
Carnitine Inborn Errors of Metabolism

Abstract
1. Introduction
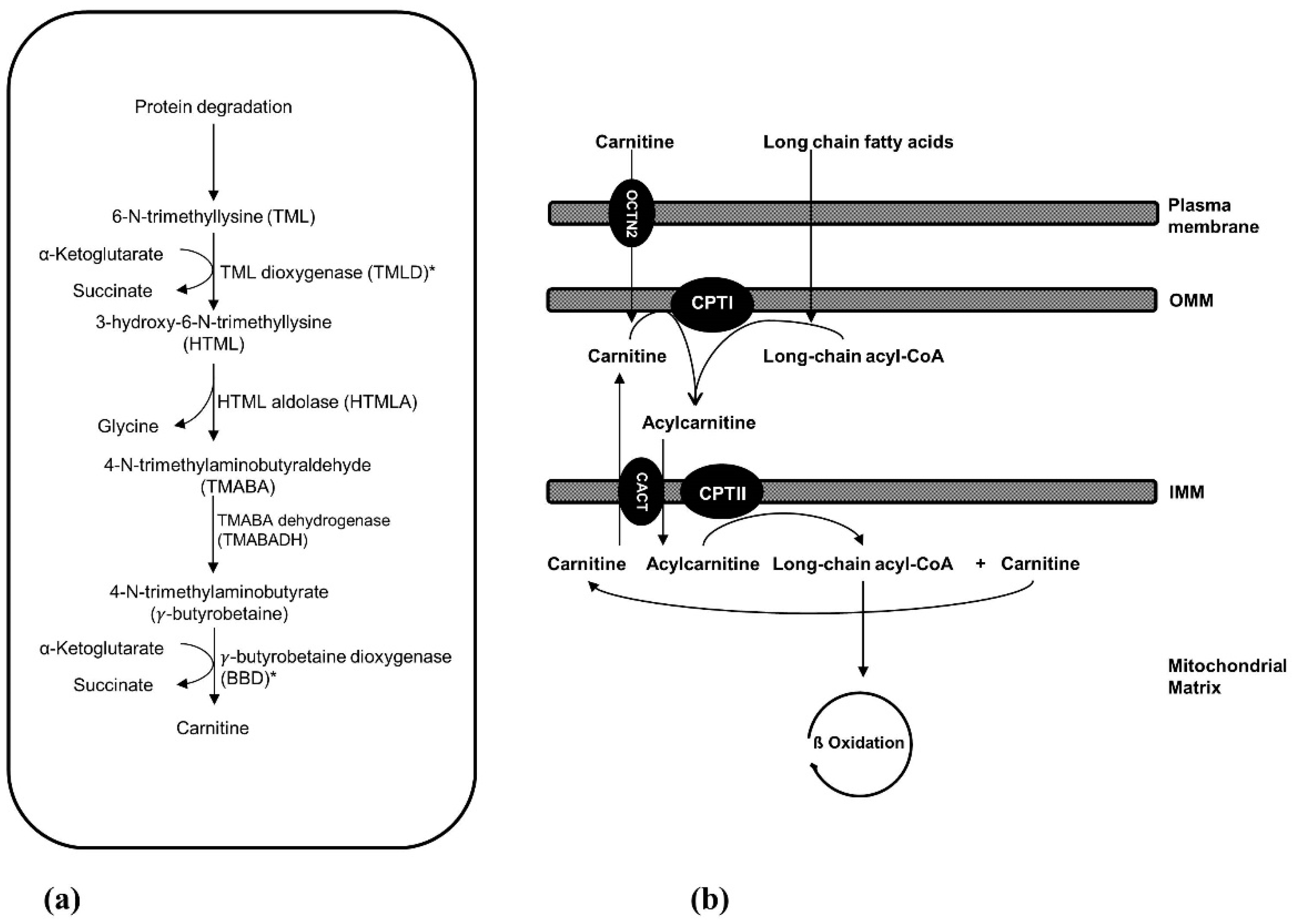
2. Carnitine Biosynthesis Disorders
2.1. 6-N-Trimethyllysine Dioxygenase (TMLD) Deficiency
2.2. γ-Butyrobetaine Dioxygenase (BBD) Deficiency
3. Carnitine Transport Defect
4. Mitochondrial Carnitine-Acylcarnitine Cycle Disorders
4.1. Carnitine Palmitoyltransferase I (CPT I) Deficiency
4.2. Carnitine-Acylcarnitine Translocase (CACT) Deficiency
4.3. Carnitine Palmitoyltransferase II (CPT II) Deficiency
4.4. Carnitine Palmitoyltransferase (CPT) Inhibitors
5. Secondary Carnitine Deficiency
6. Carnitine Use in Inborn Errors of Metabolism
7. Summery and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Bene, J.; Hadzsiev, K.; Melegh, B. Role of carnitine and its derivatives in the development and management of type 2 diabetes. Nutr. Diabetes 2018, 8, 8. [Google Scholar] [CrossRef]
- Vaz, F.M.; Wanders, R.J.A. Carnitine biosynthesis in mammals. Biochem. J. 2002, 361, 417–429. [Google Scholar] [CrossRef]
- Carter, A.L.; Abney, T.O.; Lapp, D.F. Biosynthesis and metabolism of carnitine. J. Child Neurol. 1995, 10 (Suppl. 2), S3–S7. [Google Scholar] [CrossRef]
- Ribas, G.S.; Vargas, C.R.; Wajner, M. L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene 2014, 533, 469–476. [Google Scholar] [CrossRef]
- Lee, B.-J.; Lin, J.-S.; Lin, Y.-C.; Lin, P.-T. Effects of L-carnitine supplementation on oxidative stress and antioxidant enzymes activities in patients with coronary artery disease: A randomized, placebo-controlled trial. Nutr. J. 2014, 13, 79. [Google Scholar] [CrossRef]
- Komlósi, K.; Havasi, V.; Bene, J.; Süle, N.; Pajor, L.; Nicolai, R.; Benatti, P.; Calvani, M.; Melegh, B. Histopathologic abnormalities of the lymphoreticular tissues in organic cation transporter 2 deficiency: Evidence for impaired B cell maturation. J. Pediatr. 2007, 150, 109–111.e2. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, W.; Chen, G.; Zhu, W.; Ding, W.; Ge, Z.; Tan, Y.; Ma, T.; Cui, G. L-carnitine treatment of insulin resistance: A systematic review and meta-analysis. Adv. Clin. Exp. Med. Off. Organ Wroclaw Med. Univ. 2017, 26, 333–338. [Google Scholar]
- Rebouche, C.J. Carnitine function and requirements during the life cycle. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1992, 6, 3379–3386. [Google Scholar]
- Stanley, C.A. Carnitine deficiency disorders in children. Ann. N. Y. Acad. Sci. 2004, 1033, 42–51. [Google Scholar] [CrossRef]
- Tein, I.; Bukovac, S.W.; Xie, Z.W. Characterization of the human plasmalemmal carnitine transporter in cultured skin fibroblasts. Arch. Biochem. Biophys. 1996, 329, 145–155. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Opalka, J.R.; Gellerich, F.N.; Zierz, S. Age and sex dependency of carnitine concentration in human serum and skeletal muscle. Clin. Chem. 2001, 47, 2150–2153. [Google Scholar]
- Rebouche, C.J. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann. N. Y. Acad. Sci. 2004, 1033, 30–41. [Google Scholar] [CrossRef]
- Strijbis, K.; Vaz, F.M.; Distel, B. Enzymology of the carnitine biosynthesis pathway. IUBMB Life 2010, 62, 357–362. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Disorders of carnitine biosynthesis and transport. Mol. Genet. Metab. 2015, 116, 107–112. [Google Scholar] [CrossRef]
