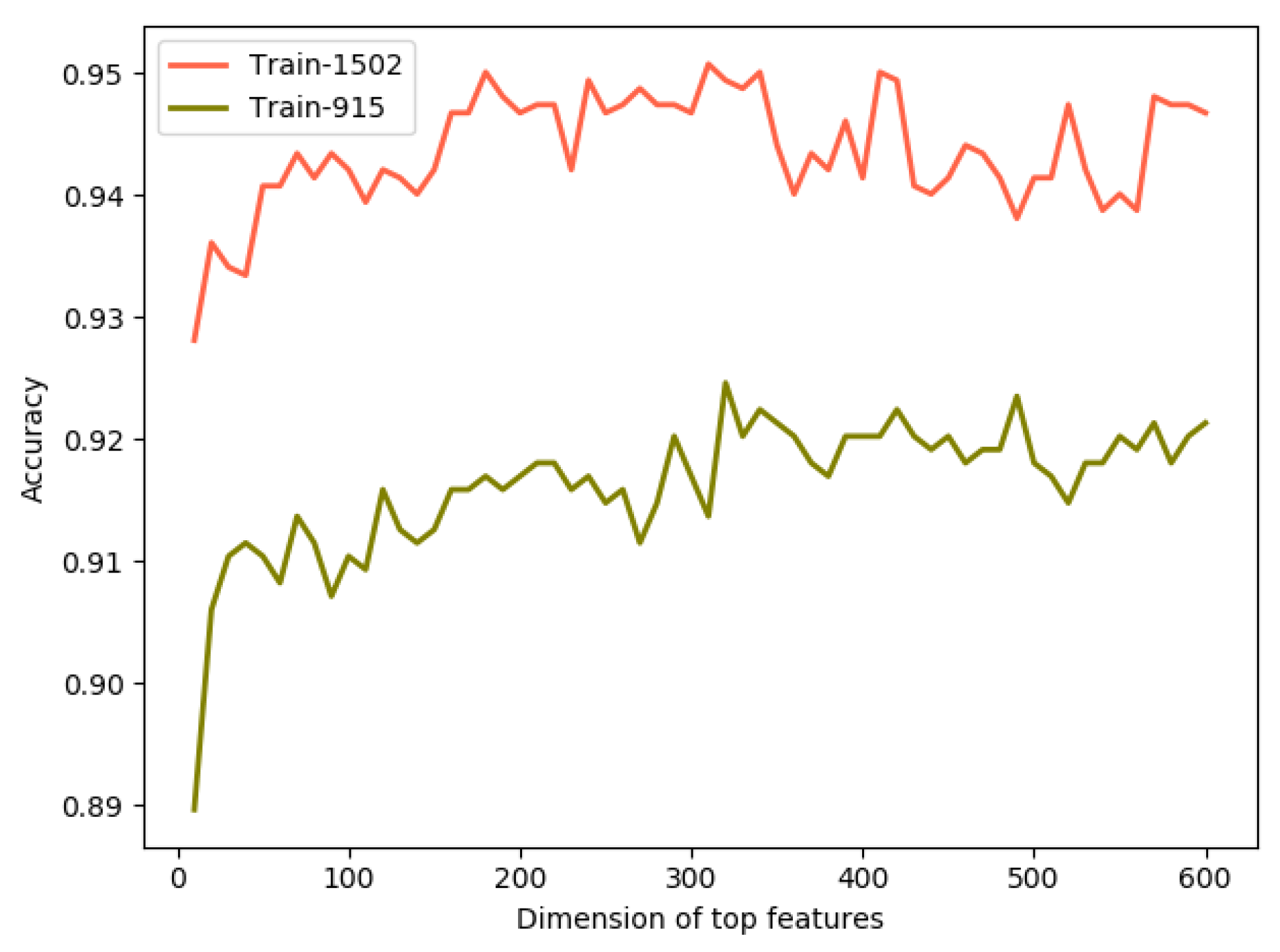
2.3. Performance Analysis of Feature Selection
2.4. Peformance Comparision with Existing Methods
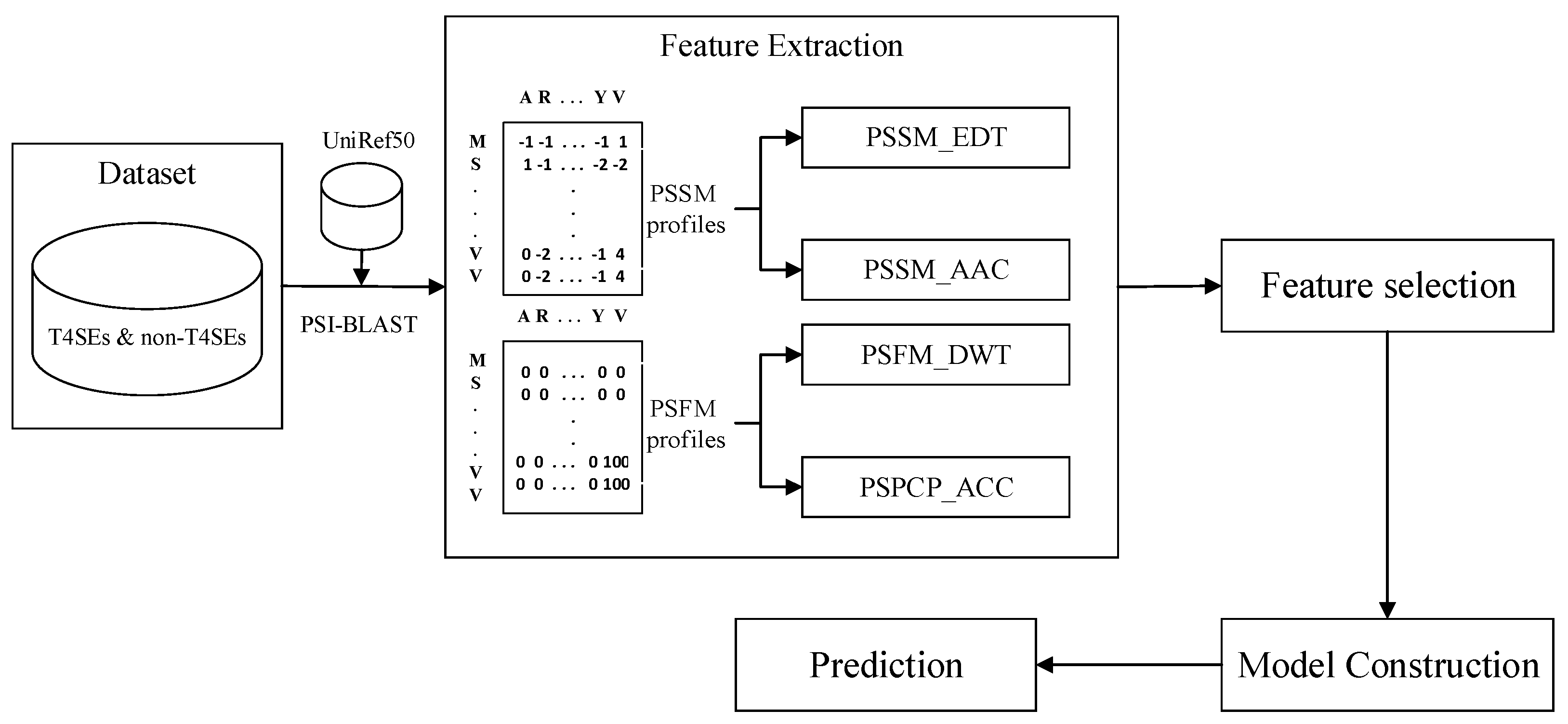
3. Materials and Methods
3.1. Datasets
3.2. Feature Extraction
3.2.1. PSI-BLAST Profiles
3.2.2. Amino Acid Composition
3.2.3. Evolutionary Difference Transformation
3.2.4. Auto-Covariance and Cross-Covariance
3.2.5. Discrete Wavelet Transform
3.3. Feature Normalization
3.4. Model Construction
3.4.1. Support Vector Machine
3.4.2. Random Forest
3.5. Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Desvaux, M.; Hebraud, M.; Talon, R.; Henderson, I.R. Secretion and subcellular localizations of bacterial proteins: A semantic awareness issue. Trends Microbiol. 2009, 17, 139–145. [Google Scholar] [CrossRef]
- Bi, D.; Liu, L.; Tai, C.; Deng, Z.; Rajakumar, K.; Ou, H.-Y. SecReT4: A web-based bacterial type IV secretion system resource. Nucleic Acids Res. 2013, 41, D660–D665. [Google Scholar] [CrossRef]
- Meyer, D.F.; Noroy, C.; Moumene, A.; Raffaele, S.; Albina, E.; Vachiery, N. Searching algorithm for type IV secretion system effectors 1.0: A tool for predicting type IV effectors and exploring their genomic context. Nucleic Acids Res. 2013, 41, 9218–9229. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, X.; Bao, H.; Liu, S.-L. Prediction of bacterial type IV secreted effectors by C-terminal features. BMC Genom. 2014, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Noroy, C.; Lefrancois, T.; Meyer, D.F. Searching algorithm for Type IV effector proteins (S4TE) 2.0: Improved tools for Type IV effector prediction, analysis and comparison in proteobacteria. PLoS Comput. Biol. 2019, 15, e1006847. [Google Scholar] [CrossRef]
- Zou, L.; Nan, C.; Hu, F. Accurate prediction of bacterial type IV secreted effectors using amino acid composition and PSSM profiles. Bioinformatics 2013, 29, 3135–3142. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, Q.; Yang, J.; Zhu, X.; Weil, D.-Q. PredT4SE-Stack: Prediction of Bacterial Type IV Secreted Effectors From Protein Sequences Using a Stacked Ensemble Method. Front. Microbiol. 2018, 9, 2571. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Tang, B.; Chen, W.; Luo, J. A deep learning framework for sequence-based bacteria type IV secreted effectors prediction. Chemom. Intell. Lab. Syst. 2018, 183, 134–139. [Google Scholar] [CrossRef]
