
The Intriguing Effects of Substituents in the N-Phenethyl Moiety of Norhydromorphone: A Bifunctional Opioid from a Set of “Tail Wags Dog” Experiments

 , ,
, ,  , , , and
, , , and
Abstract
:
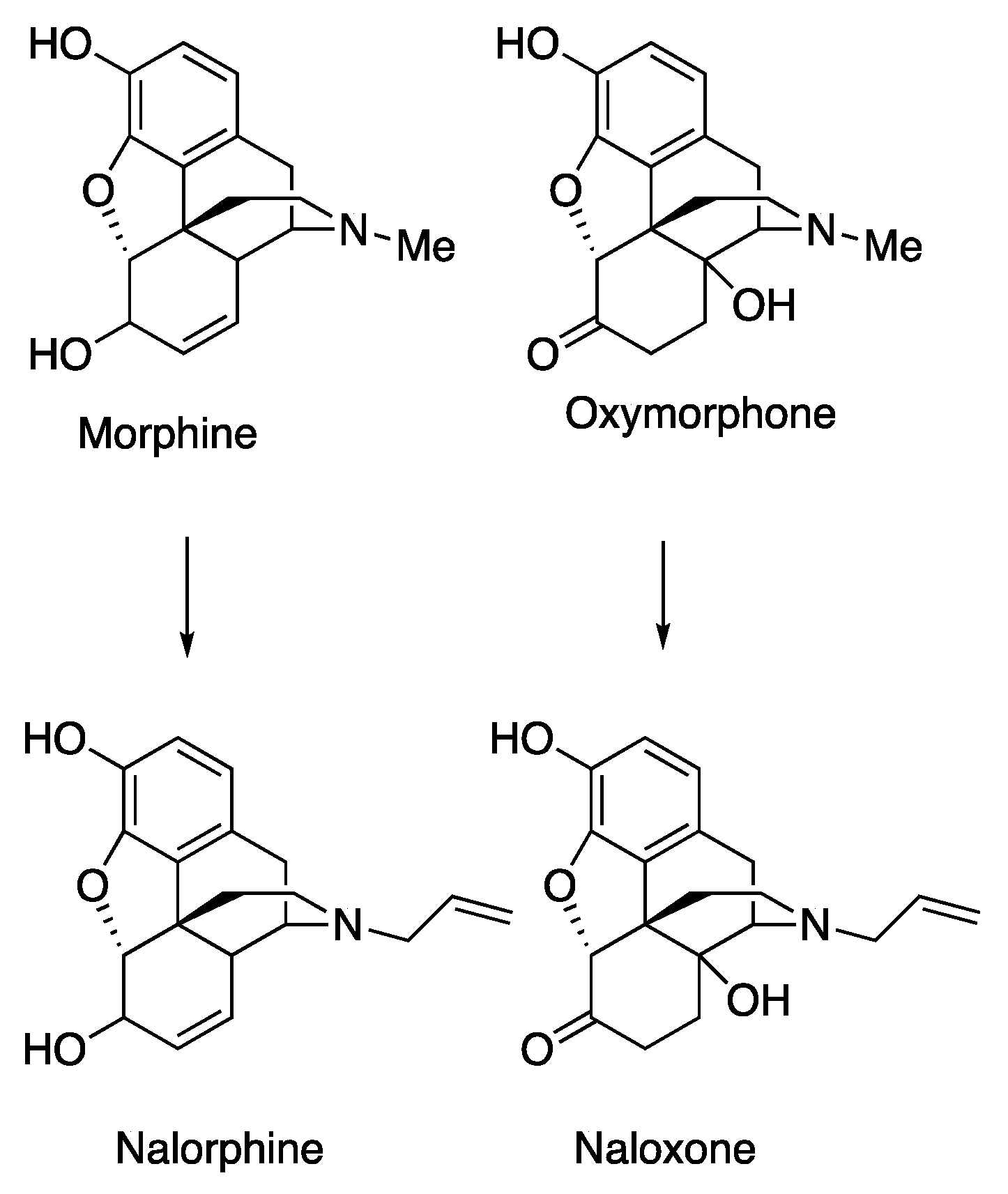
1. Introduction
2. Results and Discussion
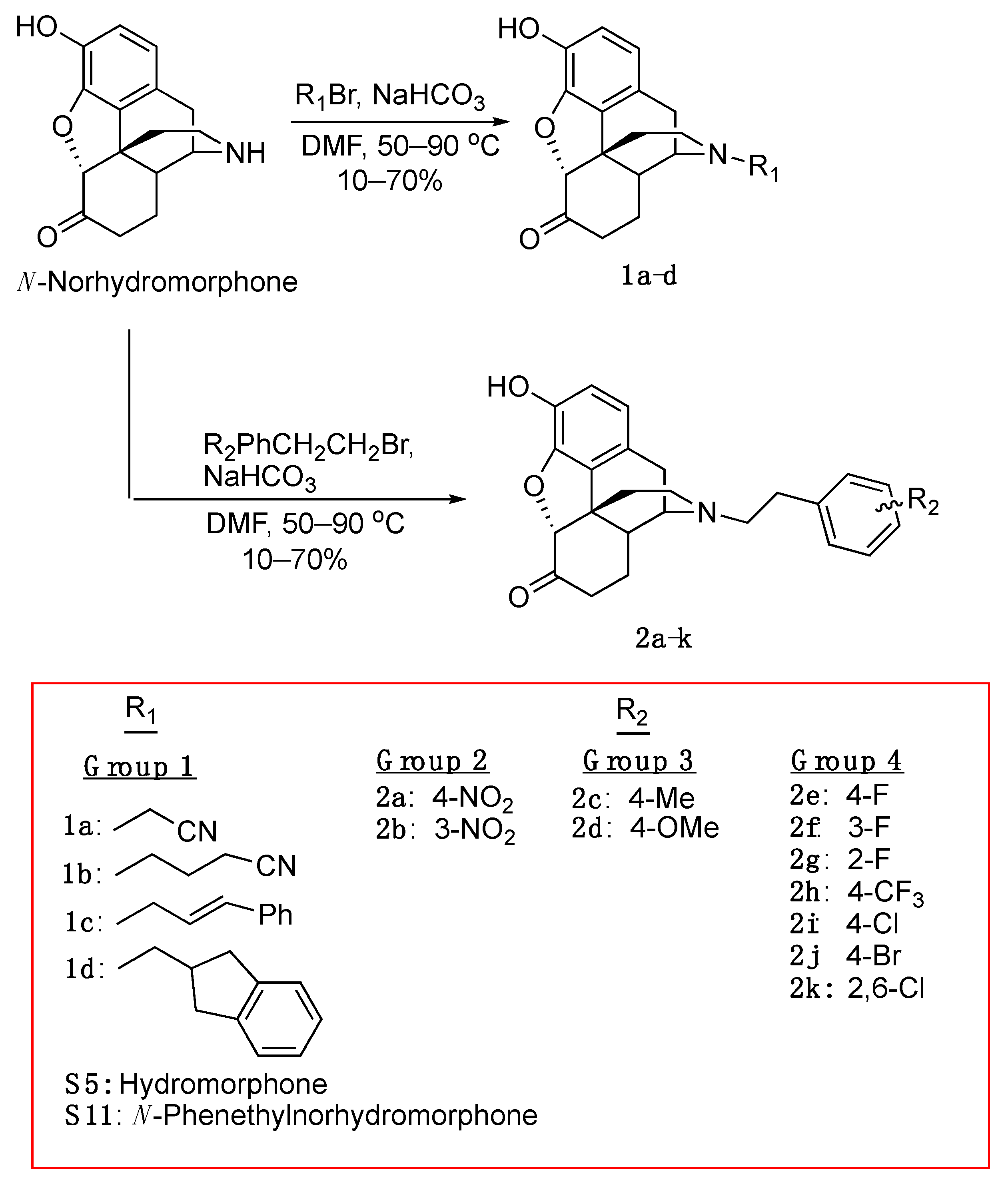
2.1. Chemistry
2.2. In Vitro Studies
2.2.1. Opioid Receptor Binding, Ligand Efficacy and Potency ([35S]GTPγS Binding Assays)
2.2.2. Ligand Potency and Efficacy Using the Forskolin-induced cAMP Accumulation Assay
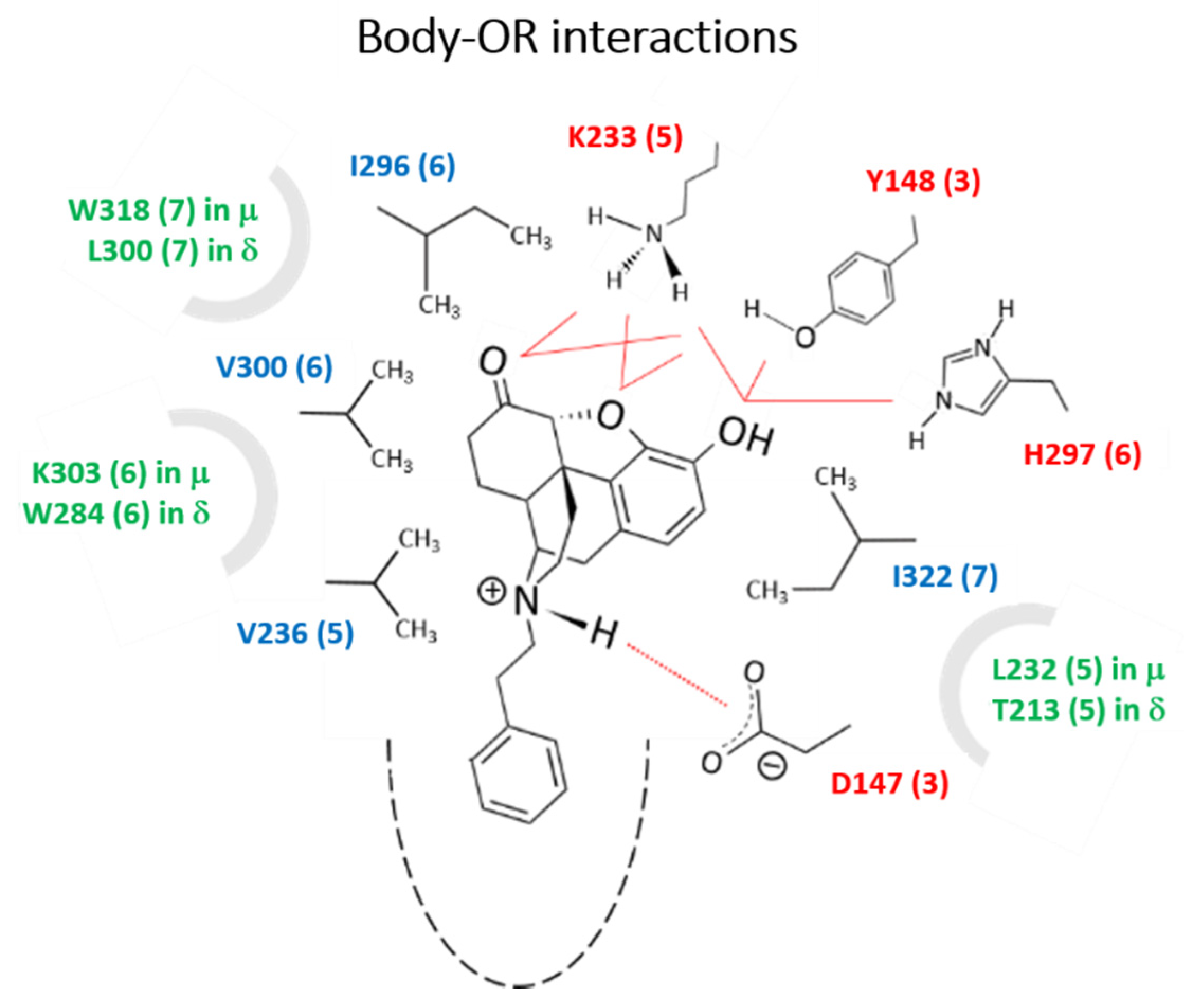
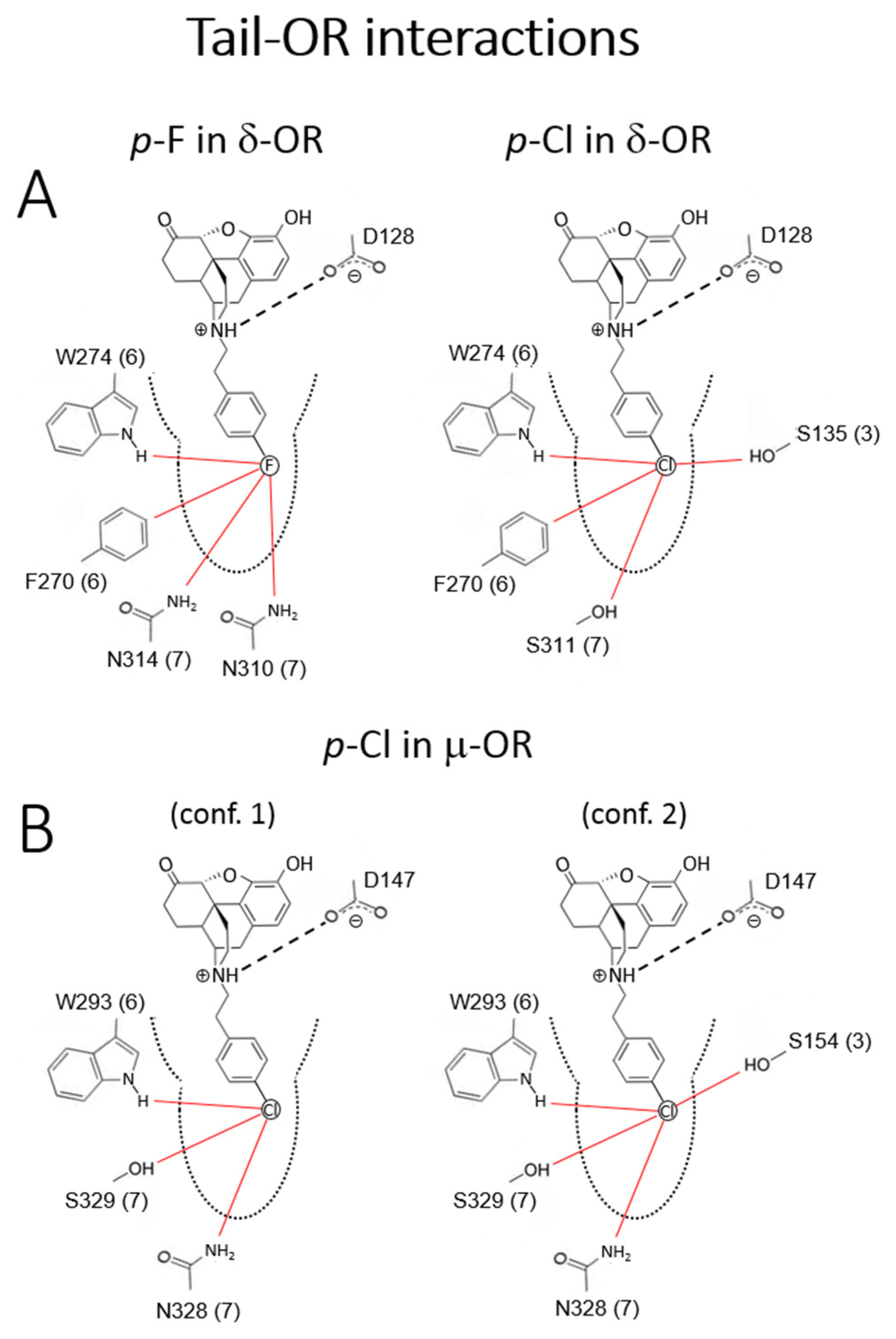
2.2.3. Molecular Modeling and Simulations.
Body-Opioid Receptor (OR) interactions
Tail-OR interactions
2.3. In Vivo Data
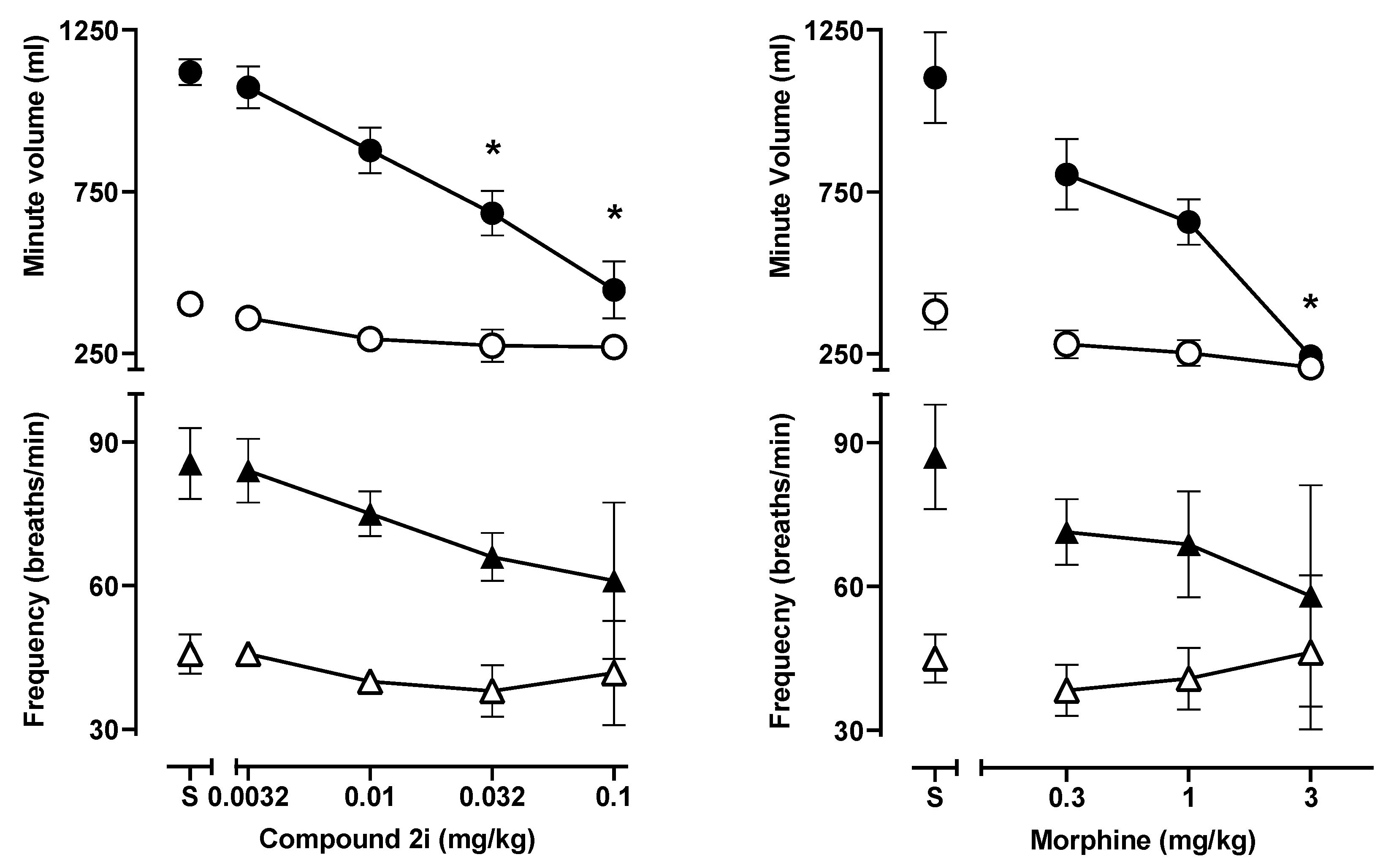
2.3.1. Respiratory Depression Assays in Mice
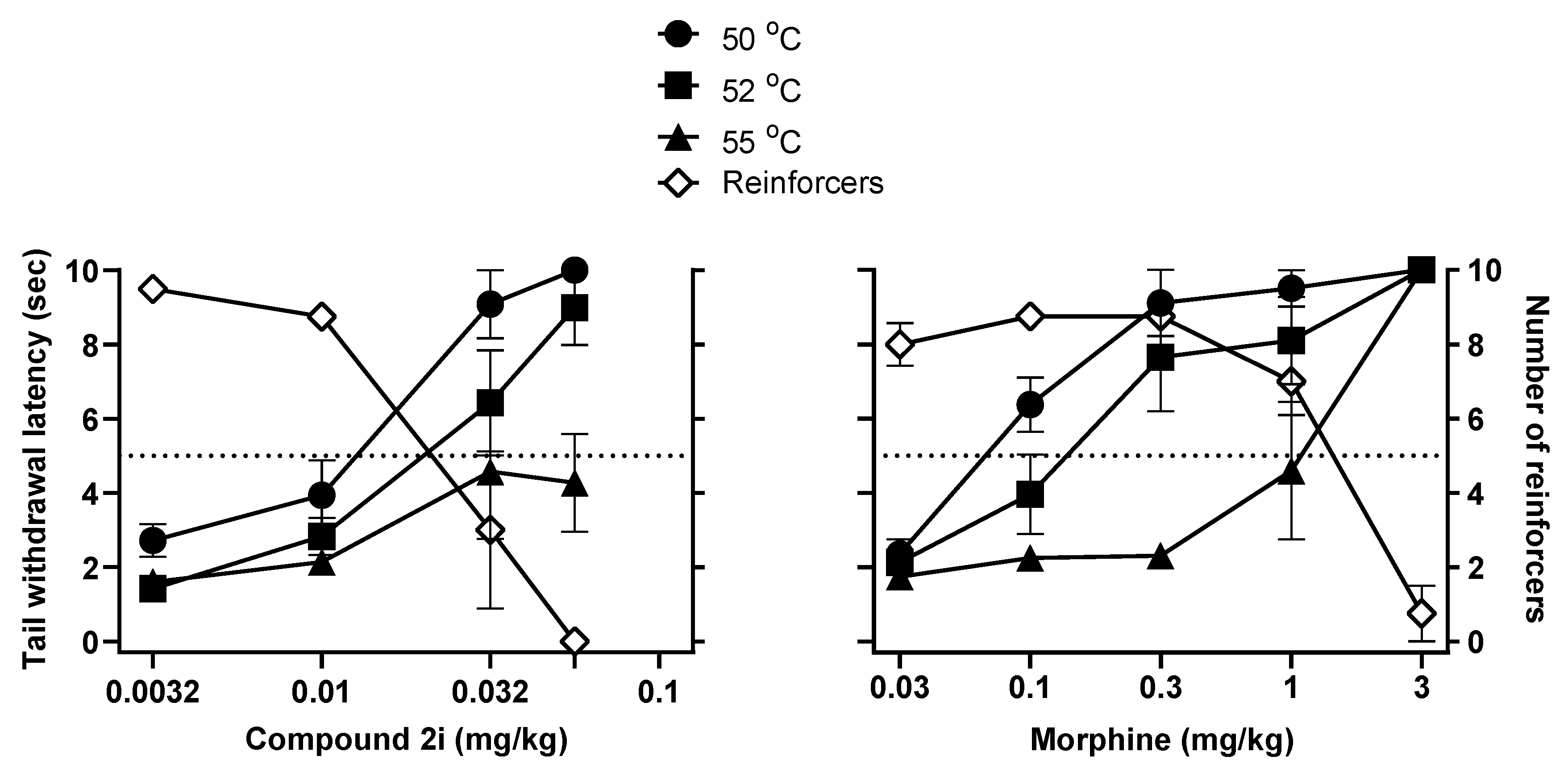
2.3.2. Antinociceptive Studies and Respiratory Depression Studies in Squirrel Monkeys