- Celestino-Soper, P.B.S.; Shaw, C.A.; Sanders, S.J.; Li, J.; Murtha, M.T.; Ercan-Sencicek, A.G.; Davis, L.; Thomson, S.; Gambin, T.; Chinault, A.C.; et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum. Mol. Genet. 2011, 20, 4360–4370. [Google Scholar] [CrossRef]
- Celestino-Soper, P.B.S.; Violante, S.; Crawford, E.L.; Luo, R.; Lionel, A.C.; Delaby, E.; Cai, G.; Sadikovic, B.; Lee, K.; Lo, C.; et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc. Natl. Acad. Sci. USA 2012, 109, 7974–7981. [Google Scholar] [CrossRef]
- Nava, C.; Lamari, F.; Héron, D.; Mignot, C.; Rastetter, A.; Keren, B.; Cohen, D.; Faudet, A.; Bouteiller, D.; Gilleron, M.; et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl. Psychiatry 2012, 2, e179. [Google Scholar] [CrossRef]
- Ziats, M.N.; Comeaux, M.S.; Yang, Y.; Scaglia, F.; Elsea, S.H.; Sun, Q.; Beaudet, A.L.; Schaaf, C.P. Improvement of regressive autism symptoms in a child with TMLHE deficiency following carnitine supplementation. Am. J. Med. Genet. A. 2015, 167A, 2162–2167. [Google Scholar] [CrossRef]
- Beaudet, A.L. Brain carnitine deficiency causes nonsyndromic autism with an extreme male bias: A hypothesis. BioEssays News Rev. Mol. Cell. Dev. Biol. 2017, 39. [Google Scholar] [CrossRef]
- Demarquoy, C.; Demarquoy, J. Autism and carnitine: A possible link. World J. Biol. Chem. 2019, 10, 7–16. [Google Scholar] [CrossRef]
- Rashidi-Nezhad, A.; Talebi, S.; Saebnouri, H.; Akrami, S.M.; Reymond, A. The effect of homozygous deletion of the BBOX1 and Fibin genes on carnitine level and acyl carnitine profile. BMC Med. Genet. 2014, 15, 75. [Google Scholar] [CrossRef]
- Lee, H.; Kim, H.-K.; Kwon, J.-T.; Park, S.; Park, H.J.; Kim, S.K.; Park, J.K.; Kang, W.S.; Kim, Y.J.; Chung, J.-H.; et al. BBOX1 is down-regulated in maternal immune-activated mice and implicated in genetic susceptibility to human schizophrenia. Psychiatry Res. 2018, 259, 197–202. [Google Scholar] [CrossRef]
- Nezu, J.; Tamai, I.; Oku, A.; Ohashi, R.; Yabuuchi, H.; Hashimoto, N.; Nikaido, H.; Sai, Y.; Koizumi, A.; Shoji, Y.; et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat. Genet. 1999, 21, 91–94. [Google Scholar] [CrossRef]
- Therrell, B.L.; Lloyd-Puryear, M.A.; Camp, K.M.; Mann, M.Y. Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol. Genet. Metab. 2014, 113, 14–26. [Google Scholar] [CrossRef]
- Koizumi, A.; Nozaki, J.; Ohura, T.; Kayo, T.; Wada, Y.; Nezu, J.; Ohashi, R.; Tamai, I.; Shoji, Y.; Takada, G.; et al. Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum. Mol. Genet. 1999, 8, 2247–2254. [Google Scholar] [CrossRef]
- Ohashi, R.; Tamai, I.; Nezu Ji, J.; Nikaido, H.; Hashimoto, N.; Oku, A.; Sai, Y.; Shimane, M.; Tsuji, A. Molecular and physiological evidence for multifunctionality of carnitine/organic cation transporter OCTN2. Mol. Pharmacol. 2001, 59, 358–366. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Huener, G.; Baykal, T.; Demirkol, M.; Duran, M.; Wanders, R.; Nezu, J.; Mayatepek, E. Silent and symptomatic primary carnitine deficiency within the same family due to identical mutations in the organic cation/carnitine transporter OCTN2. J. Inherit. Metab. Dis. 2003, 26, 613–615. [Google Scholar] [CrossRef]
- El-Hattab, A.W. Systemic Primary Carnitine Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Magoulas, P.L.; El-Hattab, A.W. Systemic primary carnitine deficiency: An overview of clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 2012, 7, 68. [Google Scholar] [CrossRef]