- Wang, J.; Yang, B.; An, Y.; Marquez-Lago, T.; Leier, A.; Wilksch, J.; Hong, Q.; Zhang, Y.; Hayashida, M.; Akutsu, T.; et al. Systematic analysis and prediction of type IV secreted effector proteins by machine learning approaches. Brief. Bioinform. 2019, 20, 931–951. [Google Scholar] [CrossRef]
- Ashari, Z.E.; Brayton, K.A.; Broschat, S.L. Prediction of T4SS Effector Proteins for Anaplasma phagocytophilum Using OPT4e, A New Software Tool. Front. Microbiol. 2019, 10, 1391. [Google Scholar] [CrossRef]
- Hong, J.; Luo, Y.; Mou, M.; Fu, J.; Zhang, Y.; Xue, W.; Xie, T.; Tao, L.; Lou, Y.; Zhu, F. Convolutional neural network-based annotation of bacterial type IV secretion system effectors with enhanced accuracy and reduced false discovery. Brief. Bioinform. 2020, 21, 1825–1836. [Google Scholar] [CrossRef]
- Chen, T.; Wang, X.; Chu, Y.; Wang, Y.; Jiang, M.; Wei, D.-Q.; Xiong, Y. T4SE-XGB: Interpretable Sequence-Based Prediction of Type IV Secreted Effectors Using eXtreme Gradient Boosting Algorithm. Front. Microbiol. 2020, 11, 580382. [Google Scholar] [CrossRef]
- Burstein, D.; Zusman, T.; Degtyar, E.; Viner, R.; Segal, G.; Pupko, T. Genome-Scale Identification of Legionella pneumophila Effectors Using a Machine Learning Approach. PLoS Pathog. 2009, 5, e1000508. [Google Scholar] [CrossRef]
- An, Y.; Wang, J.; Li, C.; Leier, A.; Marquez-Lago, T.; Wilksch, J.; Zhang, Y.; Webb, G.I.; Song, J.; Lithgow, T. Comprehensive assessment and performance improvement of effector protein predictors for bacterial secretion systems III, IV and VI. Brief. Bioinform. 2018, 19, 148–161. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.; Pu, X.; Li, M. Effective prediction of bacterial type IV secreted effectors by combined features of both C-termini and N-termini. J. Comput.-Aided Mol. Des. 2017, 31, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Z.