
Ramatroban-Based Analogues Containing Fluorine Group as Potential 18F-Labeled Positron Emission Tomography (PET) G-Protein Coupled Receptor 44 (GPR44) Tracers



Abstract
1. Introduction
1.1. Positron Emission Tomography (PET) Imaging for Beta Cell Mass (BCM)
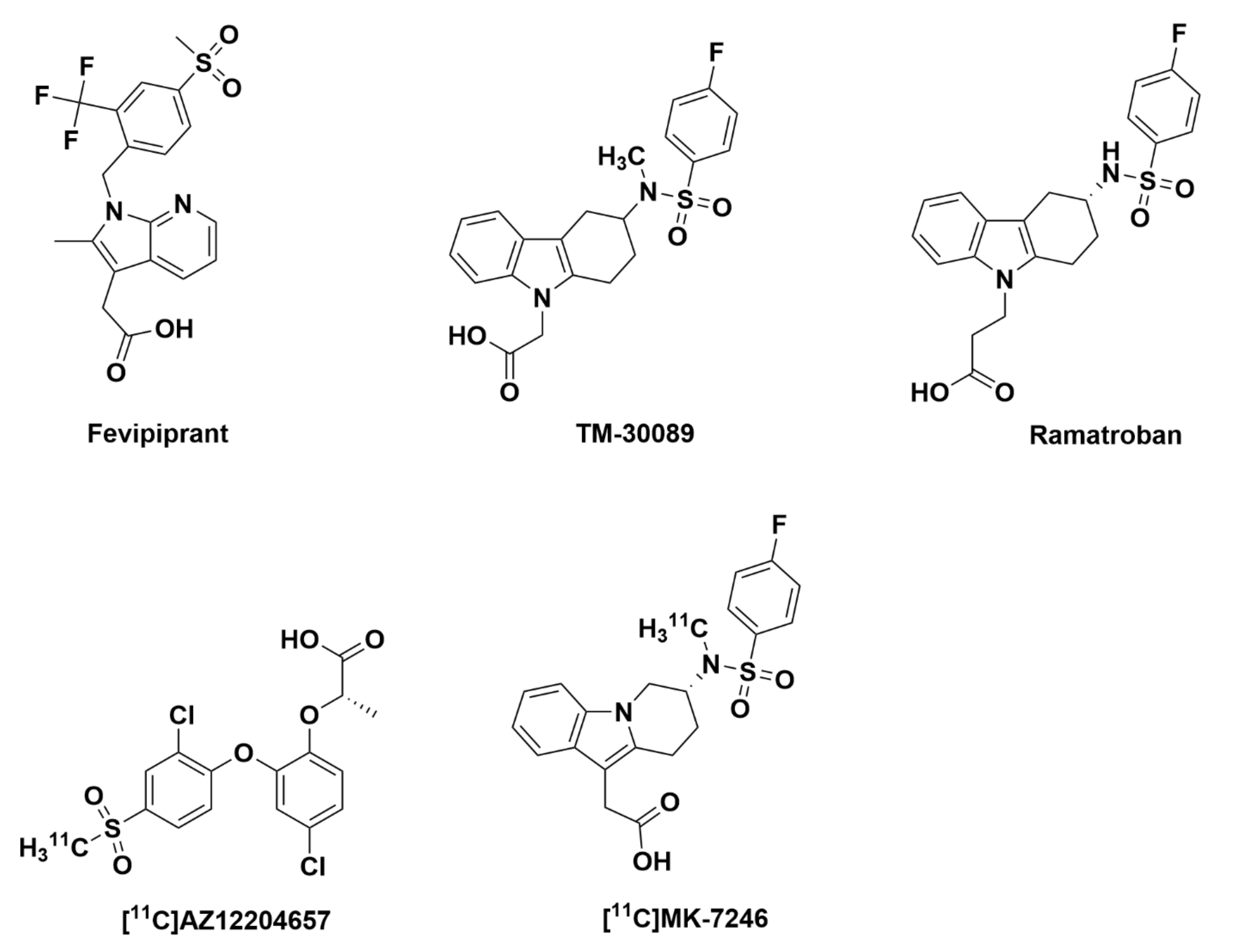
1.2. G-Protein Coupled Receptor 44 (GPR44) and Related PET Tracers
2. Ramatroban-Based Analogues for Potential GPR44 Binding
2.1. Ramatroban
2.2. Ramatroban Analogues
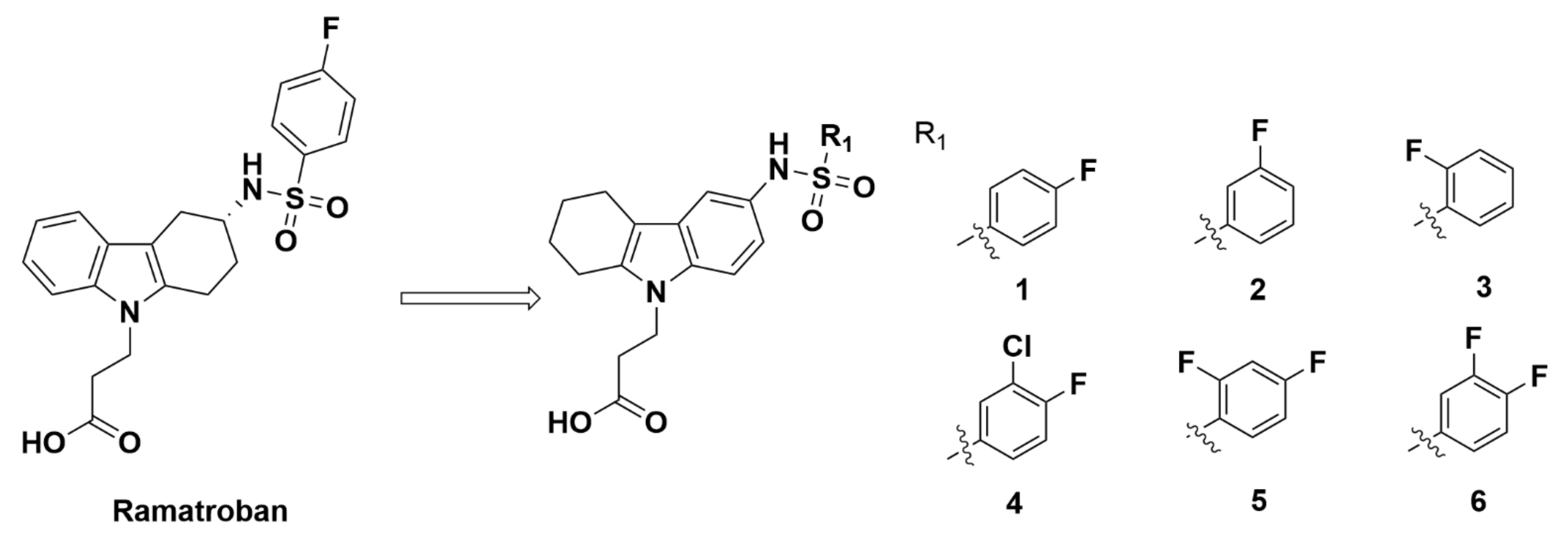
2.2.1. Athersys, Inc.
Recommendations
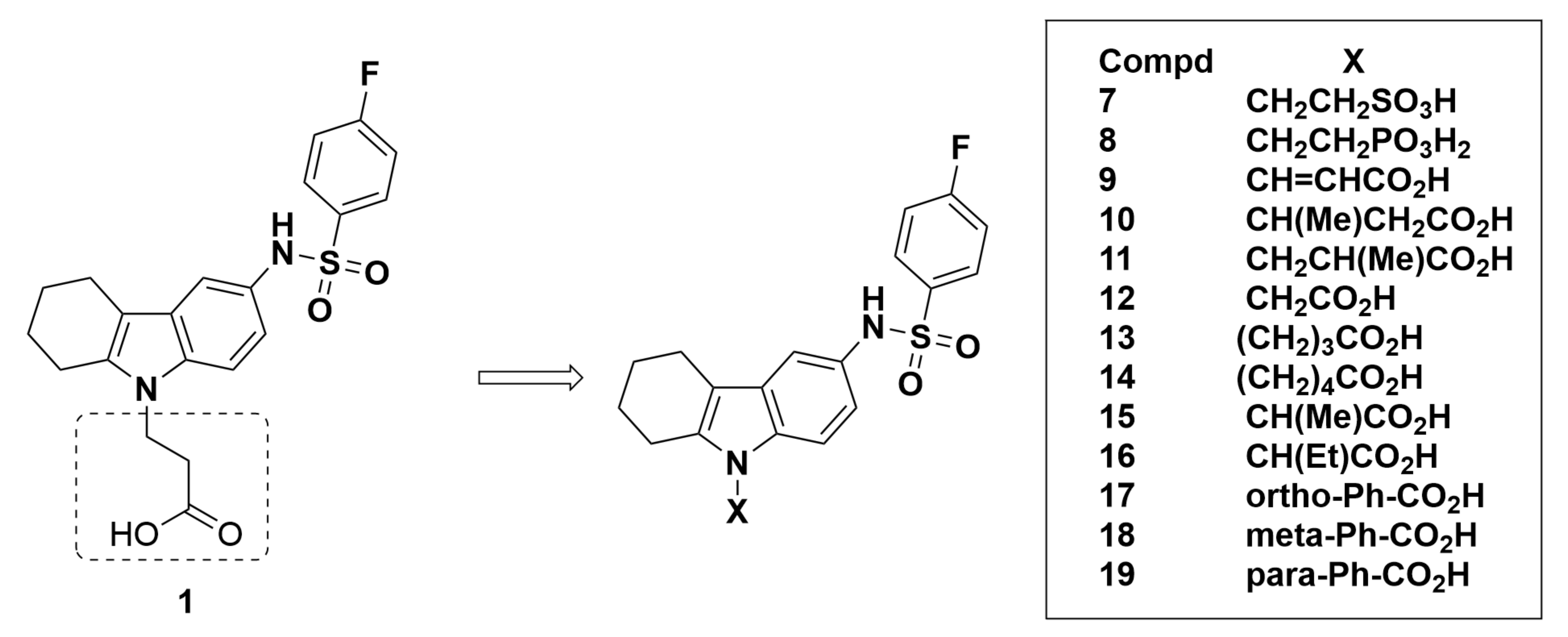
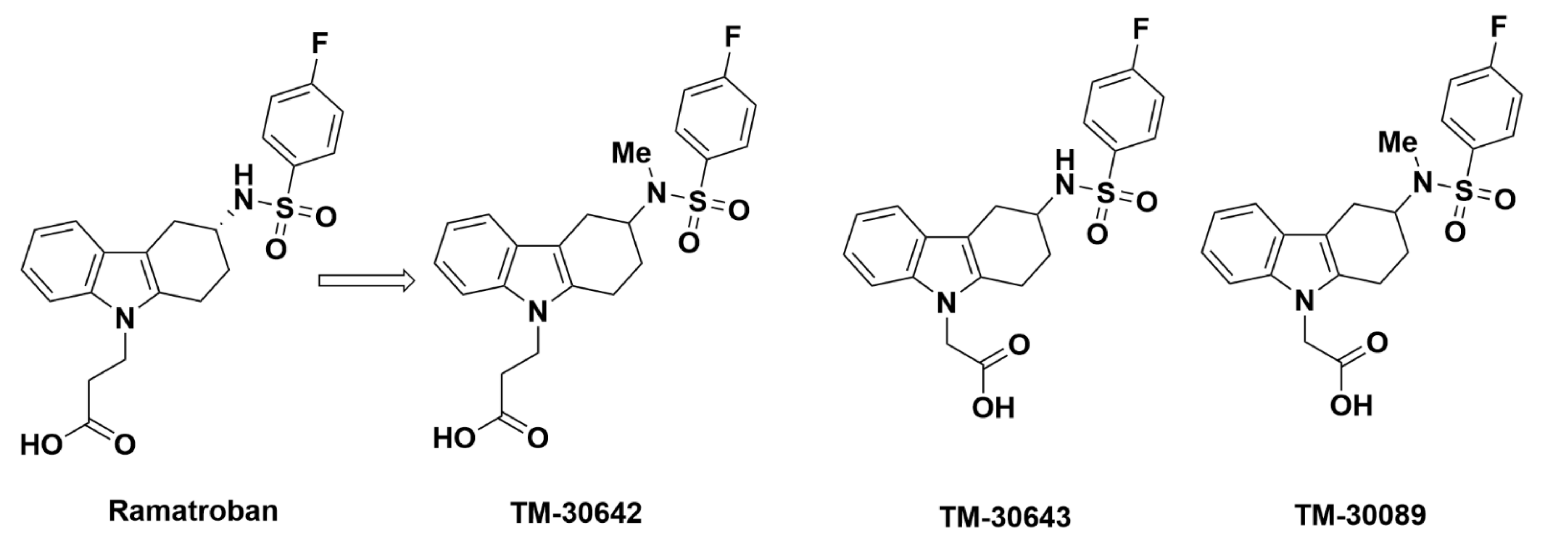
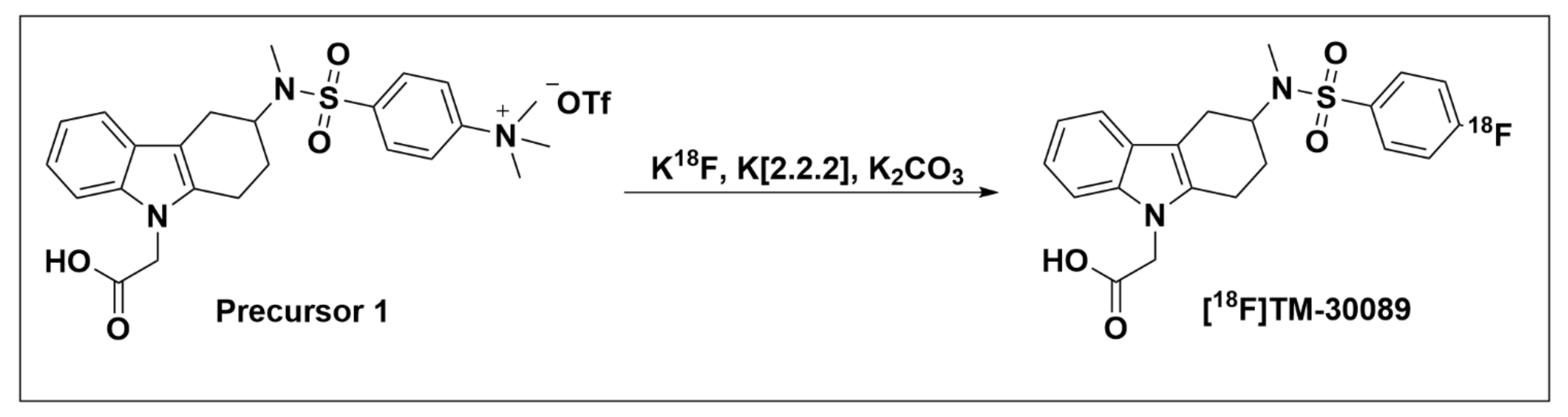
2.2.2. TM-30642, TM-30643, and TM-30089
Recommendations
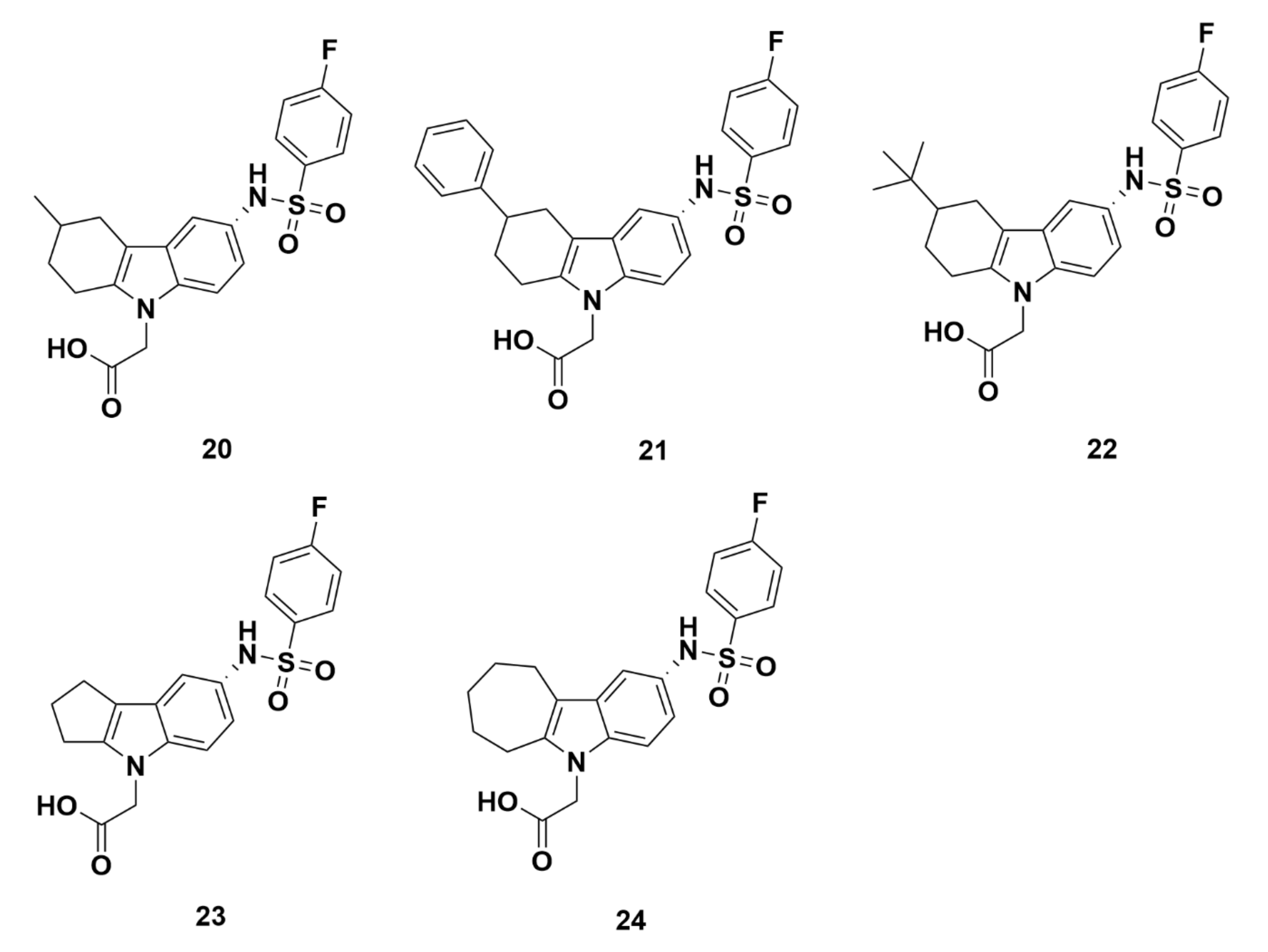
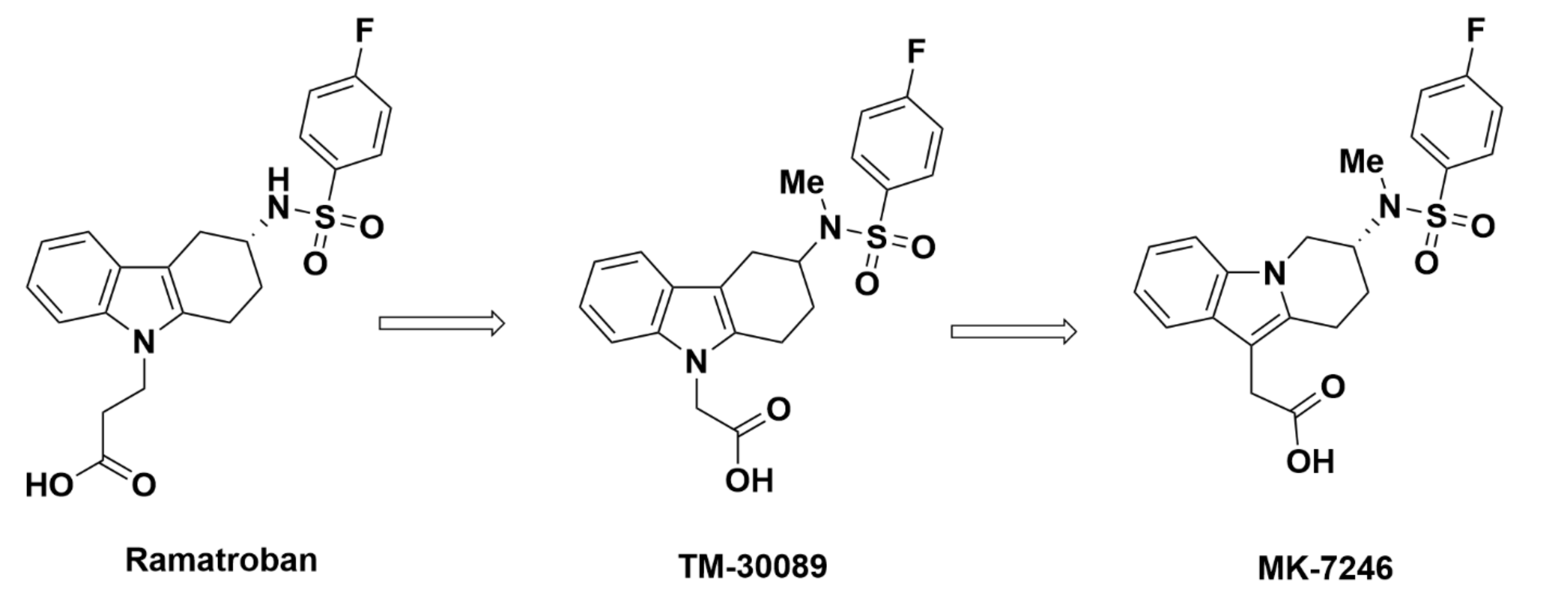
2.2.3. Merck Analogues
MK-7246
Recommendations
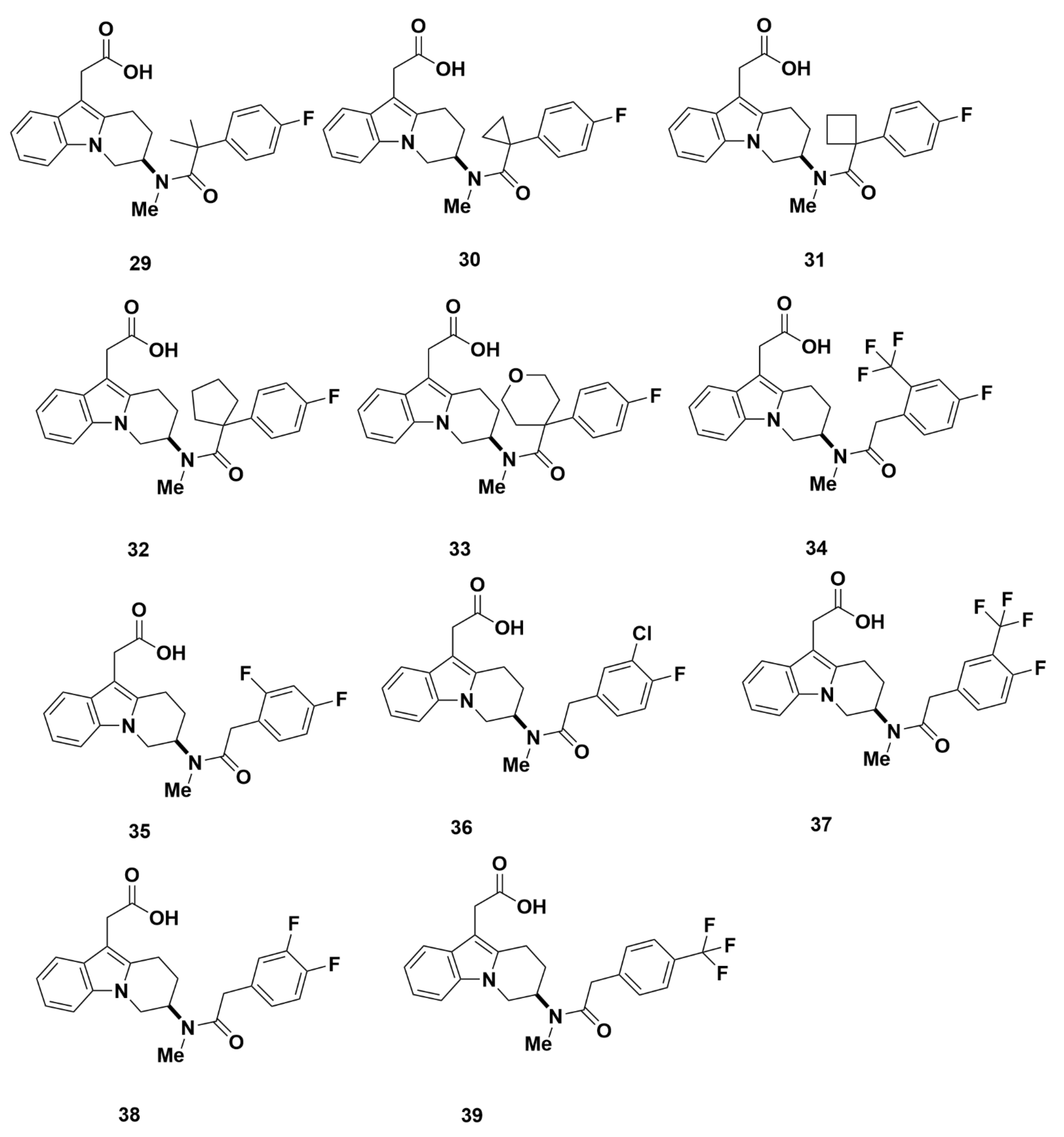
Merck Analogues in 2008
Merck Analogues in 2011a
Recommendations
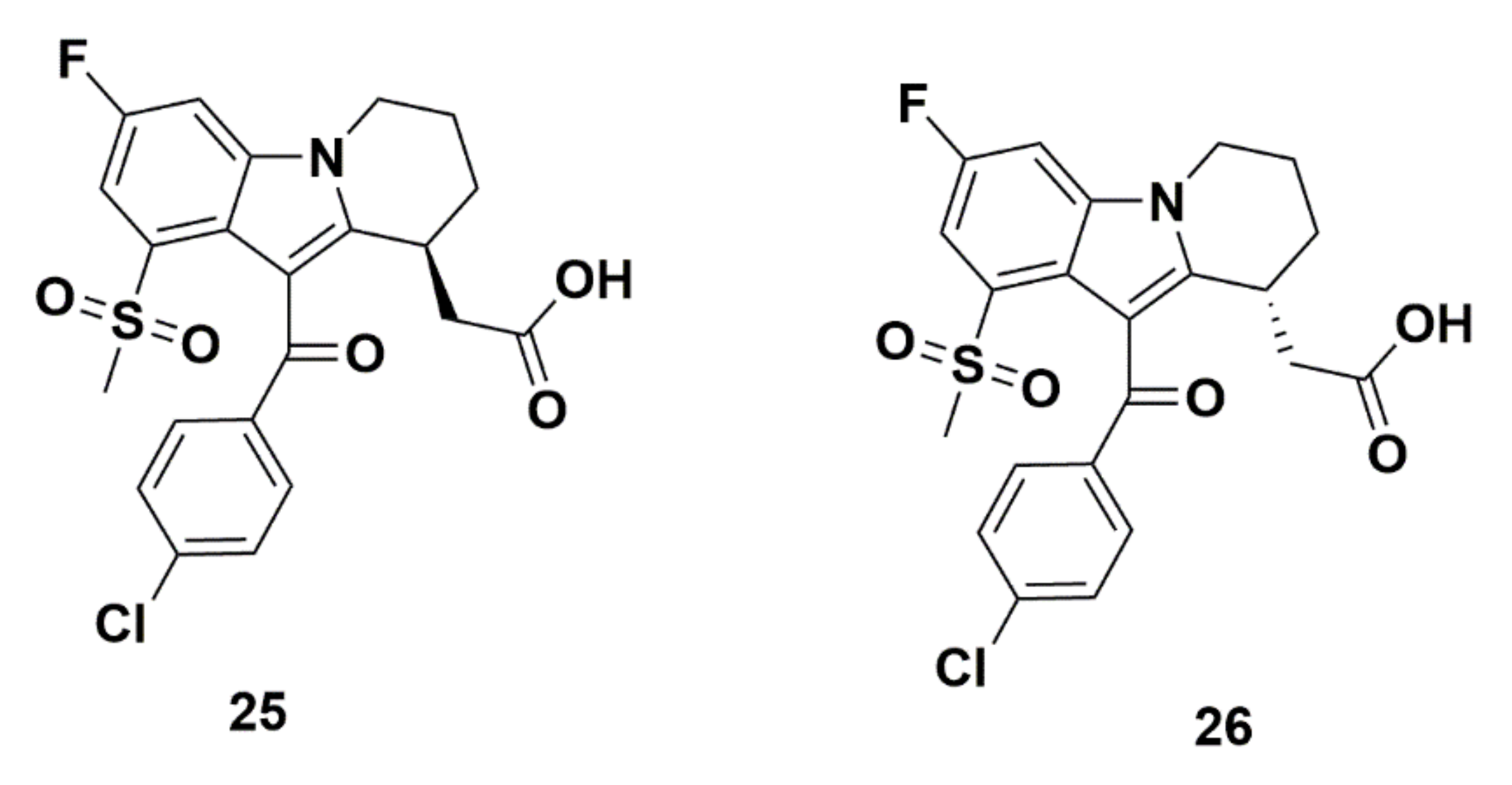
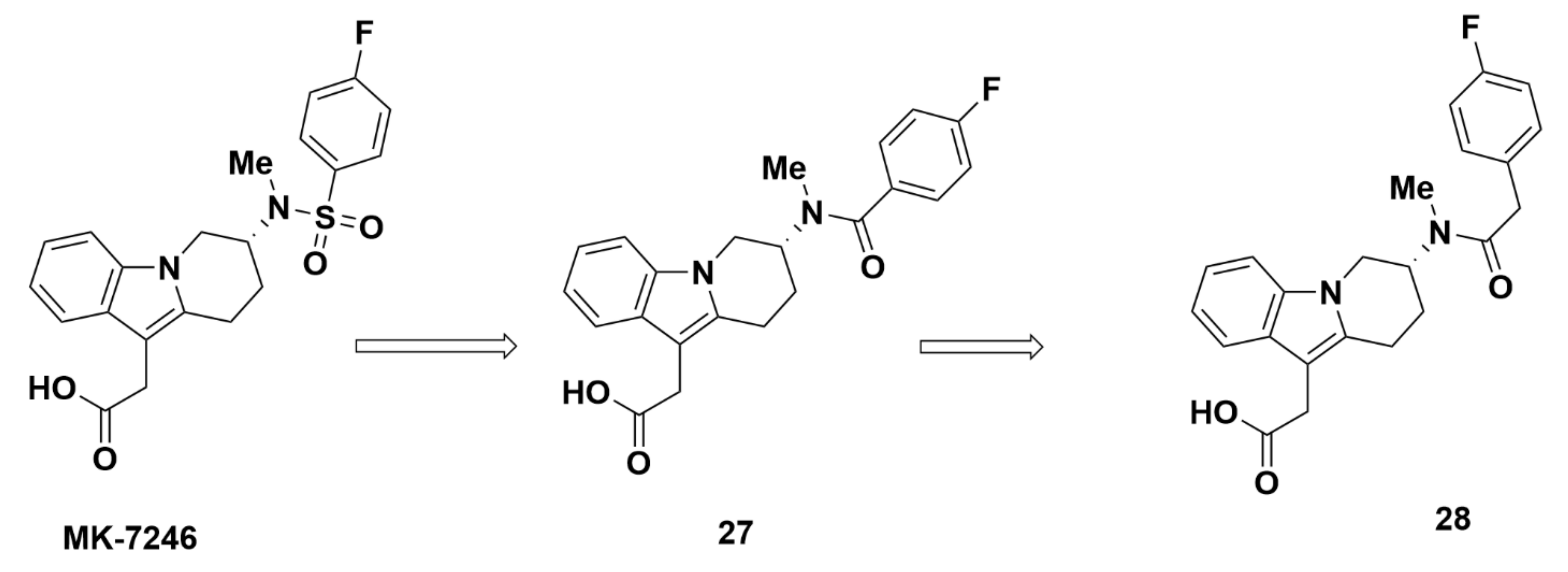
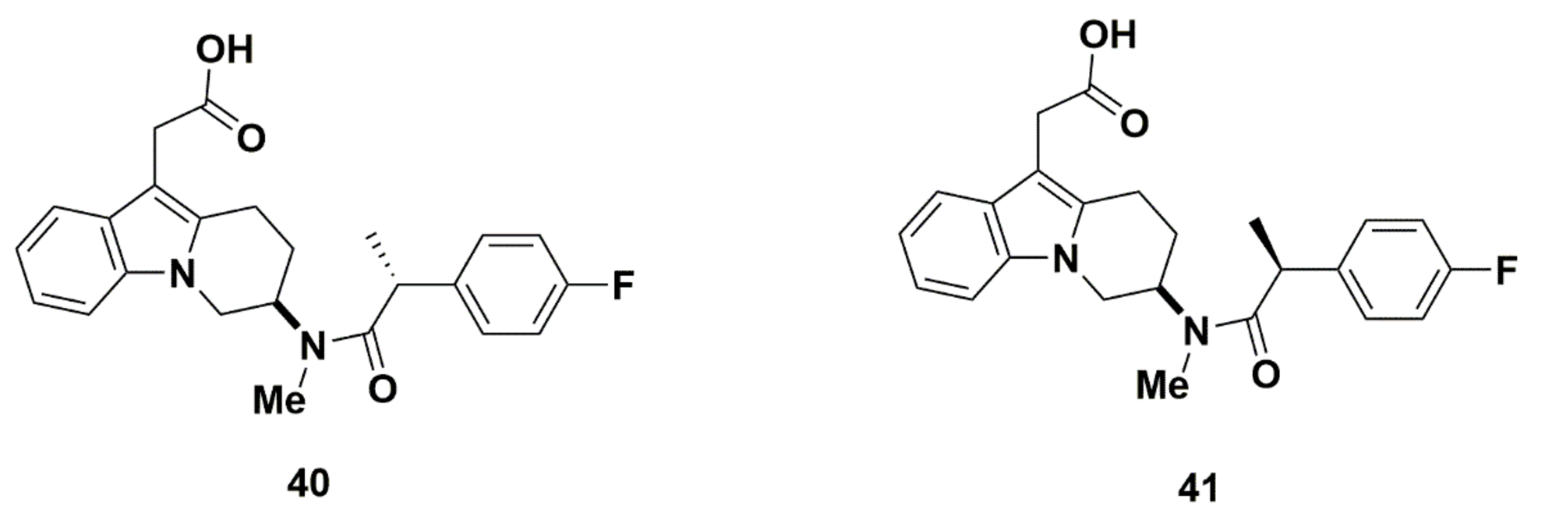
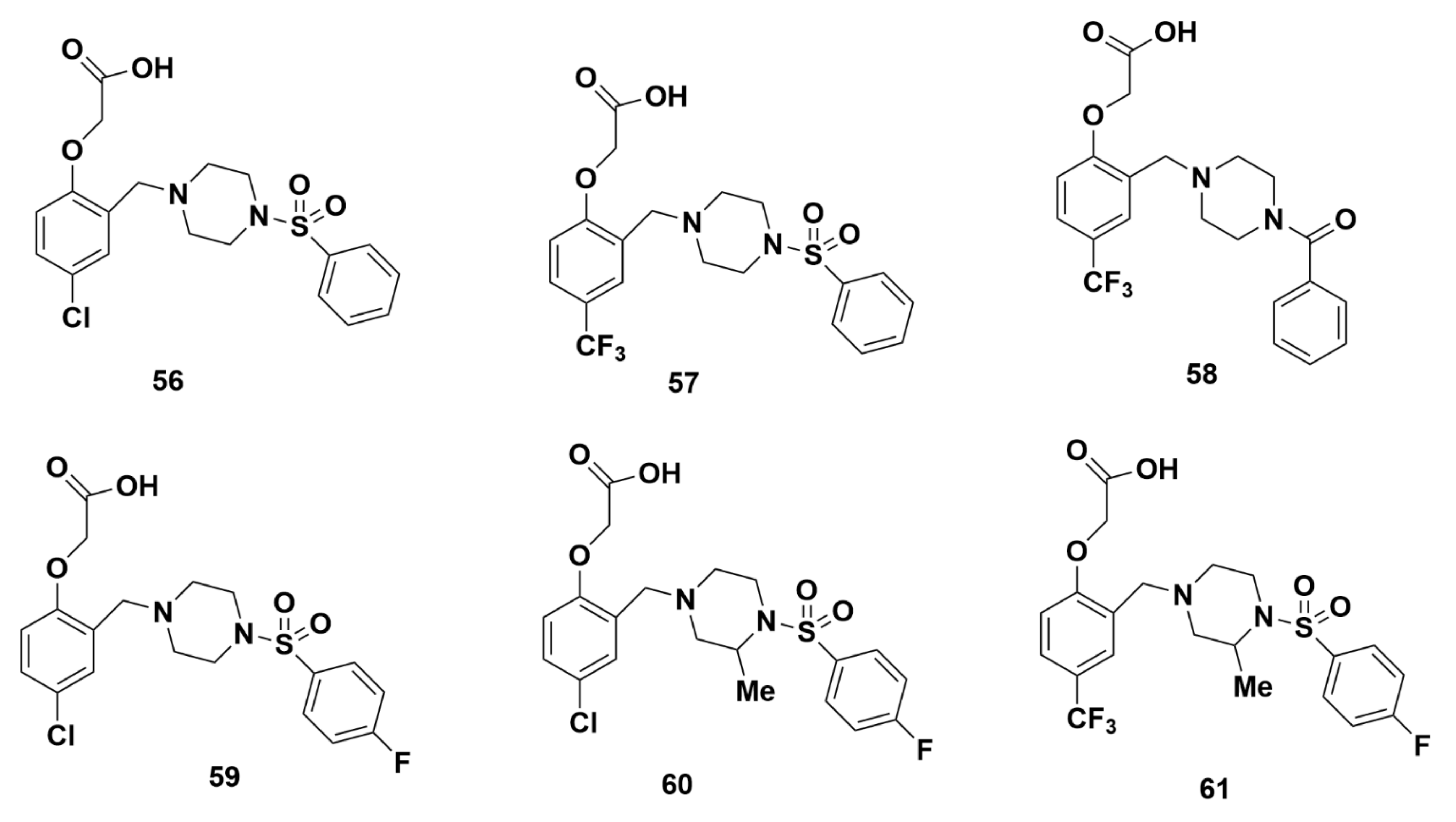
Merck Analogues in 2011b
Recommendations
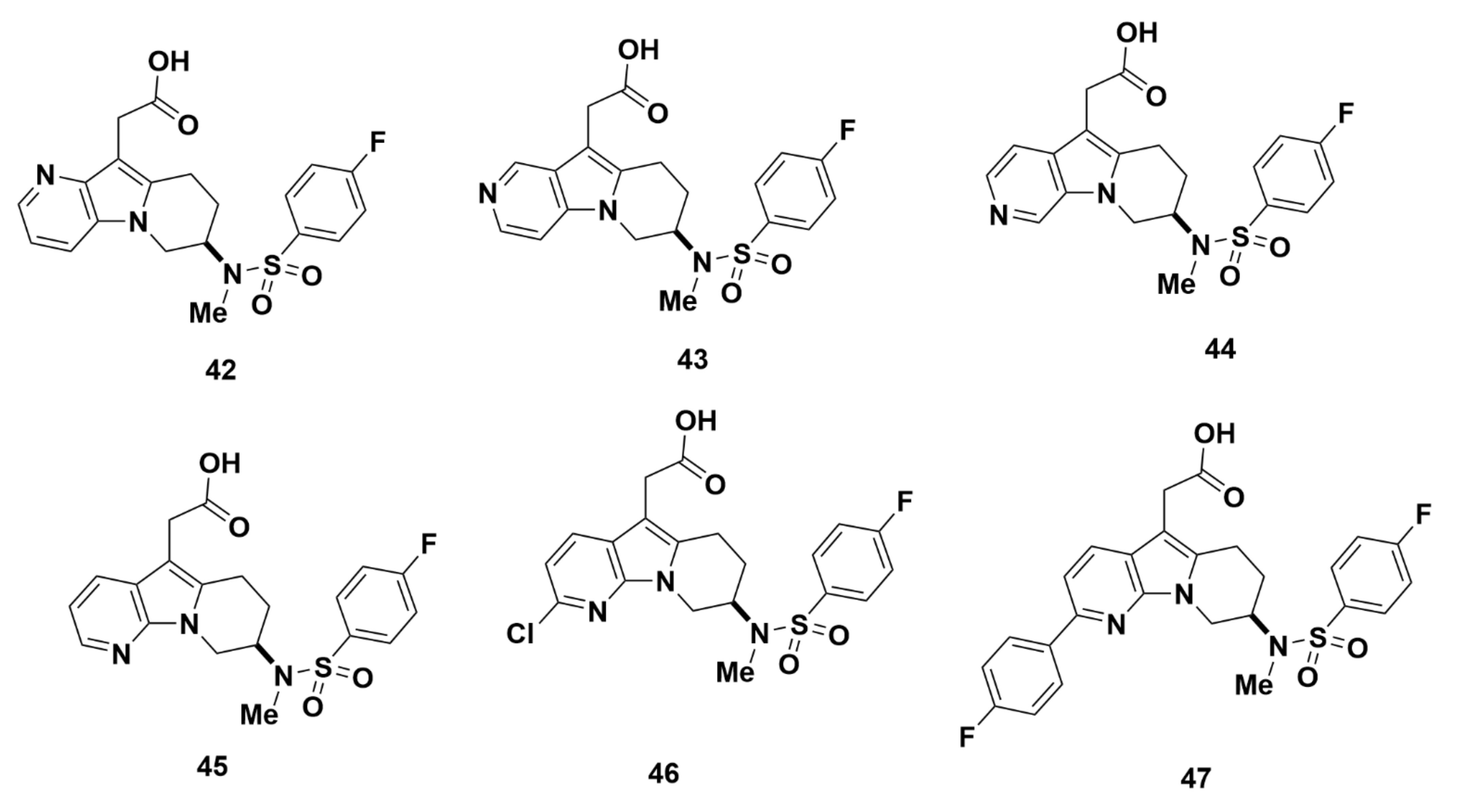
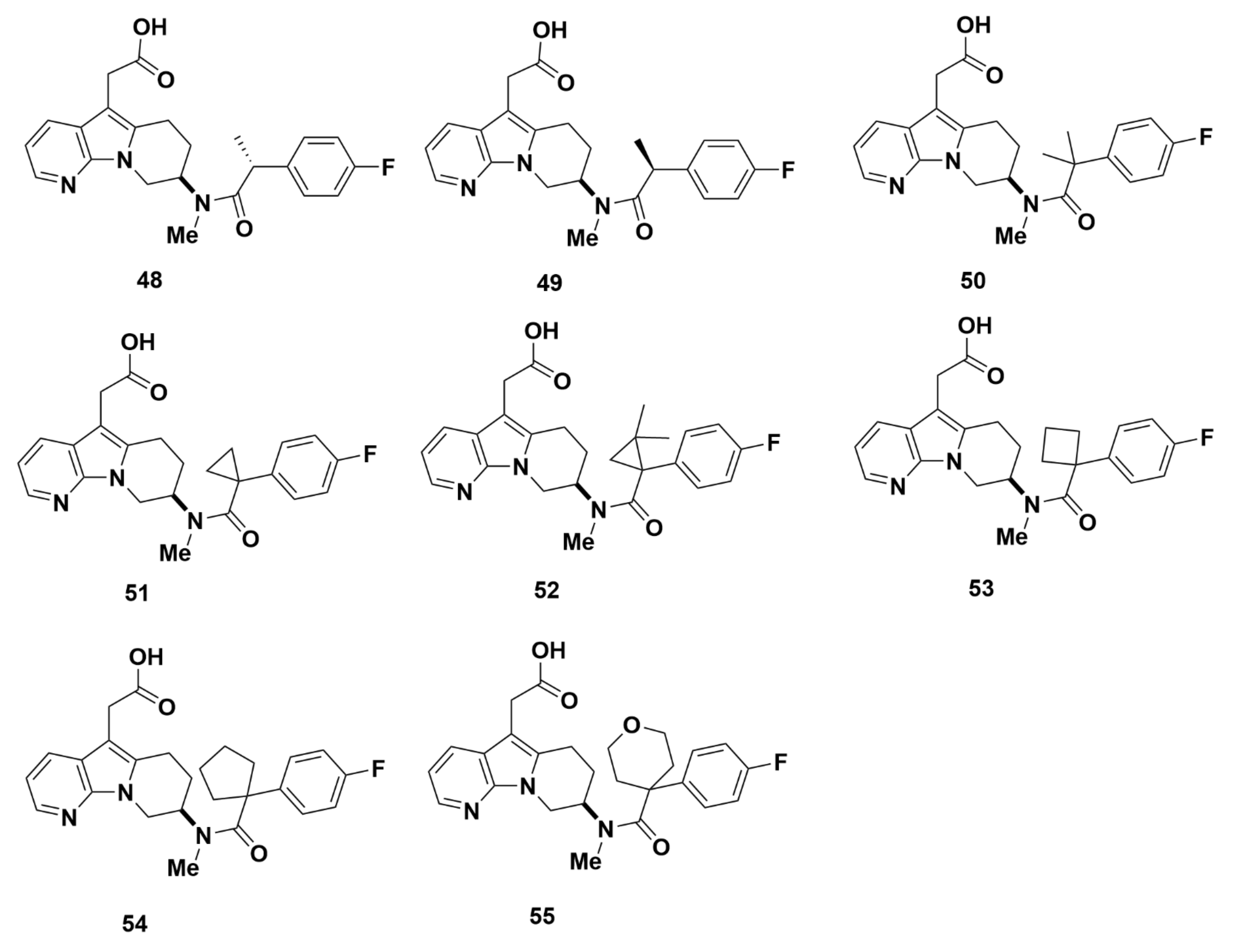
2.2.4. AstraZeneca
Recommendations
2.2.5. Actelion Pharmaceuticals Ltd.
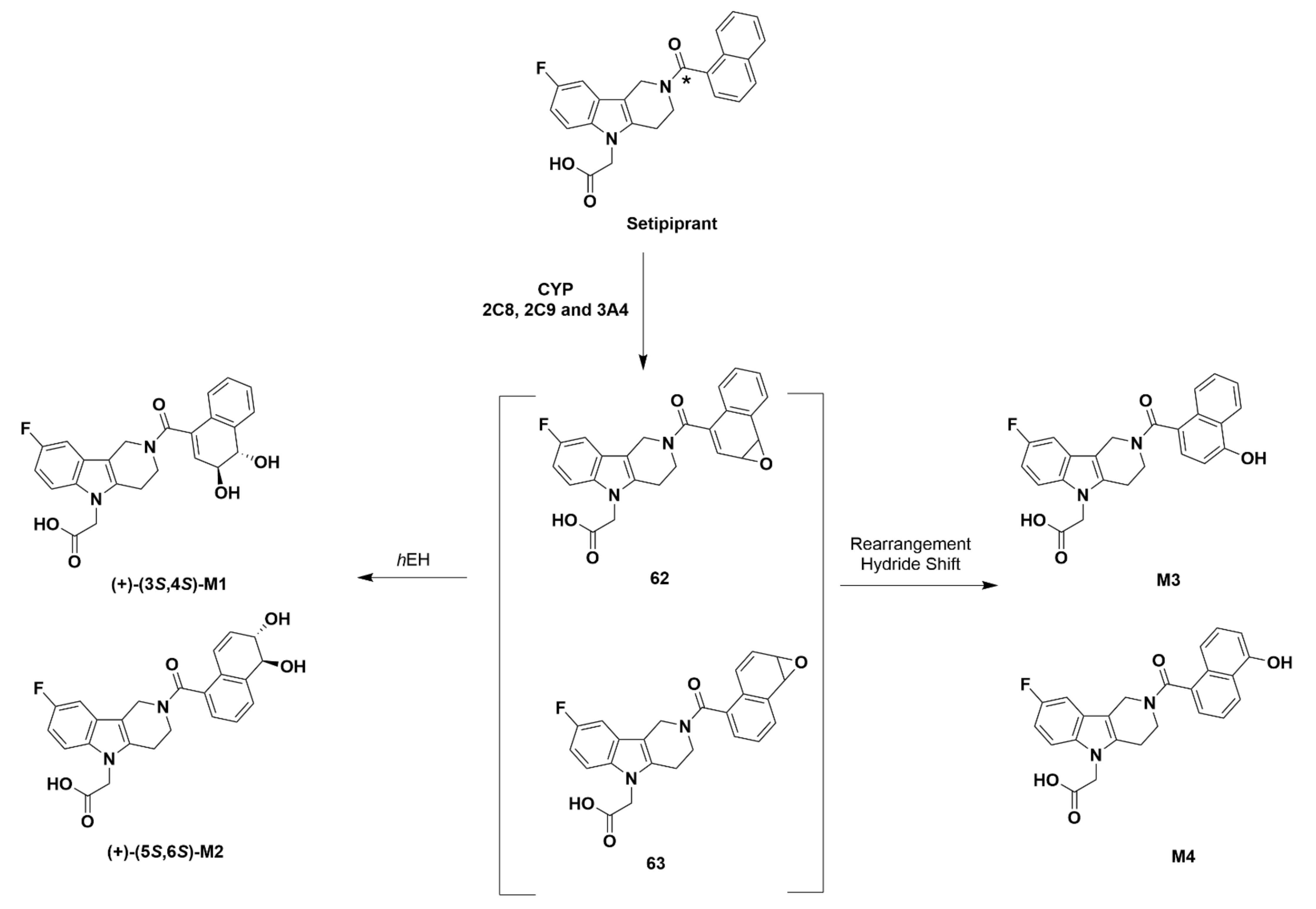
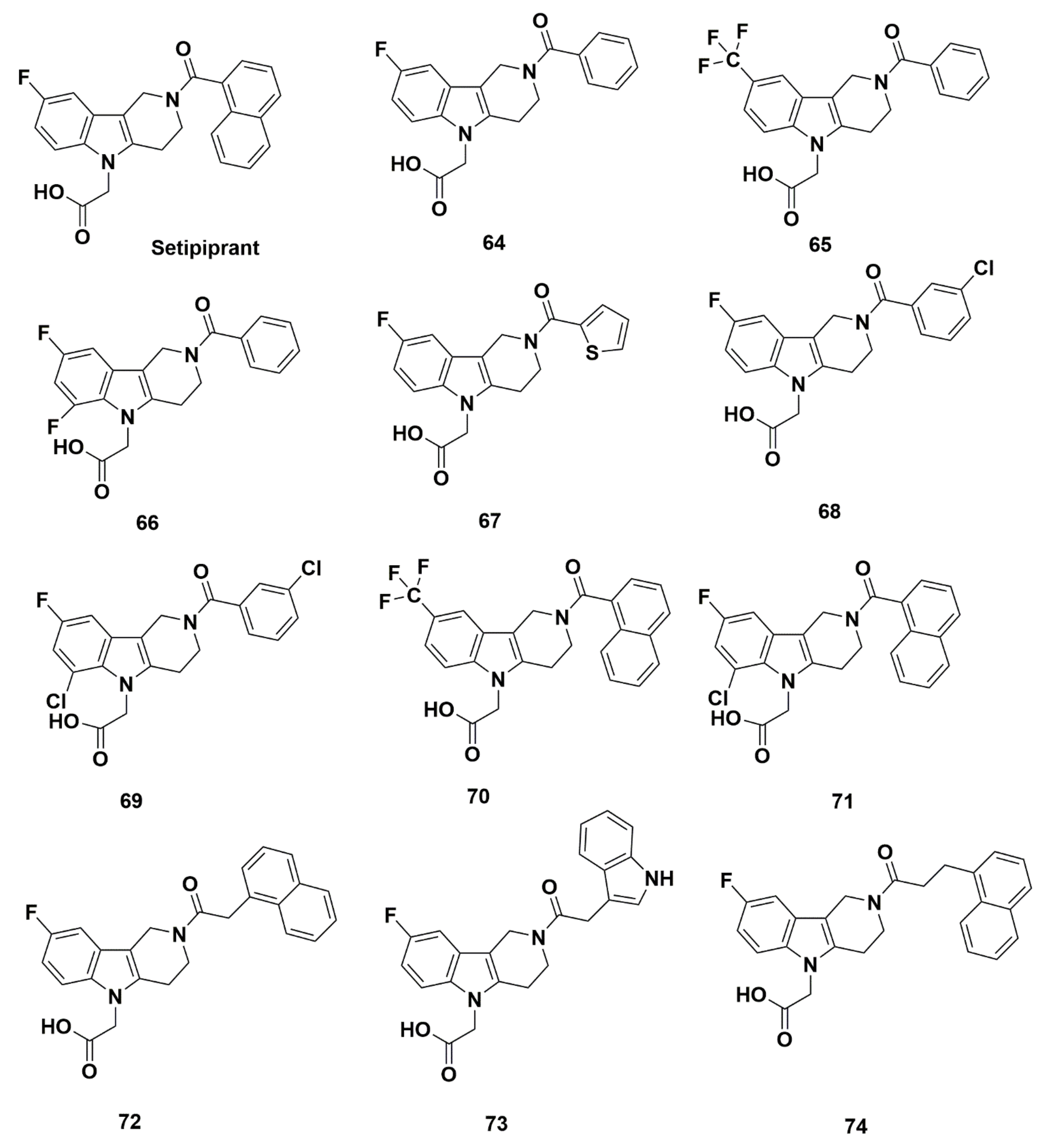
Setipiprant
Recommendations
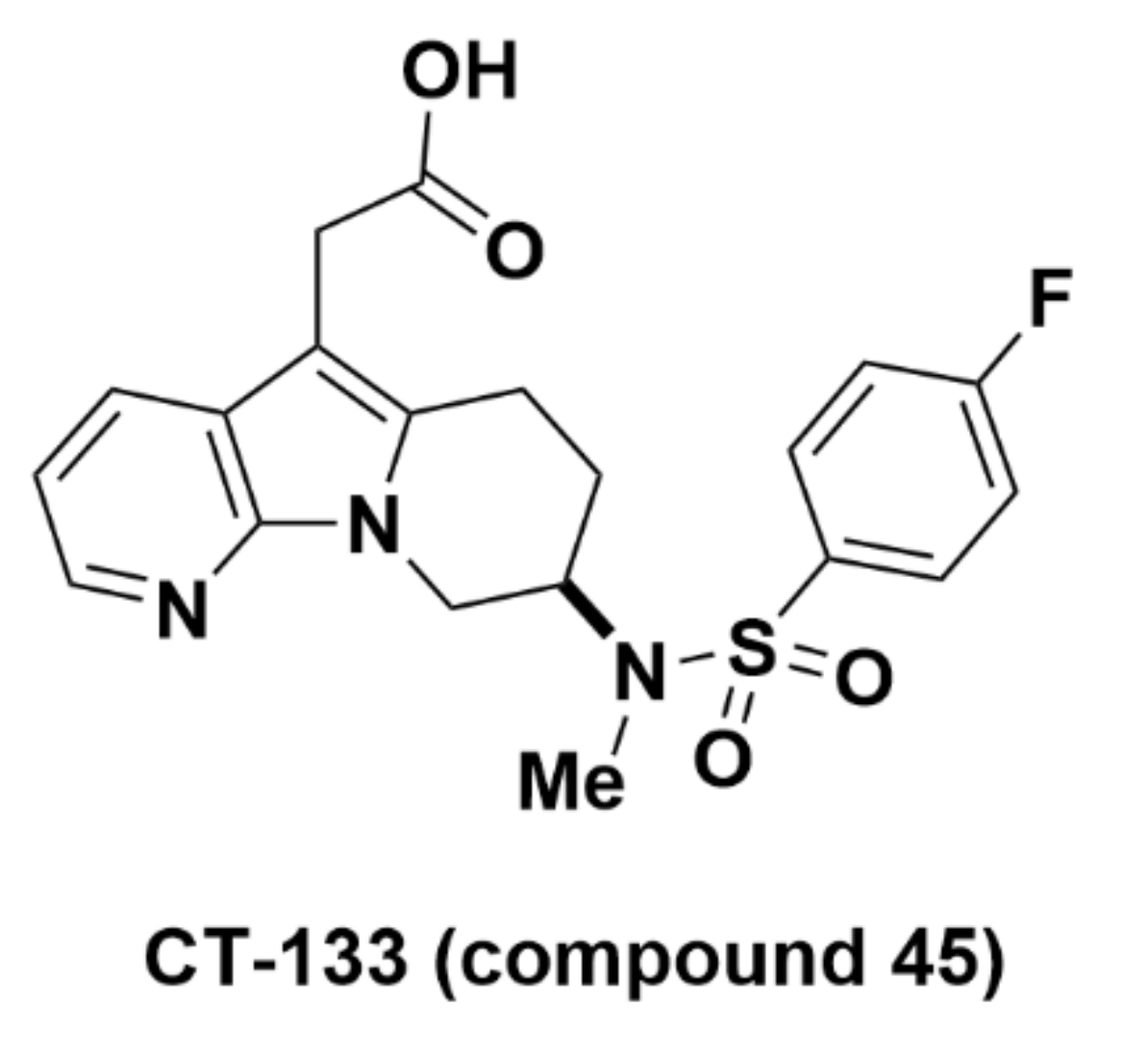
2.2.6. CT-133 (Compound 45)
Recommendations
2.3. Radiosynthesis Strategies for 18F-Labeled Ramatroban-Based Analogues
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.-F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Eriksson, O.; Laughlin, M.; Brom, M.; Nuutila, P.; Roden, M.; Hwa, A.; Bonadonna, R.; Gotthardt, M. In vivo imaging of beta cells with radiotracers: State of the art, prospects and recommendations for development and use. Diabetologia 2016, 59, 1340–1349. [Google Scholar] [CrossRef][Green Version]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Krogvold, L.; Edwin, B.; Buanes, T.; Ludvigsson, J.; Korsgren, O.; Hyöty, H.; Frisk, G.; Hanssen, K.F.; Dahl-Jørgensen, K. Pancreatic biopsy by minimal tail resection in live adult patients at the onset of type 1 diabetes: Experiences from the DiViD study. Diabetologia 2014, 57, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.-C.; Dufrane, D.; Nenquin, M. Nutrient control of insulin secretion in isolated normal human islets. Diabetes 2006, 55, 3470–3477. [Google Scholar] [CrossRef] [PubMed]
- Sweet, I.R.; Cook, D.L.; Lernmark, Å.; Greenbaum, C.J.; Krohn, K.A. Non—Invasive imaging of beta cell mass: A quantitative analysis. Diabetes Technol. Ther. 2004, 6, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ehlerding, E.B.; Lan, X.; Luo, Q.Y.; Cai, W. Molecular imaging of beta-cells: Diabetes and beyond. Adv. Drug. Deliv. Rev. 2019, 139, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O. GPR44 as a target for imaging pancreatic beta-cell mass. Curr. Diabetes Rep. 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Marchese, A.; George, S.R.; Kolakowski, L.F.; Lynch, K.R.; O’Dowd, B.F. Novel GPCRs and their endogenous ligands: Expanding the boundaries of physiology and pharmacology. Trends Pharmacol. Sci. 1999, 20, 370–375. [Google Scholar] [CrossRef]