3. Material and Methods
3.1. General Information
3.2. Synthesis. General Procedure for Formation of Tertiary Amines.
3.3. In Vitro Pharmacology.
3.3.1. Opioid Receptor Binding Affinity
3.3.2. Stimulation of [35S]GTPγS Binding
3.3.3. Forskolin-Induced cAMP Accumulation Assays
Cell Lines and Cell Culture
Assays
3.3.4. β-Arrestin-2 EFC Recruitment Assay
3.3.5. Data Analysis
3.4. In Vivo Pharmacolog
3.4.1. Measurement of Respiration Rate and Arterial Oxygen Saturation in Mice
3.4.2. Statistical Analysis
3.5. Warm-Water Squirrel Tail-Withdrawal and Operant Responding
3.6. Squirrel Monkey Ventilation
4. Computational Methods
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bohn, L.M.; Aubé, J. Seeking (and Finding) Biased Ligands of the Kappa Opioid Receptor. ACS Med. Chem. Lett. 2017, 8, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Bond, R.A.; Lucero Garcia-Rojas, E.Y.; Hegde, A.; Walker, J.K.L. Therapeutic Potential of Targeting ß-Arrestin. Front. Pharmacol. 2019, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, A.; Gillis, A.; Hill, R.; Schmidel, F.; Bailey, C.; Kelly, E.; Henderson, G.; Christie, M.J.; Schulz, S. Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Ben Haddou, T.; Béni, S.; Hosztafi, S.; Malfacini, D.; Calo, G.; Schmidhammer, H.; Spetea, M. Pharmacological investigations of N-substituent variation in morphine and oxymorphone: Opioid receptor binding, signaling and antinociceptive activity. PLoS ONE 2014, 9, e99231. [Google Scholar] [CrossRef] [Green Version]
- May, E.L.; Eddy, N.B. Structures Related to Morphine. XII.1 (±)-2′-Hydroxy-5,9-dimethyl-2-phenethyl-6,7-benzomorphan (NIH 7519) and Its Optical Forms. J. Org. Chem. 1959, 24, 1435–1437. [Google Scholar] [CrossRef]