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, B.; Revote, J.; Leier, A.; Marquez-Lago, T.T.; Webb, G.; Song, J.; Chou, K.-C.; Lithgow, T. POSSUM: A bioinformatics toolkit for generating numerical sequence feature descriptors based on PSSM profiles. Bioinformatics 2017, 33, 2756–2758. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; UniProt, C. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zheng, X.; Wang, J. Prediction of protein structural class for low-similarity sequences using support vector machine and PSI-BLAST profile. Biochimie 2010, 92, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, X.; Kong, L. Predict protein structural class for low-similarity sequences by evolutionary difference information into the general form of Chou’s pseudo amino acid composition. J. Theor. Biol. 2014, 355, 105–110. [Google Scholar] [CrossRef]
- Kidera, A.; Konishi, Y.; Oka, M.; Ooi, T.; Scheraga, H.A. Statistical analysis of the physical properties of the 20 naturally occurring amino acids. J. Protein Chem. 1985, 4, 23–55. [Google Scholar] [CrossRef]
- Du, P.; Yu, Y. SubMito-PSPCP: Predicting Protein Submitochondrial Locations by Hybridizing Positional Specific Physicochemical Properties with Pseudoamino Acid Compositions. Biomed Res. Int. 2013, 2013, 263829. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Zhou, S.; Guan, J. A new taxonomy-based protein fold recognition approach based on autocross-covariance transformation. Bioinformatics 2009, 25, 2655–2662. [Google Scholar] [CrossRef]
- Shensa, M.J. The discrete wavelet transform: Wedding the a trous and Mallat algorithms. IEEE Trans. Signal Process. 1992, 40, 2464–2482. [Google Scholar] [CrossRef]
- Nanni, L.; Brahnam, S.; Lumini, A. Wavelet images and Chou’s pseudo amino acid composition for protein classification. Amino Acids 2012, 43, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef]
- Bressin, A.; Schulte-Sasse, R.; Figini, D.; Urdaneta, E.C.; Beckmann, B.M.; Marsico, A. TriPepSVM: De novo prediction of RNA-binding proteins based on short amino acid motifs. Nucleic Acids Res. 2019, 47, 4406–4417. [Google Scholar] [CrossRef]