- Marchese, A.; Sawzdargo, M.; Nguyen, T.; Cheng, R.; Heng, H.H.; Nowak, T.; Im, D.S.; Lynch, K.R.; George, S.R.; O’Dowd, B.F. Discovery of three novel orphan G-protein-coupled receptors. Genomics 1999, 56, 12–21. [Google Scholar] [CrossRef]
- Lindskog, C.; Korsgren, O.; Pontén, F.; Eriksson, J.W.; Johansson, L.; Danielsson, A. Novel pancreatic beta cell-specific proteins: Antibody—Based proteomics for identification of new biomarker candidates. J. Proteom. 2012, 75, 2611–2620. [Google Scholar] [CrossRef]
- Jahan, M.; Johnström, P.; Selvaraju, R.K.; Svedberg, M.; Winzell, M.S.; Bernström, J.; Kingston, L.; Schou, M.; Jia, Z.; Skrtic, S.; et al. The development of a GPR44 targeting radioligand [(11)C]AZ12204657 for in vivo assessment of beta cell mass. Eur. J. Nucl. Med. Mol. Imaging Res. 2018, 8, 113. [Google Scholar]
- Hellström-Lindahl, E.; Danielsson, A.; Pontén, F.; Czernichow, P.; Korsgren, O.; Johansson, L.; Eriksson, O. GPR44 is a pancreatic protein restricted to the human beta cell. Acta Diabetol. 2015, 53, 413–421. [Google Scholar] [CrossRef]
- Hirai, H.; Tanaka, K.; Yoshie, O.; Ogawa, K.; Kenmotsu, K.; Takamori, Y.; Ichimasa, M.; Sugamura, K.; Nakamura, M.; Takano, S.; et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor Crth2. J. Exp. Med. 2001, 193, 255–262. [Google Scholar] [CrossRef]
- Monneret, G.; Gravel, S.; Diamond, M.; Rokach, J.; Powell, W.S. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood 2001, 98, 1942–1948. [Google Scholar] [CrossRef]
- Kupczyk, M.; Kuna, P. Targeting the PGD2/CRTH2/DP1 signaling pathway in asthma and allergic disease: Current status and future perspectives. Drugs 2017, 77, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Pettipher, R.; Whittaker, M. Update on the development of antagonists of chemoattractant receptor—Homologous molecule expressed on Th2 Cells (CRTH2). From lead optimization to clinical proof-of-concept in asthma and allergic rhinitis. J. Med. Chem. 2012, 55, 2915–2931. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yao, D.; Deepak, R.K.; Liu, H.; Xiao, Q.; Fan, H.; Gong, W.; Wei, Z.; Zhang, C. Structures of the human PGD2 receptor CRTH2 reveal novel mechanisms for ligand recognition. Mol. Cell 2018, 72, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O.; Johnstrom, P.; Cselenyi, Z.; Jahan, M.; Selvaraju, R.K.; Jensen-Waern, M.; Takano, A.; Sorhede Winzell, M.; Halldin, C.; Skrtic, S.; et al. In vivo visualization of beta-cells by targeting of GPR44. Diabetes 2018, 67, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Jahan, M. Development of Novel PET Radioligands for Visualizing Beta Cell Mass and Amyloid Plaques. Ph.D. Thesis, Karolinska Institutet, Stockholm, Sweden, 2016. [Google Scholar]
- Matthews, P.M.; Rabiner, E.A.; Passchier, J.; Gunn, R.N. Positron emission tomography molecular imaging for drug development. Br. J. Clin. Pharmacol. 2012, 73, 175–186. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 radiochemistry, labeling strategies and synthetic routes. Bioconj. Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef]