- Vijay, S.; Patterson, A.; Olpin, S.; Henderson, M.J.; Clark, S.; Day, C.; Savill, G.; Walter, J.H. Carnitine transporter defect: Diagnosis in asymptomatic adult women following analysis of acylcarnitines in their newborn infants. J. Inherit. Metab. Dis. 2006, 29, 627–630. [Google Scholar] [CrossRef]
- Schimmenti, L.A.; Crombez, E.A.; Schwahn, B.C.; Heese, B.A.; Wood, T.C.; Schroer, R.J.; Bentler, K.; Cederbaum, S.; Sarafoglou, K.; McCann, M.; et al. Expanded newborn screening identifies maternal primary carnitine deficiency. Mol. Genet. Metab. 2007, 90, 441–445. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Li, F.-Y.; Shen, J.; Powell, B.R.; Bawle, E.V.; Adams, D.J.; Wahl, E.; Kobori, J.A.; Graham, B.; Scaglia, F.; et al. Maternal systemic primary carnitine deficiency uncovered by newborn screening: Clinical, biochemical, and molecular aspects. Genet. Med. Off. J. Am. Coll. Med. Genet. 2010, 12, 19–24. [Google Scholar] [CrossRef]
- Rose, E.C.; di San Filippo, C.A.; Ndukwe Erlingsson, U.C.; Ardon, O.; Pasquali, M.; Longo, N. Genotype-phenotype correlation in primary carnitine deficiency. Hum. Mutat. 2012, 33, 118–123. [Google Scholar] [CrossRef]
- Rasmussen, J.; Nielsen, O.W.; Lund, A.M.; Køber, L.; Djurhuus, H. Primary carnitine deficiency and pivalic acid exposure causing encephalopathy and fatal cardiac events. J. Inherit. Metab. Dis. 2013, 36, 35–41. [Google Scholar] [CrossRef]
- Scaglia, F.; Wang, Y.; Singh, R.H.; Dembure, P.P.; Pasquali, M.; Fernhoff, P.M.; Longo, N. Defective urinary carnitine transport in heterozygotes for primary carnitine deficiency. Genet. Med. Off. J. Am. Coll. Med. Genet. 1998, 1, 34–39. [Google Scholar] [CrossRef]
- Xiaofei, E.; Wada, Y.; Dakeishi, M.; Hirasawa, F.; Murata, K.; Masuda, H.; Sugiyama, T.; Nikaido, H.; Koizumi, A. Age-associated cardiomyopathy in heterozygous carrier mice of a pathological mutation of carnitine transporter gene, OCTN2. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, B270–B278. [Google Scholar] [CrossRef]
- Takahashi, R.; Asai, T.; Murakami, H.; Murakami, R.; Tsuzuki, M.; Numaguchi, Y.; Matsui, H.; Murohara, T.; Okumura, K. Pressure overload-induced cardiomyopathy in heterozygous carrier mice of carnitine transporter gene mutation. Hypertens. Dallas Tex. 1979 2007, 50, 497–502. [Google Scholar] [CrossRef]
- Giudice, P.L.; Bonomini, M.; Arduini, A. A Moderate Carnitine Deficiency Exacerbates Isoproterenol-Induced Myocardial Injury in Rats. Cardiovasc. Drugs Ther. 2016, 30, 119–127. [Google Scholar] [CrossRef]
- Roussel, J.; Labarthe, F.; Thireau, J.; Ferro, F.; Farah, C.; Roy, J.; Horiuchi, M.; Tardieu, M.; Lefort, B.; François Benoist, J.; et al. Carnitine deficiency induces a short QT syndrome. Heart Rhythm 2016, 13, 165–174. [Google Scholar] [CrossRef]
- Wong, G.K.; Pehora, C.; Crawford, M.W. L-carnitine reduces susceptibility to bupivacaine-induced cardiotoxicity: An experimental study in rats. Can. J. Anaesth. J. Can. Anesth. 2017, 64, 270–279. [Google Scholar] [CrossRef]
- Amat di San Filippo, C.; Taylor, M.R.G.; Mestroni, L.; Botto, L.D.; Longo, N. Cardiomyopathy and carnitine deficiency. Mol. Genet. Metab. 2008, 94, 162–166. [Google Scholar] [CrossRef]