- Hashimoto, A.; Jacobson, A.E.; Rothman, R.B.; Dersch, C.M.; George, C.; Flippen-Anderson, J.L.; Rice, K.C. Probes for narcotic receptor mediated phenomena. Part 28: New opioid antagonists from enantiomeric analogues of 5-(3-hydroxyphenyl)-N-phenylethylmorphan. Bioorg. Med. Chem. 2002, 10, 3319–3329. [Google Scholar] [CrossRef]
- Truong, P.M.; Hassan, S.A.; Lee, Y.-S.; Kopajtic, T.A.; Katz, J.L.; Chadderdon, A.M.; Traynor, J.R.; Deschamps, J.R.; Jacobson, A.E.; Rice, K.C. Modulation of opioid receptor affinity and efficacy via N-substitution of 9β-hydroxy-5-(3-hydroxyphenyl)morphan: Synthesis and computer simulation study. Bioorg. Med. Chem. 2017, 25, 2406–2422. [Google Scholar] [CrossRef] [Green Version]
- Lo, K. Synthesis of N-phenethylnorhydromorphone: A Hydromorphone Analogue. Ph.D. Thesis, University of British Columbia, Vancouver, BC, Canada, 2001. Available online: http://hdl.handle.net/2429/11862 (accessed on 26 March 2020).