- Sanchez-Crespo, A. Comparison of gallium-68 and fluorine-18 imaging characteristics in positron emission tomography. Appl. Radiat. Isotopes 2013, 76, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Darius, H.; Michael-Hepp, J.; Meyer, J. Receptor binding properties of the new and specific thromboxane receptor antagonist Bay U 3405. Agents Actions Suppl. 1992, 37, 157–161. [Google Scholar] [PubMed]
- Martin-Martin, I.; Kern, O.; Brooks, S.; Smith, L.B.; Valenzuela-Leon, P.C.; Bonilla, B.; Ackerman, H.; Calvo, E. Biochemical characterization of AeD7L2 and its physiological relevance in blood feeding in the dengue mosquito vector, Aedes aegypti. Fed. Eur. Biochem. Soc. J. 2020. [Google Scholar] [CrossRef]
- Ishizuka, T.; Matsui, T.; Okamoto, Y.; Ohta, A.; Shichijo, M. Ramatroban (BAY u 3405): A novel dual antagonist of TXA2 receptor and CRTh2, a newly identified prostaglandin D2 receptor. Cardiovasc. Drug Rev. 2006, 22, 71–90. [Google Scholar] [CrossRef]
- Theis, J.G.; Dellweg, H.; Perzborn, E.; Gross, R. Binding characteristics of the new thromboxane A2/prostaglandin H2 receptor antagonist [3H]BAY U 3405 to washed human platelets and platelet membranes. Biochem. Pharmacol. 1992, 44, 495–503. [Google Scholar] [CrossRef]
- Seuter, F.; Perzborn, E.; Rosentreter, U.; Böshagen, H.; Fiedler, V.B. Inhibition of platelet aggregation in vitro and ex vivo by the new thromboxane antagonist (3R)-3-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydro-9-Carbazolepropanoic acid. Arzneimittelforschung 1989, 39, 1525–1527. [Google Scholar]
- Fiedler, V.B.; Perzborn, E.; Seuter, F.; Rosentreter, U.; Böshagen, H. Reduction of in vivo coronary artery thrombosis by the novel thromboxane antagonist (3R)-3-(4-fluorophenylsulfonamido)-1,2,3,4-tetrahydro-9-Carbazolepropanoic acid. Arzneimittelforschung 1989, 39, 1527–1530. [Google Scholar]
- Perzborn, E.; Fiedler, V.B.; Seuter, F.; Stasch, J.P.; Weber, H.; Sander, E.; Böshagen, H.; Rosentreter, U. Characterization of Bay U 3405, a novel thromboxane A2/endoperoxide receptor antagonist. Stroke 1990, 21, IV143–IV145. [Google Scholar]
- Pieters, H.; Roodt, J.P.; Badenhorst, P.N.; van Wyk, V.; Schall, R.; Lötter, M.G.; Hundt, H.K.; Nel, C.J. Antithrombotic activity of Bay u3405, a thromboxane A2-antagonist, in patients with Dacron aortic grafts: A random controlled clinical trial. Thromb. Haemost. 1993, 70, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Escolar, G.; Albors, M.; Garrido, M.; Bioque, G.; Díaz Ricart, M.; Carretero, M.; Ordinas, A. Inhibition of platelet-vessel wall interactions by thromboxane receptor antagonism in a human in vitro system: Potentiation of antiplatelet effects of aspirin. Eur. J. Clin. Investig. 1998, 28, 562–568. [Google Scholar] [CrossRef]
- Mitsuhashi, M.; Tanaka, A.; Fujisawa, C.; Kawamoto, K.; Itakura, A.; Takaku, M.; Hironaka, T.; Sawada, S.; Matsuda, H. Necessity of thromboxane A2for initiation of platelet—Mediated contact sensitivity: Dual activation of platelets and vascular endothelial cells. J. Immunol. 2001, 166, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Ulrych, T.; Böhm, A.; Polzin, A.; Daum, G.; Nüsing, R.M.; Geisslinger, G.; Hohlfeld, T.; Schrör, K.; Rauch, B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost. 2011, 9, 790–798. [Google Scholar] [CrossRef]