- Garg, A.; Singhal, N.; Kumar, R.; Kumar, M. mRNALoc: A novel machine-learning based in-silico tool to predict mRNA subcellular localization. Nucleic Acids Res. 2020, 48, W239–W243. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Jiang, P.; Wu, H.; Wang, W.; Ma, W.; Sun, X.; Lu, Z. MiPred: Classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res. 2007, 35, W339–W344. [Google Scholar] [CrossRef]
- Hooghe, B.; Broos, S.; van Roy, F.; De Bleser, P. A flexible integrative approach based on random forest improves prediction of transcription factor binding sites. Nucleic Acids Res. 2012, 40, e106. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y. Random Forest for Bioinformatics. In Ensemble Machine Learning: Methods and Applications; Zhang, C., Ma, Y., Eds.; Springer US: Boston, MA, USA, 2012; pp. 307–323. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Feature | Acc | Sen | Spe | Pre | F-Score | MCC | AUC |
|---|---|---|---|---|---|---|---|---|
| SVM | PSSM_AAC (20D) | 0.908 | 0.836 | 0.944 | 0.882 | 0.858 | 0.791 | 0.928 |
| PSPCP_ACC (1000D) | 0.834 | 0.645 | 0.929 | 0.820 | 0.722 | 0.616 | 0.868 | |
| PSFM_DWT (1040D) | 0.902 | 0.793 | 0.957 | 0.902 | 0.844 | 0.777 | 0.927 | |
| PSSM_EDT (1600D) | 0.920 | 0.845 | 0.957 | 0.908 | 0.876 | 0.818 | 0.943 | |
| All features (3660D) | 0.919 | 0.845 | 0.955 | 0.905 | 0.874 | 0.816 | 0.935 | |
| RF | PSSM_AAC (20D) | 0.904 | 0.809 | 0.952 | 0.894 | 0.850 | 0.782 | 0.933 |
| PSPCP_ACC (1000D) | 0.803 | 0.563 | 0.922 | 0.785 | 0.656 | 0.537 | 0.834 | |
| PSFM_DWT (1040D) | 0.893 | 0.777 | 0.952 | 0.890 | 0.830 | 0.757 | 0.924 | |
| PSSM_EDT (1600D) | 0.900 | 0.806 | 0.947 | 0.884 | 0.843 | 0.772 | 0.940 | |
| All features (3660D) | 0.907 | 0.816 | 0.952 | 0.895 | 0.854 | 0.788 | 0.937 |
| Dataset | Feature | Acc | Sen | Spe | Pre | F-Score | MCC |
|---|---|---|---|---|---|---|---|
| Train-915 | All features (3660D) | 0.919 | 0.845 | 0.955 | 0.905 | 0.874 | 0.816 |
| Optimal subset (320D) | 0.924 | 0.845 | 0.963 | 0.921 | 0.882 | 0.828 | |
| Train-1502 | All features (3660D) | 0.954 | 0.879 | 0.981 | 0.942 | 0.909 | 0.880 |