- Portoghese, P.S.; Sultana, M.; Nagase, H.; Takemori, A.E. Application of the message-address concept in the design of highly potent and selective non-peptide delta opioid receptor antagonists. J. Med. Chem. 1988, 31, 281–282. [Google Scholar] [CrossRef]
- Bender, A.M.; Clark, M.J.; Agius, M.P.; Traynor, J.R.; Mosberg, H.I. Synthesis and evaluation of 4-substituted piperidines and piperazines as balanced affinity μ opioid receptor (MOR) agonist/δ opioid receptor (DOR) antagonist ligands. Bioorg. Med. Chem. Lett. 2014, 24, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Wade, P.R.; Palmer, J.M.; McKenney, S.; Kenigs, V.; Chevalier, K.; Moore, B.A.; Mabus, J.R.; Saunders, P.R.; Wallace, N.H.; Schneider, C.R.; et al. Modulation of gastrointestinal function by MuDelta, a mixed µ opioid receptor agonist/ µ opioid receptor antagonist. Br. J. Pharmacol. 2012, 167, 1111–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, K.M.; Keresztes, A.; Tashiro, J.K.; Daconta, L.V.; Hruby, V.J.; Streicher, J.M. Synthesis and Evaluation of a Novel Bivalent Selective Antagonist for the Mu-Delta Opioid Receptor Heterodimer that Reduces Morphine Withdrawal in Mice. J. Med. Chem. 2018, 61, 6075–6086. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Mosberg, H.I.; Porreca, F. Modulation of the Potency and Efficacy of Mu-Mediated Antinociception by Delta-Agonists in the Mouse. J. Pharmacol. Exp. Ther. 1990, 254, 683–689. [Google Scholar] [PubMed]
- Ananthan, S. Opioid Ligands with mixed mu/delta opioid receptor interactions: An emerging approach to novel analgesics. AAPS J. 2006, 8, E118–E125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, J.J.; Raymond, T.J.; Giuvelis, D.; Bidlack, J.M.; Polt, R.; Bilsky, E.J. In vivo characterization of MMP-2200, a mixed δ/μ opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011, 336, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.-F.; McNutt, R.W.; Chang, K.-J. Delta-Opioid Ligands Reverse Alfentanil-Induced Respiratory Depression but Not Antinociception. J. Pharmacol. Exp. Ther. 1998, 287, 815–823. [Google Scholar]
- Montandon, G.; Slutsky, A.S. Solving the Opioid Crisis: Respiratory Depression by Opioids as Critical End Point. CHEST 2019, 156, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Bertha, C.M.; Flippen-Anderson, J.L.; Rothman, R.B.; Porreca, F.; Davis, P.; Xu, H.; Becketts, K.; Cha, X.-Y.; Rice, K.C. Probes for Narcotic Receptor-Mediated Phenomena. 20. Alteration of Opioid Receptor Subtype Selectivity of the 5-(3-Hydroxyphenyl)morphans by Application of the Message-Address Concept: Preparation of delta.-Opioid Receptor Ligands. J. Med. Chem. 1995, 38, 1523–1537. [Google Scholar] [CrossRef]
- Bertha, C.M.; Ellis, M.; Flippen-Anderson, J.L.; Porreca, F.; Rothman, R.B.; Davis, P.; Xu, H.; Becketts, K.; Rice, K.C. Probes for Narcotic Receptor-Mediated Phenomena. 21. Novel Derivatives of 3-(1,2,3,4,5,11-Hexahydro-3-methyl-2,6-methano-6H-azocino[4¨CC5-b]indol-6-yl)- phenols with Improved Œ¥ Opioid Receptor Selectivity. J. Med. Chem. 1996, 39, 2081–2086. [Google Scholar] [CrossRef]
- Breslin, H.J.; Miskowski, T.A.; Rafferty, B.M.; Coutinho, S.V.; Palmer, J.M.; Wallace, N.H.; Schneider, C.R.; Kimball, E.S.; Zhang, S.P.; Li, J.; et al. Rationale, design, and synthesis of novel phenyl imidazoles as opioid receptor agonists for gastrointestinal disorders. J. Med. Chem. 2004, 47, 5009–5020. [Google Scholar] [CrossRef]
- Zhang, Q.; Keenan, S.M.; Peng, Y.Y.; Nair, A.C.; Yu, S.J.; Howells, R.D.; Welsh, W.J. Discovery of novel triazole-based opioid receptor antagonists. J. Med. Chem. 2006, 49, 4044–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthan, S.; Saini, S.; Dersch, C.; Xu, H.; McGlinchey, N.; Giuvelis, D.; Bilsky, E.J.; Rothman, R.B. 14-Alkoxy- and 14-acyloxypyridomorphinans: Mu agonist/delta antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 2012, 55, 8350–8363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslin, H.; Diamond, C.; Kavash, R.; Cai, C.; Dyatkin, A. Identification of a dual delta OR antagonist/mu OR agonist as a potential therapeutic for diarrhea-predominant Irritable Bowel Syndrome (IBS-d). Bioorg. Med. Chem. Lett. 2012, 22, 4869–4872. [Google Scholar] [CrossRef] [PubMed]