- Aizawa, H.; Takata, S.; Shigyo, M.; Matsumoto, K.; Koto, H.; Inoue, H.; Hara, N. Effect of BAY u3405, a thromboxane A2 receptor antagonist, on neuro-effector transmission in canine tracheal tissue. Prostaglandins Leukot. Essent. Fat. Acids 1995, 53, 213–217. [Google Scholar] [CrossRef]
- Braun, M.; Schrör, K. Bay U 3405 inhibits cerebral vasospasm induced by authentic thromboxane A2. Stroke 1990, 21, IV152–IV154. [Google Scholar]
- Aizawa, H.; Inoue, H.; Matsumoto, K.; Koto, H.; Nakano, H.; Hara, N. Thromboxane A2 antagonist inhibits leukotriene D4-induced smooth muscle contraction in guinea-pig lung parenchyma, but not in trachea. Prostaglandins Leukot. Essent. Fat. Acids 1996, 55, 437–440. [Google Scholar] [CrossRef]
- Seuter, F.; Perzborn, E.; Fiedler, V.B. Effect of Bay U 3405, a new thromboxane antagonist, on collagen–induced thromboembolism in rabbits. Stroke 1990, 21, IV146–IV148. [Google Scholar] [PubMed]
- Seuter, F.; Perzborn, E.; Fiedler, V.B.; Rosentreter, U.; Böshagen, H. Effect of BAY U 3405, a new thromboxane antagonist, on sudden death in rabbits. J. Lipid Mediat. 1991, 3, 283–288. [Google Scholar]
- Ma, X.L.; Karasawa, A.; Lefer, A.M. Mechanism of the protective action of Bay U 3405, a new specific thromboxane receptor antagonist, in arachidonate–induced sudden death. Methods Find. Exp. Clin. Pharmacol. 1991, 13, 105–110. [Google Scholar]
- Fiedler, V.B.; Seuter, F.; Perzborn, E. Effects of the novel thromboxane antagonist Bay U 3405 on experimental coronary artery disease. Stroke 1990, 21, IV149–IV151. [Google Scholar] [PubMed]
- Squadrito, F.; Ioculano, M.; Altavilla, D.; Zingarelli, B.; Canale, P.; Campo, G.M.; Saitta, A.; Oriti, S.; Faggiotto, A.; Caputi, A.P. Reduction of myocardial leukocyte accumulation and myocardial infarct size following administration of BAY U 3405, a thromboxane A2 receptor antagonist, in myocardial ischaemia–reperfusion injury. Inflamm. Res. 1993, 39, 143–149. [Google Scholar] [CrossRef]
- Fiedler, V.B.; Perzborn, E.; Seuter, F. Protective effect of a novel thromboxane antagonist, BAY-U3405, on canine myocardial damage after coronary artery occlusion and reperfusion. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1991, 11, 77–84. [Google Scholar]
- Canale, P.; Squadrito, F.; Altavilla, D.; Ioculano, M.; Campo, G.M.; Squadrito, G.; Urna, G.; Sardella, A.; Caputi, A.P. Beneficial effects of BAY U 3405, a novel thromboxane A2 receptor antagonist, in splanchnic artery occlusion shock. Pharmacology 1994, 49, 376–385. [Google Scholar] [CrossRef]
- Kotzé, H.F.; Lamprecht, S.; Badenhorst, P.N.; van Wyk, V.; Roodt, J.P.; Alexander, K. In vivo inhibition of acute platelet-dependent thrombosis in a baboon model by Bay U 3405, a thromboxane A2-receptor antagonist. Thromb. Haemost. 1993, 70, 672–675. [Google Scholar] [PubMed]
- Rote, W.E.; Mu, D.X.; Lucchesi, B.R. Thromboxane antagonism in experimental canine carotid artery thrombosis. Stroke 1993, 24, 820–827. [Google Scholar] [CrossRef][Green Version]
- Cissé-Thiam, M.; Drouet, L. Comparative study of the antithrombotic effect of aspirin and Bay U3405, antagonist of a thromboxane A2 receptor. Dakar Med. 1999, 44, 25–27. [Google Scholar] [PubMed]