- Wilcken, B.; Wiley, V.; Sim, K.G.; Carpenter, K. Carnitine transporter defect diagnosed by newborn screening with electrospray tandem mass spectrometry. J. Pediatr. 2001, 138, 581–584. [Google Scholar] [CrossRef]
- Winter, S.C.; Linn, L.S.; Helton, E. Plasma carnitine concentrations in pregnancy, cord blood, and neonates and children. Clin. Chim. Acta Int. J. Clin. Chem. 1995, 243, 87–93. [Google Scholar] [CrossRef]
- Gallant, N.M.; Leydiker, K.; Wilnai, Y.; Lee, C.; Lorey, F.; Feuchtbaum, L.; Tang, H.; Carter, J.; Enns, G.M.; Packman, S.; et al. Biochemical characteristics of newborns with carnitine transporter defect identified by newborn screening in California. Mol. Genet. Metab. 2017, 122, 76–84. [Google Scholar] [CrossRef]
- Yan, S.; Yang, X.-F.; Liu, H.-L.; Fu, N.; Ouyang, Y.; Qing, K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: An update. World J. Gastroenterol. 2015, 21, 3492–3498. [Google Scholar] [CrossRef]
- Watkins, P.A.; Maiguel, D.; Jia, Z.; Pevsner, J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J. Lipid Res. 2007, 48, 2736–2750. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Hong, D.; Cong, L.; Zhong, S.; Liu, L.; Xu, Y.; Zhang, J. A novel CPT1C variant causes pure hereditary spastic paraplegia with benign clinical course. Ann. Clin. Transl. Neurol. 2019, 6, 610–614. [Google Scholar] [CrossRef]
- Rinaldi, C.; Schmidt, T.; Situ, A.J.; Johnson, J.O.; Lee, P.R.; Chen, K.-L.; Bott, L.C.; Fadó, R.; Harmison, G.H.; Parodi, S.; et al. Mutation in CPT1C Associated with Pure Autosomal Dominant Spastic Paraplegia. JAMA Neurol. 2015, 72, 561–570. [Google Scholar] [CrossRef]
- Lindner, M.; Hoffmann, G.F.; Matern, D. Newborn screening for disorders of fatty-acid oxidation: Experience and recommendations from an expert meeting. J. Inherit. Metab. Dis. 2010, 33, 521–526. [Google Scholar] [CrossRef]
- Collins, S.A.; Sinclair, G.; McIntosh, S.; Bamforth, F.; Thompson, R.; Sobol, I.; Osborne, G.; Corriveau, A.; Santos, M.; Hanley, B.; et al. Carnitine palmitoyltransferase 1A (CPT1A) P479L prevalence in live newborns in Yukon, Northwest Territories, and Nunavut. Mol. Genet. Metab. 2010, 101, 200–204. [Google Scholar] [CrossRef]
- Prasad, C.; Johnson, J.P.; Bonnefont, J.P.; Dilling, L.A.; Innes, A.M.; Haworth, J.C.; Beischel, L.; Thuillier, L.; Prip-Buus, C.; Singal, R.; et al. Hepatic carnitine palmitoyl transferase 1 (CPT1 A) deficiency in North American Hutterites (Canadian and American): Evidence for a founder effect and results of a pilot study on a DNA-based newborn screening program. Mol. Genet. Metab. 2001, 73, 55–63. [Google Scholar] [CrossRef]
- Bennett, M.J.; Santani, A.B. Carnitine Palmitoyltransferase 1A Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef]
- Olpin, S.E.; Allen, J.; Bonham, J.R.; Clark, S.; Clayton, P.T.; Calvin, J.; Downing, M.; Ives, K.; Jones, S.; Manning, N.J.; et al. Features of carnitine palmitoyltransferase type I deficiency. J. Inherit. Metab. Dis. 2001, 24, 35–42. [Google Scholar] [CrossRef]
- Bonnefont, J.-P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol. Aspects Med. 2004, 25, 495–520. [Google Scholar] [CrossRef]
- Innes, A.M.; Seargeant, L.E.; Balachandra, K.; Roe, C.R.; Wanders, R.J.; Ruiter, J.P.; Casiro, O.; Grewar, D.A.; Greenberg, C.R. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr. Res. 2000, 47, 43–45. [Google Scholar] [CrossRef]