- Healy, J.R.; Bezawada, P.; Shim, J.; Jones, J.W.; Kane, M.A.; MacKerell, A.D.; Coop, A.; Matsumoto, R.R. Synthesis, Modeling, and Pharmacological Evaluation of UMB 425, a Mixed μ Agonist/δ Antagonist Opioid Analgesic with Reduced Tolerance Liabilities. ACS Chem. Neurosci. 2013, 4, 1256–1266. [Google Scholar] [CrossRef] [Green Version]
- Yadlapalli, J.S.K.; Ford, B.M.; Ketkar, A.; Wan, A.; Penthala, N.R.; Eoff, R.L.; Prather, P.L.; Dobretsov, M.; Crooks, P.A. Antinociceptive effects of the 6-O-sulfate ester of morphine in normal and diabetic rats: Comparative role of mu- and delta-opioid receptors. Pharmacol. Res. 2016, 113 Pt A, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, G.W.; Luginbuhl, A.; Dunbar, C.; LaVigne, J.; Dutra, J.; Atherton, P.; Bell, B.; Cone, K.; Giuvelis, D.; Polt, R.; et al. The mixed-action delta/mu opioid agonist MMP-2200 does not produce conditioned place preference but does maintain drug self-administration in rats, and induces in vitro markers of tolerance and dependence. Pharmacol. Biochem. Behav. 2015, 132, 49–55. [Google Scholar] [CrossRef]
- Przybyl, A.K.; Flippen-Anderson, J.L.; Jacobson, A.E.; Rice, K.C. Practical and High-Yield Syntheses of Dihydromorphine from Tetrahydrothebaine and Efficient Syntheses of (8S)-8-Bromomorphide. J. Org. Chem. 2003, 68, 2010–2013. [Google Scholar] [CrossRef]
- Csuk, R.; Vasileva, G.; Barthel, A. Towards an Efficient Preparation of Hydromorphone. Synthesis 2012, 44, 2840–2842. [Google Scholar] [CrossRef]
- Iijima, I.; Minamikawa, J.; Jacobson, A.E.; Brossi, A.; Rice, K.C. Studies in the (+)-morphinan series. 5. Synthesis and biological properties of (+)-naloxone. J. Med. Chem. 1978, 21, 398–400. [Google Scholar] [CrossRef]
- Purington, L.C.; Sobczyk-Kojiro, K.; Pogozheva, I.D.; Traynor, J.R.; Mosberg, H.I. Development and in vitro characterization of a novel bifunctional μ-agonist/δ-antagonist opioid tetrapeptide. ACS Chem. Biol. 2011, 6, 1375–1381. [Google Scholar] [CrossRef] [Green Version]
- Beck, T.C.; Hapstack, M.A.; Beck, K.R.; Dix, T.A. Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitrascuta, M.; Bermudez, M.; Ben Haddou, T.; Guerrieri, E.; Schläfer, L.; Ritsch, A.; Hosztafi, S.; Lantero, A.; Kreutz, C.; Massotte, D.; et al. N-Phenethyl Substitution in 14-Methoxy-N-methylmorphinan-6-ones Turns Selective µ Opioid Receptor Ligands into Dual µ/δ Opioid Receptor Agonists. Sci. Rep. 2020, 10, 5653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. Fluorine-Centered Halogen Bonding: A Factor in Recognition Phenomena and Reactivity. Cryst. Growth Des. 2011, 11, 4238–4246. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [Green Version]
- Matter, H.; Nazaré, M.; Güssregen, S.; Will, D.W.; Schreuder, H.; Bauer, A.; Urmann, M.; Ritter, K.; Wagner, M.; Wehner, V. Evidence for C-Cl/C-Br⋅⋅⋅π Interactions as an Important Contribution to Protein–Ligand Binding Affinity. Angew. Chem. Int. Ed. Engl. 2009, 48, 2911–2916. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Wang, J.; Palczewski, K.; Filipek, S.; Vogel, H.; Liu, Z.-J.; Yuan, S. Exploring a new ligand binding site of G protein-coupled receptors. Chem. Sci. 2018, 9, 6480–6489. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.O.; Akil, H.; Woods, J.H.; Traynor, J.R. Differential binding properties of oripavines at cloned mu- and delta-opioid receptors. Eur. J. Pharmacol. 1999, 378, 323–330. [Google Scholar] [CrossRef]
- Husbands, S.M.; Neilan, C.L.; Broadbear, J.; Grundt, P.; Breeden, S.; Aceto, M.D.; Woods, J.H.; Lewis, J.W.; Traynor, J.R. BU74, a complex oripavine derivative with potent kappa opioid receptor agonism and delayed opioid antagonism. Eur. J. Pharmacol. 2005, 509, 117–125. [Google Scholar] [CrossRef]
- Traynor, J.R.; Nahorski, S.R. Modulation of mu-opioid agonists of guanisine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neurobastoma SH-SY5Y cells. J. Mol. Pharmacol. 1995, 47, 848–854. [Google Scholar]
- Crowley, R.S.; Riley, A.P.; Sherwood, A.M.; Groer, C.E.; Shivaperumal, N.; Biscaia, M.; Paton, K.; Schneider, S.; Provasi, D.; Kivell, B.M.; et al. Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Identification of a Potent and Centrally Acting μ Opioid Analgesic with Reduced Abuse Liability. J. Med. Chem. 2016, 59, 11027–11038. [Google Scholar] [CrossRef] [Green Version]