- Seuter, F.; Perzborn, E.; Fiedler, V.B. Effect of BAY U 3405, a new thromboxane antagonist, on arachidonic acid induced thromboembolism. Adv. Prostaglandin Thromboxane Leukot. Res. 1991, 21, 355–358. [Google Scholar]
- Rounding, H.; Fiedler, V.B. Improved coronary thrombolysis by tissue-type plasminogen activator in the presence of BAY U 3405. Eur. J. Pharmacol. 1991, 198, 207–210. [Google Scholar] [CrossRef]
- Chakraborty, R.; Bhullar, R.P.; Dakshinamurti, S.; Hwa, J.; Chelikani, P. Inverse agonism of SQ 29,548 and ramatroban on thromboxane A2 receptor. PLoS ONE 2014, 9, e85937. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Chen, S.; Yuan, X.; Han, S.; Zhang, H.; Xia, W.; Xu, Y.; Zhao, Q.; Wu, B. Structural basis for ligand recognition of the human thromboxane A2 receptor. Nat. Chem. Biol. 2018, 15, 27–33. [Google Scholar] [CrossRef]
- Mckenniff, M.G.; Norman, P.; Cuthbert, N.J.; Gardiner, P.J. BAY U3405, a potent and selective thromboxane A2 receptor antagonist on airway smooth muscle in vitro. Br. J. Pharmacol. 1991, 104, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Shichijo, M.; Okano, M.; Bacon, K.B. CRTH2-specific binding characteristics of [3H] ramatroban and its effects on PGD2, 15-deoxy-Δ12, 14-PGJ2 and indomethacin-induced agonist responses. Eur. J. Pharmacol. 2005, 524, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, J.M.; Ulven, T.; Martini, L.; Gerlach, L.O.; Heinemann, A.; Kostenis, E. Identification of indole derivatives exclusively interfering with a G protein-independent signaling pathway of the prostaglandin D2 receptor CRTH2. Mol. Pharmacol. 2005, 68, 393–402. [Google Scholar] [CrossRef]
- Mathiesen, J.M.; Christopoulos, A.; Ulven, T.; Royer, J.F.; Campillo, M.; Heinemann, A.; Pardo, L.; Kostenis, E. On the mechanism of interaction of potent surmountable and insurmountable antagonists with the prostaglandin D2 receptor CRTH2. Mol. Pharmacol. 2006, 69, 1441–1453. [Google Scholar] [CrossRef]
- Robarge, M.J.; Bom, D.C.; Tumey, L.N.; Varga, N.; Gleason, E.; Silver, D.; Song, J.; Murphy, S.M.; Ekema, G.; Doucette, C.; et al. Isosteric ramatroban analogs: Selective and potent CRTH-2 antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 1749–1753. [Google Scholar] [CrossRef]
- Sugimoto, H.; Shichijo, M.; Iino, T.; Manabe, Y.; Watanabe, A.; Shimazaki, M.; Gantner, F.; Bacon, K.B. An orally bioavailable small molecule antagonist of CRTH2, ramatroban (BAY U3405), inhibits prostaglandin D2-Induced eosinophil migration in vitro. J. Pharmacol. Exp. Ther. 2003, 305, 347–352. [Google Scholar] [CrossRef]
- Vinall, S.L.; Townsend, E.R.; Pettipher, R. A paracrine role for chemoattractant receptor-homologous molecule expressed on T helper type 2 cells (CRTH2) in mediating chemotactic activation of CRTH2+ CD4+T helper type 2 lymphocytes. J. Immunol. 2007, 121, 577–584. [Google Scholar] [CrossRef]
- Schratl, P.; Royer, J.F.; Kostenis, E.; Ulven, T.; Sturm, E.M.; Waldhoer, M.; Hoefler, G.; Schuligoi, R.; Lippe, I.T.; Peskar, B.A.; et al. The role of the prostaglandin D2 receptor, DP, in eosinophil trafficking. J. Immunol. 2007, 179, 4792–4799. [Google Scholar] [CrossRef]