- de Sain-van der Velden, M.G.M.; Diekman, E.F.; Jans, J.J.; van der Ham, M.; Prinsen, B.H.C.M.T.; Visser, G.; Verhoeven-Duif, N.M. Differences between acylcarnitine profiles in plasma and bloodspots. Mol. Genet. Metab. 2013, 110, 116–121. [Google Scholar] [CrossRef]
- Primassin, S.; Spiekerkoetter, U. ESI-MS/MS measurement of free carnitine and its precursor γ-butyrobetaine in plasma and dried blood spots from patients with organic acidurias and fatty acid oxidation disorders. Mol. Genet. Metab. 2010, 101, 141–145. [Google Scholar] [CrossRef]
- Korman, S.H.; Waterham, H.R.; Gutman, A.; Jakobs, C.; Wanders, R.J.A. Novel metabolic and molecular findings in hepatic carnitine palmitoyltransferase I deficiency. Mol. Genet. Metab. 2005, 86, 337–343. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Mol. Aspects Med. 2011, 32, 223–233. [Google Scholar] [CrossRef]
- Rubio-Gozalbo, M.E.; Bakker, J.A.; Waterham, H.R.; Wanders, R.J.A. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol. Aspects Med. 2004, 25, 521–532. [Google Scholar] [CrossRef]
- Lopriore, E.; Gemke, R.J.; Verhoeven, N.M.; Jakobs, C.; Wanders, R.J.; Roeleveld-Versteeg, A.B.; Poll-The, B.T. Carnitine-acylcarnitine translocase deficiency: Phenotype, residual enzyme activity and outcome. Eur. J. Pediatr. 2001, 160, 101–104. [Google Scholar] [CrossRef]
- Morris, A.A.; Olpin, S.E.; Brivet, M.; Turnbull, D.M.; Jones, R.A.; Leonard, J.V. A patient with carnitine-acylcarnitine translocase deficiency with a mild phenotype. J. Pediatr. 1998, 132, 514–516. [Google Scholar] [CrossRef]
- Wieser, T. Carnitine Palmitoyltransferase II Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Nance, J.R.; Mammen, A.L. Diagnostic evaluation of rhabdomyolysis. Muscle Nerve 2015, 51, 793–810. [Google Scholar] [CrossRef]
- Wieser, T.; Deschauer, M.; Olek, K.; Hermann, T.; Zierz, S. Carnitine palmitoyltransferase II deficiency: Molecular and biochemical analysis of 32 patients. Neurology 2003, 60, 1351–1353. [Google Scholar] [CrossRef]
- Deschauer, M.; Wieser, T.; Zierz, S. Muscle carnitine palmitoyltransferase II deficiency: Clinical and molecular genetic features and diagnostic aspects. Arch. Neurol. 2005, 62, 37–41. [Google Scholar] [CrossRef]
- Anichini, A.; Fanin, M.; Vianey-Saban, C.; Cassandrini, D.; Fiorillo, C.; Bruno, C.; Angelini, C. Genotype-phenotype correlations in a large series of patients with muscle type CPT II deficiency. Neurol. Res. 2011, 33, 24–32. [Google Scholar] [CrossRef]
- Vladutiu, G.D.; Bennett, M.J.; Fisher, N.M.; Smail, D.; Boriack, R.; Leddy, J.; Pendergast, D.R. Phenotypic variability among first-degree relatives with carnitine palmitoyltransferase II deficiency. Muscle Nerve 2002, 26, 492–498. [Google Scholar] [CrossRef]
- Malik, S.; Paldiwal, A.A.; Korday, C.S.; Jadhav, S.S. Neonatal Carnitine Palmitoyltransferase II Deficiency: A Lethal Entity. J. Clin. Diagn. Res. JCDR 2015, 9, SD01–SD02. [Google Scholar] [CrossRef]
- Vladutiu, G.D.; Quackenbush, E.J.; Hainline, B.E.; Albers, S.; Smail, D.S.; Bennett, M.J. Lethal neonatal and severe late infantile forms of carnitine palmitoyltransferase II deficiency associated with compound heterozygosity for different protein truncation mutations. J. Pediatr. 2002, 141, 734–736. [Google Scholar] [CrossRef]