- Riley, A.P.; Groer, C.E.; Young, D.; Ewald, A.W.; Kivell, B.M.; Prisinzano, T.E. Synthesis and κ-opioid receptor activity of furan-substituted salvinorin A analogues. J. Med. Chem. 2014, 57, 10464–10475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, R.; Disney, A.; Conibear, A.; Sutcliffe, K.; Dewey, W.; Husbands, S.; Bailey, C.; Kelly, E.; Henderson, G. The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol. 2018, 175, 2653–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Withey, S.L.; Paronis, C.A.; Bergman, J. Concurrent Assessment of the Antinociceptive and Behaviorally Disruptive Effects of Opioids in Squirrel Monkeys. J. Pain 2018, 19, 728–740. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Mehler, E.L.; Hassan, S.A.; Kortagere, S.; Weinstein, H. Ab initio computational modeling of loops in G-protein-coupled receptors: Lessons from the crystal structure of rhodopsin. Proteins 2006, 64, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Mehler, E.L.; Periole, X.; Hassan, S.A.; Weinstein, H. Key issues in the computational simulation of GPCR function: Representation of loop domains. J. Comput. Aided Mol. Des. 2002, 16, 841–853. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor Binding (Ki, nM) | [35S]GTPγS | ||||||
|---|---|---|---|---|---|---|---|
| Compound | Structure | MOR Binding | DOR Binding | KOR Binding | MOR EC50 c, nM (% Stimulation) | DOR EC50 c, nM (% Stimulation) | KOR EC50 c, nM (% Stimulation) |
| Group 1. N-Cyanoalkyl Compounds and Compounds Lacking Substituents on the Aromatic Ring | |||||||
| 1a |  | 4.51 ± 0.27 | 141 ± 2.8 | 90.3 ± 6.6 | 425 ± 133 (47.1 ± 3.3) | NT | NT |
| 1b |  | 4.25 ± 0.29 | 72.2 ± 1.2 | 2.71 ± 0.22 | DNS | NT | 24.3 ± 5.0 (89.6 ± 0.9) |
| 1c | 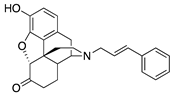 | 90.5 ± 11.0 | NT | NT | NT | NT | NT |
| 1d | 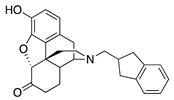 | 40.1 ± 5.9 | 844 ± 107 | NT | NT | NT | NT |
| Group 2. Nitro Substituents | |||||||
| 2a | 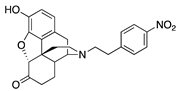 | 0.52 ± 0.01 | 6.07 ± 0.86 | 56.0 ± 8.2 | 1.9 ± 0.5 (26.6 ± 1.4) | 227 ± 53 (64.3 ± 3.3) | NT |
| 2b | 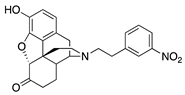 | 5.88 ± 0.38 | 92.5 ± 7.3 | 65.4 ± 7.7 | DNS | NT | NT |
| Group 3. Alkyl and Alkoxy Substituents | |||||||
| 2c | 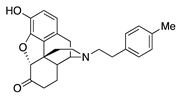 | 0.86 ± 0.12 | 6.34 ± 0.65 | 44.0 ± 3.8 | 1.9 ± 0.7 (55.6 ± 2.1) | 23.1 ± 6.9 (95.4 ± 3.8) | 38.1 ± 10.6 (31.4 ± 4.2) |
| 2d | 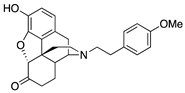 | 0.67 ± 0.05 | 5.43 ± 0.47 | 26.3 ± 1.9 | DNS | 33.6 ± 6.4 (48.9 ± 3.2) | 5.9 ± 1.7 (21.8 ± 0.8) |
| Group 4. Halides and Trifluromethyl Substituents | |||||||
| 2e | 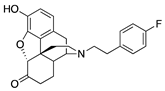 | 0.87 ± 0.10 | 13.4 ± 0.9 | 44.3 ± 6.0 | 2.5 ± 0.3 (42.6 ± 2.9) | 64.2 ± 9.9 (243 ± 49) | DNS |
| 2f |  | 0.49 ± 0.07 | 4.48 ± 0.43 | 30.2 ± 2.3 | 2.4 ± 0.3 (56.8 ± 7.5) | 54.1 ± 4.7 (86.7 ± 8.0) | DNS |
| 2g |  | 0.32 ± 0.04 | 3.57 ± 0.49 | 23.2 ± 2.8 | 2.1 ± 0.7 (100 ± 0.3) | 92.7 ± 17.0 (83.0 ± 3.3) | DNS |
| 2h | 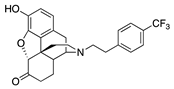 | 2.70 ± 0.17 | 16.4 ± 0.4 | NT | 3.4 ± 1.2 (72.2 ± 5.5) | 35.7 ± 12.3 (82.3 ± 2.3) | NT |
| 2i | 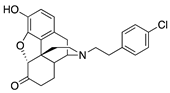 | 1.09 ± 0.06 | 7.89 ± 1.10 | NT | 2.0 ± 1.0 (39.0 ± 2.1) | 2.4 ± 0.7 (83.8 ± 3.7) | NT |
| Standard | |||||||
| - | Morphine | 3.26 ± 0.39 | NT | 145 ± 15 | 194 ± 21 (57 ± 5) [31] | NT | NT |
| Compound | Structure | MOR cAMP Agonist Potency ± SEM (nM) (% Efficacy) | MOR Mediated β-arrestin Recruitment (% Control, Emax DAMGO), nM | Bias Factor | DOR cAMP Agonist Potency ± SEM (nM) (% Efficacy) | KOR cAMP Agonist Potency ± SEM (nM) (% Efficacy) |
|---|---|---|---|---|---|---|
| Group 1. N-Cyanoalkyl Compounds and Compounds Lacking Substituents on the Aromatic Ring | ||||||
| 1a | 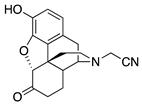 | 4.16 ± 0.64 (101.6 ± 1.1) | 424 ± 69 (33.4 ± 0.25) | 2.18 | 254 ± 167 (77 ± 4) | 109.4 ± 45.4 (99.3 ± 4.0) |
| 1c |  | 76.7 ± 7.4 (103 ± 0.9) | 584 ± 286 (4.1 ± 0.1) | 1.35 | 1699 ± 847 (96.5 ± 5.4) | >10,000 |
| 1d | 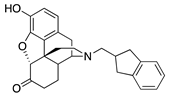 | 18.29 ± 4.70 (95.2 ± 1.2) | 130.4 ± 37.5 (1.9 ± 0.4) | 2.53 | 596 ± 92 (94.4 ± 1.4) | >10,000 |
| S5 Hydromorphone |  | 1.67 ± 0.30 (102 ± 1) | 159 ± 28 (19.8 ± 3) | 3.48 | 134.3 ± 31.4 (95.4 ± 0.8) | 176.1 ± 32.7 (80.4 ± 3.9) |
| S11 N-Phenethylnor-hydromorphone | 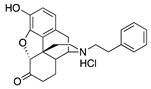 | 0.04 ± 0.01 (102.1 ± 0.8) | 1.71 ± 0.66 (61.9 ± 5.2) | 0.47 | 1.54 ± 0.22 (95.1 ± 0.8) | 22.7 ± 4.8 (80.6 ± 18.3) |