- Whelan, C.J. Evidence that 13–14 di-hydro, 15-keto prostaglandin D2-induced airway eosinophilia in guinea-pigs is independent of interleukin-5. Inflamm. Res. 2009, 58, 103–108. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lou, H.-Q.; Ying, Y.-F.; Hu, Y. CRTH2 antagonist ameliorates airway inflammation in rats with asthma. J. Zhejiang Univ. Med. Sci. 2010, 39, 64–70. [Google Scholar]
- Boehm, E.; Sturm, G.J.; Weiglhofer, I.; Sandig, H.; Shichijo, M.; McNamee, A.; Pease, J.E.; Kollroser, M.; Peskar, B.A.; Heinemann, A. 11-Dehydro-thromboxane B2, a stable thromboxane metabolite, is a full agonist of chemoattractant receptor-homologous molecule expressed on TH2 cells (CRTH2) in human eosinophils and basophils. J. Biol. Chem. 2004, 279, 7663–7670. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Asano, K.; Nakajima, T.; Oguma, T.; Suzuki, Y.; Shiomi, T.; Sayama, K.; Niimi, K.; Wakaki, M.; Kagyo, J.; et al. Prostaglandin D2-induced eosinophilic airway inflammation is mediated by CRTH2 receptor. J. Pharmacol. Exp. Ther. 2005, 312, 954–960. [Google Scholar] [CrossRef]
- Gyles, S.L.; Xue, L.; Townsend, E.R.; Wettey, F.; Pettipher, R. A dominant role for chemoattractant receptor-homologous molecule expressed on T helper type 2 (Th2) cells (CRTH2) in mediating chemotaxis of CRTH2+ CD4+Th2 lymphocytes in response to mast cell supernatants. Immunology 2006, 119, 362–368. [Google Scholar] [CrossRef]
- Ulven, T.; Kostenis, E. Minor structural modifications convert the dual TP/CRTH2 antagonist ramatroban into a highly selective and potent CRTH2 antagonist. J. Med. Chem. 2005, 48, 897–900. [Google Scholar] [CrossRef]
- Uller, L.; Mathiesen, J.M.; Alenmyr, L.; Korsgren, M.; Ulven, T.; Högberg, T.; Andersson, G.; A Persson, C.G.; Kostenis, E. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir. Res. 2007, 8, 16. [Google Scholar] [CrossRef]
- Kawaguchi, C.; Shintani, N.; Hayata-Takano, A.; Hatanaka, M.; Kuromi, A.; Nakamura, R.; Yamano, Y.; Shintani, Y.; Nagai, K.; Tsuchiya, S.; et al. Lipocalin-type prostaglandin D synthase regulates light-induced phase advance of the central circadian rhythm in mice. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Yan, X.; Nagata, N.; Aritake, K.; Katsumata, Y.; Matsuhashi, T.; Nakamura, M.; Hirai, H.; Urade, Y.; Asano, K.; et al. PGD2-CRTH2 Pathway Promotes Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2012, 23, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Gallant, M.; Beaulieu, C.; Berthelette, C.; Colucci, J.; Crackower, M.A.; Dalton, C.; Denis, D.; Ducharme, Y.; Friesen, R.W.; Guay, D.; et al. Discovery of MK-7246, a selective CRTH2 antagonist for the treatment of respiratory diseases. Bioorg. Med. Chem. Lett. 2011, 21, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Gervais, F.G.; Sawyer, N.; Stocco, R.; Hamel, M.; Krawczyk, C.; Sillaots, S.; Denis, D.; Wong, E.; Wang, Z.; Gallant, M.; et al. Pharmacological characterization of MK-7246, a potent and selective CRTH2 (chemoattractant receptor-homologous molecule expressed on T-helper type 2 Cells) Antagonist. Mol. Pharmacol. 2010, 79, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.; Roy, T.; Sawadjoon, S.; Bachmann, K.; Sköld, C.; Larhed, M.; Weis, J.; Selvaraju, R.K.; Korsgren, O.; Eriksson, O.; et al. Synthesis and preclinical evaluation of the CRTH2 antagonist [11C]MK-7246 as a novel PET tracer and potential surrogate marker for pancreatic beta-cell mass. Nucl. Med. Biol. 2019, 71, 1–10. [Google Scholar] [CrossRef]