- Boemer, F.; Deberg, M.; Schoos, R.; Caberg, J.-H.; Gaillez, S.; Dugauquier, C.; Delbecque, K.; François, A.; Maton, P.; Demonceau, N.; et al. Diagnostic pitfall in antenatal manifestations of CPT II deficiency. Clin. Genet. 2016, 89, 193–197. [Google Scholar] [CrossRef]
- Isackson, P.J.; Bennett, M.J.; Lichter-Konecki, U.; Willis, M.; Nyhan, W.L.; Sutton, V.R.; Tein, I.; Vladutiu, G.D. CPT2 gene mutations resulting in lethal neonatal or severe infantile carnitine palmitoyltransferase II deficiency. Mol. Genet. Metab. 2008, 94, 422–427. [Google Scholar] [CrossRef]
- Tajima, G.; Hara, K.; Tsumura, M.; Kagawa, R.; Okada, S.; Sakura, N.; Maruyama, S.; Noguchi, A.; Awaya, T.; Ishige, M.; et al. Newborn screening for carnitine palmitoyltransferase II deficiency using (C16+C18:1)/C2: Evaluation of additional indices for adequate sensitivity and lower false-positivity. Mol. Genet. Metab. 2017, 122, 67–75. [Google Scholar] [CrossRef]
- Ceccarelli, S.M.; Chomienne, O.; Gubler, M.; Arduini, A. Carnitine palmitoyltransferase (CPT) modulators: A medicinal chemistry perspective on 35 years of research. J. Med. Chem. 2011, 54, 3109–3152. [Google Scholar] [CrossRef]
- Foley, J.E. Rationale and application of fatty acid oxidation inhibitors in treatment of diabetes mellitus. Diabetes Care 1992, 15, 773–784. [Google Scholar] [CrossRef]
- Conti, R.; Mannucci, E.; Pessotto, P.; Tassoni, E.; Carminati, P.; Giannessi, F.; Arduini, A. Selective reversible inhibition of liver carnitine palmitoyl-transferase 1 by teglicar reduces gluconeogenesis and improves glucose homeostasis. Diabetes 2011, 60, 644–651. [Google Scholar] [CrossRef]
- Wang, W.; Lopaschuk, G.D. Metabolic therapy for the treatment of ischemic heart disease: Reality and expectations. Expert Rev. Cardiovasc. Ther. 2007, 5, 1123–1134. [Google Scholar] [CrossRef]
- Lionetti, V.; Linke, A.; Chandler, M.P.; Young, M.E.; Penn, M.S.; Gupte, S.; d’Agostino, C.; Hintze, T.H.; Stanley, W.C.; Recchia, F.A. Carnitine palmitoyl transferase-I inhibition prevents ventricular remodeling and delays decompensation in pacing-induced heart failure. Cardiovasc. Res. 2005, 66, 454–461. [Google Scholar] [CrossRef]
- Rupp, H.; Zarain-Herzberg, A.; Maisch, B. The use of partial fatty acid oxidation inhibitors for metabolic therapy of angina pectoris and heart failure. Herz 2002, 27, 621–636. [Google Scholar] [CrossRef]
- Holubarsch, C.J.F.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. Lond. Engl. 1979 2007, 113, 205–212. [Google Scholar] [CrossRef]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- Scaglia, F.; Longo, N. Primary and secondary alterations of neonatal carnitine metabolism. Semin. Perinatol. 1999, 23, 152–161. [Google Scholar] [CrossRef]
- Flanagan, J.L.; Simmons, P.A.; Vehige, J.; Willcox, M.D.; Garrett, Q. Role of carnitine in disease. Nutr. Metab. 2010, 7, 30. [Google Scholar] [CrossRef]
- Lombard, K.A.; Olson, A.L.; Nelson, S.E.; Rebouche, C.J. Carnitine status of lactoovovegetarians and strict vegetarian adults and children. Am. J. Clin. Nutr. 1989, 50, 301–306. [Google Scholar] [CrossRef]
- Limketkai, B.N.; Zucker, S.D. Hyperammonemic Encephalopathy Caused by Carnitine Deficiency. J. Gen. Intern. Med. 2008, 23, 210–213. [Google Scholar] [CrossRef]
- Clark, M.A.; Stein, R.E.K.; Silver, E.J.; Khalid, S.; Fuloria, M.; Esteban-Cruciani, N.V. Carnitine deficiency in preterm infants: A national survey of knowledge and practices. J. Neonatal-Perinat. Med. 2017, 10, 381–386. [Google Scholar] [CrossRef]