| Group 2. Nitro Substituents | ||||||
| 2a | 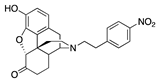 | 0.05 ± 0.02 (102 ± 0.4) | 6.38 ± 0.59 (26.2 ± 2.8) | 4.51 | 0.53 ± 0.03 (96 ± 1.5) | 74.6 ± 5.6 (63 ± 1.5) |
| 2b | 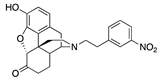 | 5.22 ± 1.20 (102.3 ± 0.6) | 116 ± 21 (6 ± 0.1) | 3.38 | 46.7 ± 17.6 (95.3 ± 1.3) | >10,000 |
| Group 3. Alkyl and Alkoxy Substituents | ||||||
| 2c | 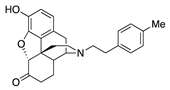 | 0.08 ± 0.02 (101.5 ± 0.2) | 5.6 ± 1.4 (46.5 ± 3) | 1.12 | 1.0 ± 0.3 (95.0 ± 1.3) | 8.71 ± 0.09 (99.4 ± 0.8) |
| 2d |  | 0.13 ± 0.04 (103 ± 2) | 11.2 ± 1.8 (23 ± 2.2) | 2.69 | 2.71 ± 0.93 (97.5 ± 0.7) | 7.38 ± 2.10 (96.2 ± 1.0) |
| Group 4. Halides and Trifluromethyl Substituents | ||||||
| 2e |  | 0.17 ± 0.08 (100.8 ± 1.1) | 15.5 ± 2.1 (45.8 ± 2.4) | 1.39 | 2.58 ± 1.60 (98.1 ± 1.1) | 19.9 ± 1.3 (80.0 ± 3.8) |
| 2f | 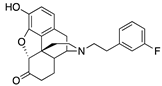 | 0.06 ± 0.01 (101.7 ± 0.3) | 6.2 ± 1.1 (40.3 ± 2.3) | 1.94 | 1.64 ± 0.11 (96.9 ± 1.6) | 56.5 ± 21.1 (58.0 ± 8.4) |
| 2g | 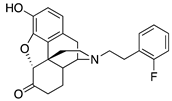 | 0.010 ± 0.003 (101.2 ± 0.1) | 1.7 ± 0.6 (73.2 ± 2.1) | 1.37 | 0.26 ± 0.13 (93.8 ± 2.1) | 19.9 ± 0.6 (74.0 ± 5.2) |
| 2h | 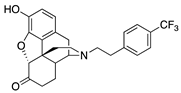 | 0.21 ± 0.04 (102 ± 0.2) | 10.3 ± 1.8 (62.4 ± 1.5) | 0.56 | 2.41 ± 0.47 (94.3 ± 1.8) | 98.2 ± 7.7 (95.7 ± 0.6) |
| 2i | 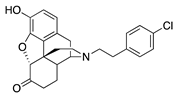 | 0.05 ± 0.03 (98.8 ± 2.1) | 2.44 ± 0.45 (44.8 ± 1.8) | 0.82 | 0.53 ± 0.18 (84.8 ±5.6) | 55.2 ± 26.1 (76.6 ± 11.3) |
| 2j | 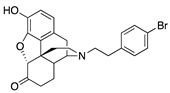 | 0.15 ± 0.04 (103 ± 1.5) | 4.9 ± 0.3 (53 ± 0.4) | 0.43 | 0.97 ± 0.22 (96 ± 1.1) | 20.1 ± 3.3 (99.04 ± 3.74) |
| 2k | 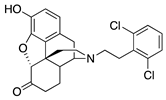 | 0.37 ± 0.09 (101.3 ± 0.5) | 13.13 ± 0.19 (24.6 ± 3.9) | 1.04 | 21.89 ± 0.87 (95.8 ±1.1) | 1881 ± 687 (73.97 ± 5.49) |
| Standards | ||||||
| - | DAMGO | 0.3 ± 0.04 (101.5 ± 0.5) | 44.1 ± 3.9 (103 ± 0.4) | 1.0 | - | |
| - | U-69593 | - | - | - | - | 0.7 ± 0.3 (101.2 ± 1.4) |
| - | Leu-Enkephalin | - | - | - | 0.04 ± 0.10 (95 ± 2) | - |
| - | Morphine | 4.7 ± 0.6 (102.9 ± 0.6) | 378 ± 41 (25.7 ± 0.6) | 2.27 | - | - |
| Behavioral Measures | 2i | Morphine |
|---|---|---|
| ED50 (mg/kg) | ||
| Antinociception (52 °C) a | 0.015 | 0.14 |
| Behavioral Disruption b | 0.022 | 1.30 |
| Respiratory Depression c | 0.057 | 1.06 |
| Potency Ratios | ||
| Behavioral Disruption: Antinociception | 1.5 | 9.0 |
| Respiratory Depression: Antinociception | 3.9 | 7.3 |
| Respiratory Depression: Behavioral Disruption | 2.6 | 0.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Irvin, T.C.; Herdman, C.A.; Hanna, R.D.; Hassan, S.A.; Lee, Y.-S.; Kaska, S.; Crowley, R.S.; Prisinzano, T.E.; Withey, S.L.; et al. The Intriguing Effects of Substituents in the N-Phenethyl Moiety of Norhydromorphone: A Bifunctional Opioid from a Set of “Tail Wags Dog” Experiments. Molecules 2020, 25, 2640. https://doi.org/10.3390/molecules25112640
Wang M, Irvin TC, Herdman CA, Hanna RD, Hassan SA, Lee Y-S, Kaska S, Crowley RS, Prisinzano TE, Withey SL, et al. The Intriguing Effects of Substituents in the N-Phenethyl Moiety of Norhydromorphone: A Bifunctional Opioid from a Set of “Tail Wags Dog” Experiments. Molecules. 2020; 25(11):2640. https://doi.org/10.3390/molecules25112640
Chicago/Turabian StyleWang, Meining, Thomas C. Irvin, Christine A. Herdman, Ramsey D. Hanna, Sergio A. Hassan, Yong-Sok Lee, Sophia Kaska, Rachel Saylor Crowley, Thomas E. Prisinzano, Sarah L. Withey, and et al. 2020. "The Intriguing Effects of Substituents in the N-Phenethyl Moiety of Norhydromorphone: A Bifunctional Opioid from a Set of “Tail Wags Dog” Experiments" Molecules 25, no. 11: 2640. https://doi.org/10.3390/molecules25112640
APA StyleWang, M., Irvin, T. C., Herdman, C. A., Hanna, R. D., Hassan, S. A., Lee, Y.-S., Kaska, S., Crowley, R. S., Prisinzano, T. E., Withey, S. L., Paronis, C. A., Bergman, J., Inan, S., Geller, E. B., Adler, M. W., Kopajtic, T. A., Katz, J. L., Chadderdon, A. M., Traynor, J. R., ... Rice, K. C. (2020). The Intriguing Effects of Substituents in the N-Phenethyl Moiety of Norhydromorphone: A Bifunctional Opioid from a Set of “Tail Wags Dog” Experiments. Molecules, 25(11), 2640. https://doi.org/10.3390/molecules25112640