- Beaulieu, C.; Guay, D.; Wang, Z.; Leblanc, Y.; Roy, P.; Dufresne, C.; Zamboni, R.; Berthelette, C.; Day, S.; Tsou, N.; et al. Identification of prostaglandin D2 receptor antagonists based on a tetrahydropyridoindole scaffold. Bioorg. Med. Chem. Lett. 2008, 18, 2696–2700. [Google Scholar] [CrossRef]
- Zaghdane, H.; Boyd, M.; Colucci, J.; Simard, D.; Berthelette, C.; Leblanc, Y.; Wang, Z.; Houle, R.; Lévesque, J.F.; Molinaro, C.; et al. New indole amide derivatives as potent CRTH2 receptor antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 3471–3474. [Google Scholar] [CrossRef]
- Simard, D.; Leblanc, Y.; Berthelette, C.; Zaghdane, M.H.; Molinaro, C.; Wang, Z.; Gallant, M.; Lau, S.; Thao, T.; Hamel, M.; et al. Azaindoles as potent CRTH2 receptor antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 841–845. [Google Scholar] [CrossRef]
- Luker, T.; Bonnert, R.; Paine, S.W.; Schmidt, J.; Sargent, C.; Cook, A.R.; Cook, A.; Gardiner, P.; Hill, S.; Weyman-Jones, C.; et al. Zwitterionic CRTh2 Antagonists. J. Med. Chem. 2011, 54, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Fretz, H.; Valdenaire, A.; Pothier, J.; Hilpert, K.; Gnerre, C.; Peter, O.; Leroy, X.; Riederer, M.A. Identification of 2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid (Setipiprant/ACT-129968), a potent, selective, and orally bioavailable chemoattractant receptor-homologous molecule expressed on Th2 Cells (CRTH2) Antagonist. J. Med. Chem. 2013, 56, 4899–4911. [Google Scholar] [CrossRef]
- Sidharta, P.N.; Diamant, Z.; Dingemanse, J. Single and multiple dose tolerability and pharmacokinetics of the CRTH2 antagonist setipiprant in healthy male subjects. Fundam. Clin. Pharmacol. 2014, 28, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Baldoni, D.; Mackie, A.; Gutierrez, M.; Theodor, R.; Dingemanse, J. Setipiprant, a Selective oral antagonist of human CRTH2: Relative bioavailability of a capsule and a tablet formulation in healthy female and male subjects. Clin. Ther. 2013, 35, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- Diamant, Z.; Sidharta, P.N.; Singh, D.; O’Connor, B.J.; Zuiker, R.; Leaker, B.R.; Silkey, M.; Dingemanse, J. Setipiprant, a selective CRTH2 antagonist, reduces allergen–induced airway responses in allergic asthmatics. Clin. Exp. Allergy 2014, 44, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Ratner, P.; Andrews, C.P.; Hampel, F.C.; Martin, B.; Mohar, D.E.; Bourrelly, D.; Danaietash, P.; Mangialaio, S.; Dingemanse, J.; Hmissi, A.; et al. Efficacy and safety of setipiprant in seasonal allergic rhinitis: Results from phase 2 and phase 3 randomized, double-blind, placebo- and active-referenced studies. Allergy Asthma Clin. Immunol. 2017, 13, 1–15. [Google Scholar] [CrossRef]
- Actelion Provides Update on CRTH2 Program. Available online: https://pipelinereview.com/index.php/2012040247547/Small-Molecules/Actelion-provides-update-on-CRTH2-program.html (accessed on 17 April 2020).