- Schmidt-Sommerfeld, E.; Penn, D. Carnitine and total parenteral nutrition of the neonate. Biol. Neonate 1990, 58 (Suppl. 1), 81–88. [Google Scholar] [CrossRef]
- Van Aerde, J.E. In preterm infants, does the supplementation of carnitine to parenteral nutrition improve the following clinical outcomes: Growth, lipid metabolism and apneic spells? Paediatr. Child Health 2004, 9, 573. [Google Scholar] [CrossRef]
- Salguero Olid, A.; Blanco Sánchez, G.; Alonso Ojembarrena, A. A systematic review about prophylactic L-carnitine administration in parenteral nutrition of extremely preterm infants. Farm. Hosp. Organo Expresion Cient. Soc. Espanola Farm. Hosp. 2018, 42, 168–173. [Google Scholar]
- Lheureux, P.E.R.; Hantson, P. Carnitine in the treatment of valproic acid-induced toxicity. Clin. Toxicol. Phila. Pa 2009, 47, 101–111. [Google Scholar] [CrossRef]
- Kobayashi, H.; Fukuda, S.; Yamada, K.; Hasegawa, Y.; Takahashi, T.; Purevsuren, J.; Yamaguchi, S. Clinical Features of Carnitine Deficiency Secondary to Pivalate-Conjugated Antibiotic Therapy. J. Pediatr. 2016, 173, 183–187. [Google Scholar] [CrossRef]
- Pochini, L.; Scalise, M.; Indiveri, C. Inactivation by omeprazole of the carnitine transporter (OCTN2) reconstituted in liposomes. Chem. Biol. Interact. 2009, 179, 394–401. [Google Scholar] [CrossRef]
- Hirano, T.; Yasuda, S.; Osaka, Y.; Kobayashi, M.; Itagaki, S.; Iseki, K. Mechanism of the inhibitory effect of zwitterionic drugs (levofloxacin and grepafloxacin) on carnitine transporter (OCTN2) in Caco-2 cells. Biochim. Biophys. Acta 2006, 1758, 1743–1750. [Google Scholar] [CrossRef]
- Hu, C.; Lancaster, C.S.; Zuo, Z.; Hu, S.; Chen, Z.; Rubnitz, J.E.; Baker, S.D.; Sparreboom, A. Inhibition of OCTN2-mediated transport of carnitine by etoposide. Mol. Cancer Ther. 2012, 11, 921–929. [Google Scholar] [CrossRef]
- Evans, A. Dialysis-related carnitine disorder and levocarnitine pharmacology. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2003, 41, S13–S26. [Google Scholar] [CrossRef]
- Evans, A.M.; Faull, R.J.; Nation, R.L.; Prasad, S.; Elias, T.; Reuter, S.E.; Fornasini, G. Impact of hemodialysis on endogenous plasma and muscle carnitine levels in patients with end-stage renal disease. Kidney Int. 2004, 66, 1527–1534. [Google Scholar] [CrossRef]
- Hedayati, S.S. Dialysis-related carnitine disorder. Semin. Dial. 2006, 19, 323–328. [Google Scholar] [CrossRef]
- Wasserstein, A.G. L-carnitine supplementation in dialysis: Treatment in quest of disease. Semin. Dial. 2013, 26, 11–15. [Google Scholar] [CrossRef]
- Arduini, A.; Bonomini, M.; Savica, V.; Amato, A.; Zammit, V. Carnitine in metabolic disease: Potential for pharmacological intervention. Pharmacol. Ther. 2008, 120, 149–156. [Google Scholar] [CrossRef]
- Merritt, J.L.; Norris, M.; Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 2018, 6. [Google Scholar] [CrossRef]
- Baumgartner, M.R.; Hörster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130. [Google Scholar] [CrossRef]
- Nasser, M.; Javaheri, H.; Fedorowicz, Z.; Noorani, Z. Carnitine supplementation for inborn errors of metabolism. Cochrane Database Syst. Rev. 2012, CD006659. [Google Scholar] [CrossRef]