| Optimal subset (180D) | 0.950 | 0.861 | 0.981 | 0.941 | 0.899 | 0.867 |
| Method | Acc | Sen | Spe | Pre | F-Score | MCC |
|---|---|---|---|---|---|---|
| Wang et al.’s method [15] | 0.853 | 0.907 | 0.848 | 0.366 | 0.521 | 0.518 |
| PredT4SE-Stack (SVM) | 0.945 | 0.867 | 0.952 | 0.637 | 0.734 | 0.715 |
| PredT4SE-Stack (LR) | 0.944 | 0.880 | 0.950 | 0.629 | 0.733 | 0.715 |
| iT4SE-EP (320D) | 0.956 | 0.893 | 0.962 | 0.697 | 0.783 | 0.766 |
| Method | Acc | Sen | Spe | Pre | F-Score | MCC |
|---|---|---|---|---|---|---|
| T4SEpre_psAac | 0.889 | 0.367 | 0.993 | 0.917 | 0.523 | 0.537 |
| T4SEpre_Joint | 0.906 | 0.500 | 0.987 | 0.882 | 0.638 | 0.620 |
| T4SEpre_bpbAac | 0.889 | 0.433 | 0.980 | 0.813 | 0.565 | 0.541 |
| Bastion4 | 0.950 | 0.967 | 0.947 | 0.784 | 0.865 | 0.842 |
| CNN-PSSSA | 0.956 | 0.767 | 0.993 | 0.958 | 0.851 | 0.833 |
| CNN-Onehot | 0.967 | 0.800 | 1.000 | 1.000 | 0.888 | 0.877 |
| CNN-PSSM | 0.989 | 0.967 | 0.993 | 0.967 | 0.966 | 0.960 |
| iT4SE-EP (180D) | 0.966 | 1.000 | 0.96 | 0.833 | 0.909 | 0.894 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, H.; Ding, C.; Cheng, X.; Sang, X.; Liu, T. iT4SE-EP: Accurate Identification of Bacterial Type IV Secreted Effectors by Exploring Evolutionary Features from Two PSI-BLAST Profiles. Molecules 2021, 26, 2487. https://doi.org/10.3390/molecules26092487
Han H, Ding C, Cheng X, Sang X, Liu T. iT4SE-EP: Accurate Identification of Bacterial Type IV Secreted Effectors by Exploring Evolutionary Features from Two PSI-BLAST Profiles. Molecules. 2021; 26(9):2487. https://doi.org/10.3390/molecules26092487
Chicago/Turabian StyleHan, Haitao, Chenchen Ding, Xin Cheng, Xiuzhi Sang, and Taigang Liu. 2021. "iT4SE-EP: Accurate Identification of Bacterial Type IV Secreted Effectors by Exploring Evolutionary Features from Two PSI-BLAST Profiles" Molecules 26, no. 9: 2487. https://doi.org/10.3390/molecules26092487
APA StyleHan, H., Ding, C., Cheng, X., Sang, X., & Liu, T. (2021). iT4SE-EP: Accurate Identification of Bacterial Type IV Secreted Effectors by Exploring Evolutionary Features from Two PSI-BLAST Profiles. Molecules, 26(9), 2487. https://doi.org/10.3390/molecules26092487