- Risch, P.; Pfeifer, T.; Segrestaa, J.; Fretz, H.; Pothier, J. Verification of the major metabolic oxidation path for the Naphthoyl group in chemoattractant receptor-homologous molecule expressed on Th2 Cells (CRTh2) antagonist 2-(2-(1-Naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid (Setipiprant/ACT-129968). J. Med. Chem. 2015, 58, 8011–8035. [Google Scholar] [CrossRef]
- Hoch, M.; Wank, J.; Kluge, I.; Wagner-Redeker, W.; Dingemanse, J. Disposition and metabolism of setipiprant, a selective oral CRTH2 antagonist, in humans. Drugs Res. Dev. 2013, 13, 253–269. [Google Scholar] [CrossRef][Green Version]
- Pees, A.; Windhorst, A.D.; Vosjan, M.J.W.D.; Tadino, V.; Vugts, D.J. Synthesis of [18 F]Fluoroform with high molar activity. Eur. J. Org. Chem. 2020, 2020, 1177–1185. [Google Scholar] [CrossRef]
- Guo, D.; Liu, L.Y. The in vivo profile of CT133, a potent, well tolerated, and selective CRTH2 antagonist for the treatment of allergic asthma and rhinitis. J. Allergy Clin. Immunol. 2015, 135, Ab3. [Google Scholar] [CrossRef]
- Hussain, M.; Xu, C.; Wu, X.; Lu, M.; Tang, L.; Wu, F.; Wu, X.; Wu, J. A CRTH2 antagonist, CT-133, suppresses NF-kappaB signalling to relieve lipopolysaccharide-induced acute lung injury. Eur. J. Pharmacol. 2019, 854, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Xu, C.; Yao, M.; Zhang, Q.; Wu, J.; Wu, X.; Lu, M.; Tang, L.; Wu, F.; Wu, X. CRTH2 antagonist, CT-133, effectively alleviates cigarette smoke-induced acute lung injury. Life Sci. 2019, 216, 156–167. [Google Scholar] [CrossRef]
- Issahaku, A.R.; Agoni, C.; Kumi, R.O.; Olotu, F.A.; Soliman, M.E.S.; A Fisayo, O. Lipid-Embedded molecular dynamics simulation model for exploring the reverse prostaglandin D2 agonism of CT-133 towards CRTH2 in the treatment of Type-2 inflammation dependent diseases. Chem. Biodivers. 2020, 17, 1900548. [Google Scholar] [CrossRef]
- Preshlock, S.M.; Tredwell, M.; Gouverneur, V. 18F-Labeling of arenes and heteroarenes for applications in positron emission tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef]
- Van Der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Late-stage [18F] Fluorination: New solutions to old problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef]
- Chen, W.; Huang, Z.; Tay, N.E.S.; Giglio, B.; Wang, M.; Wang, H.; Wu, Z.; Nicewicz, D.A.; Li, Z. Direct arene C-H fluorination with (18)F(-) via organic photoredox catalysis. Science 2019, 364, 1170–1174. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | hCRTH2 a (Ki) | cAMP b (IC50) | EOS c,d (IC50) |
|---|---|---|---|
| 27 | 340 nM | 2500 nM | |
| 28 | 7.4 nM | 12 nM | 4.8 nM |
| 29 | 3.0 nM | 6.2 nM | 6.0 nM |
| 30 | 2.0 nM | 2.0 nM | 4.4 nM |
| 31 | 1.8 nM | 4.0 nM | |
| 32 | 1.7 nM | 4.0 nM | 1.4 nM |
| 33 | 4.3 nM | 8.0 nM | 2.4 nM |
| 34 | 141 nM | ||
| 35 | 70 nM | ||
| 36 | 22 nM | ||
| 37 | 126 nM | ||
| 38 | 19 nM | ||
| 39 | 408 nM | ||
| 40 | 4.0 nM | 3.0 nM | 0.96 nM |
| 41 | 39 nM | 30 nM |
| Compd. | hCRTH2 a (Ki) | cAMP b (IC50) | EOS c (IC50) | DP a (Ki) | TP a (Ki) |
|---|---|---|---|---|---|
| 42 | 7189 nM | > 1300 nM | > 7000 nM | ||
| 43 | 6725 nM | > 3400 nM | > 6800 nM | ||
| 44 | 139 nM | 351 nM | > 12,000 nM | > 22,000 nM | |
| 45 | 3.3 nM | 3.4 nM | 7.0 nM | > 47,000 nM | > 22,000 nM |
| 46 | 1.8 nM | 3.2 nM | 3.3 nM | > 18,000 nM | > 27,000 nM |
| 47 | 1.9 nM | 3.6 nM | 15.8 nM | > 3800 nM | > 1000 nM |
| 48 | 3.4 nM | 5.7 nM | 1.2 nM | > 38,000 nM | > 71,000 nM |
| 49 | 21.1 nM | 73.8 nM | > 12,000 nM | > 22,000 nM | |
| 50 | 4.7 nM | 3.5 nM | 2.3 nM | > 10,000 nM | > 21,000 nM |
| 51 | 3.6 nM | 7.5 nM | 3.1 nM | > 3700 nM | > 6800 nM |
| 52d | 5.1 nM | 4.4 nM | 2.4 nM | > 4000 nM | > 7200 nM |
| 53 | 3.4 nM | 4.7 nM | 1.2 nM | > 4000 nM | > 7200 nM |
| 54 | 3.9 nM | 4.5 nM | 3.4 nM | > 4000 nM | > 7200 nM |
| 55 | 11.5 nM | 7.0 nM | > 4000 nM | > 7200 nM |
| Compd. | CRTH2 Binding (IC50 a) | Log D7.4 | Rat Hep Clint b | Hum Mic Clint c | Agonism EOS Shape Change d | CRTH2 Ca2+ (IC50e) |
|---|---|---|---|---|---|---|
| 57 | 18 nM | 0.4 | <4 | 6 | IA | 517 nM |
| 58 | 316 nM | <3 | 5 | |||
| 59 | 10 nM | 1.0 | <3 | 11 | IA | |
| 60 | 1.0 nM | <3 | <3 | IA | 9 nM | |
| 61 | 0.3 nM | 0.8 | <3 | <3 | yes |
| Variable | Capsule a | Tablet a | Tablet: Capsule b |
|---|---|---|---|
| Cmax, µg/mL | 6.44 (5.46–7.58) | 6.04 (4.72–7.74) | 0.94 (0.79–1.12) |
| tmax, h | 3.00 (1.50–5.00) | 3.50 (1.00–5.00) | 0.00 (−0.50–1.00) |
| t1/2, h | 11.12 (9.76–12.67) | 11.40 (10.54–12.34) | 1.03 (0.93–1.13) |
| AUC0−∞, µg∙h/mL | 31.50 (26.52–37.40) | 31.88 (26.54–38.31) | 1.01 (0.92–1.12) |
| Compd. | hCRTH2 Receptor Interaction | Prostanoid Receptor Interaction | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Binding in | Ca2+ Flux a | cAMP | hESC | hDP1 | fSel c | hEP2 | hEP4 | |||
| Buffer IC50 d | HSA IC50 d | fHSA b | IC50 d | IC50 d | IC50 d | Binding IC50 d | β-Arrestin IC50 d | β-Arrestin IC50 d | ||
| Setipiprant | 6 | 340 | 57 | 30 | 80 | 235 | 1290 | 215 | 2600 | >10,000 |
| 65 | 9 | 60 | 6 | 50 | 160 | 130 | >10,000 | >1100 | >10,000 | >10,000 |
| 67 | 4 | 12 | 3 | 40 | 70 | 34 | >10,000 | >2500 | >10,000 | >10,000 |
| 68 | 9 | 60 | 7 | 40 | 95 | 60 | >10,000 | >1100 | >10,000 | >10,000 |
| 69 | 190 | 180 | 1 | 1280 | nd | nd | 4100 | 22 | >10,000 | >10,000 |
| 70 | 11 | 70 | 6 | 20 | 380 | 180 | 2200 | 200 | >10,000 | >10,000 |
| 71 | 70 | 530 | 7 | 660 | nd | nd | 310 | 4.1 | 2700 | >10,000 |
| 72 | 4 | 180 | 45 | 70 | 160 | 275 | >10,000 | >2500 | >10,000 | >10,000 |
| 73 | 5 | 35 | 7 | 15 | 40 | 50 | >10,000 | >2000 | >10,000 | >10,000 |
| 74 | 10 | 30 | 3 | 65 | 160 | 210 | >10,000 | >1000 | 9500 | >10,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.A.; Huang, K.X.; Tu, J.; Kandeel, F.; Li, J. Ramatroban-Based Analogues Containing Fluorine Group as Potential 18F-Labeled Positron Emission Tomography (PET) G-Protein Coupled Receptor 44 (GPR44) Tracers. Molecules 2021, 26, 1433. https://doi.org/10.3390/molecules26051433
Huang LA, Huang KX, Tu J, Kandeel F, Li J. Ramatroban-Based Analogues Containing Fluorine Group as Potential 18F-Labeled Positron Emission Tomography (PET) G-Protein Coupled Receptor 44 (GPR44) Tracers. Molecules. 2021; 26(5):1433. https://doi.org/10.3390/molecules26051433
Chicago/Turabian StyleHuang, Lina A., Kelly X. Huang, Jui Tu, Fouad Kandeel, and Junfeng Li. 2021. "Ramatroban-Based Analogues Containing Fluorine Group as Potential 18F-Labeled Positron Emission Tomography (PET) G-Protein Coupled Receptor 44 (GPR44) Tracers" Molecules 26, no. 5: 1433. https://doi.org/10.3390/molecules26051433
APA StyleHuang, L. A., Huang, K. X., Tu, J., Kandeel, F., & Li, J. (2021). Ramatroban-Based Analogues Containing Fluorine Group as Potential 18F-Labeled Positron Emission Tomography (PET) G-Protein Coupled Receptor 44 (GPR44) Tracers. Molecules, 26(5), 1433. https://doi.org/10.3390/molecules26051433