- Watanabe, K.; Yamada, K.; Sameshima, K.; Yamaguchi, S. Two siblings with very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency suffered from rhabdomyolysis after l-carnitine supplementation. Mol. Genet. Metab. Rep. 2018, 15, 121–123. [Google Scholar] [CrossRef]
- Liepinsh, E.; Makrecka-Kuka, M.; Volska, K.; Kuka, J.; Makarova, E.; Antone, U.; Sevostjanovs, E.; Vilskersts, R.; Strods, A.; Tars, K.; et al. Long-chain acylcarnitines determine ischaemia/reperfusion-induced damage in heart mitochondria. Biochem. J. 2016, 473, 1191–1202. [Google Scholar] [CrossRef]
- Ferro, F.; Ouillé, A.; Tran, T.-A.; Fontanaud, P.; Bois, P.; Babuty, D.; Labarthe, F.; Le Guennec, J.-Y. Long-Chain Acylcarnitines Regulate the hERG Channel. PLoS ONE 2012, 7, e41686. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Lindner, M.; Santer, R.; Grotzke, M.; Baumgartner, M.R.; Boehles, H.; Das, A.; Haase, C.; Hennermann, J.B.; Karall, D.; et al. Treatment recommendations in long-chain fatty acid oxidation defects: Consensus from a workshop. J. Inherit. Metab. Dis. 2009, 32, 498–505. [Google Scholar] [CrossRef]
- Parikh, S.; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R. A Modern Approach to the Treatment of Mitochondrial Disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef]
- DiMauro, S.; Hirano, M.; Schon, E.A. Approaches to the treatment of mitochondrial diseases. Muscle Nerve 2006, 34, 265–283. [Google Scholar] [CrossRef]
- Vallance, H.D.; Koochin, A.; Branov, J.; Rosen-Heath, A.; Bosdet, T.; Wang, Z.; Hazen, S.L.; Horvath, G. Marked elevation in plasma trimethylamine-N-oxide (TMAO) in patients with mitochondrial disorders treated with oral l-carnitine. Mol. Genet. Metab. Rep. 2018, 15, 130–133. [Google Scholar] [CrossRef]
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef]


{kind=link}
| Category | Disease | Gene | Clinical Features | Diagnostic Markers | Long Term Management | References |
|---|---|---|---|---|---|---|
| Carnitine biosynthesis disorders | TMLD deficiency | TMLHE | • Risk factor for autism | • Low 3-hydroxy-6-N-trimethyllysine (HTML) and γ-butyrobetaine • High TML and TML/γ-butyrobetaine ratio | • Carnitine supplementation | [16,17,18,19,20] |
| BBD deficiency | BBOX1 | • Microcephaly, speech delay, poor growth and some dysmorphic features a • Schizophrenia susceptibility | • Not available | • Not available | [23,24] | |
| Carnitine transport defect | Primary carnitine deficiency | SLC22A5 | • Metabolic decompensations precipitated by fasting and intercurrent illnesses • Cardio(myopathy) • Easy fatigability • Asymptomatic | • Low free carnitine | • Carnitine supplementation (100–200 mg/kg/day) | [12,16,29,30,31,32,33,34,35,56] |
| Mitochondrial carnitine–acylcarnitine cycle disorders | CPT IA deficiency | CPT1A | • Metabolic decompensations precipitated by fasting and intercurrent illnesses • Heart and skeletal muscle involvement is less common • Acute fatty liver of pregnancy (AFLP) reported in heterozygous mothers carrying an affected fetus | • High free carnitine and C0/C16+C18 ratio | • Frequent feeding, avoidance of fasting • Low fat and high carbohydrates diet • Medium-chain triglycerides (MCT) | [55,56,57,58,59] |
| CACT deficiency | SLC25A20 | • Neonatal presentation: hypoketotic hypoglycemia, respiratory distress, arrhythmia, cardiomyopathy, liver disease, and sudden death • Milder phenotype with metabolic decompensations precipitated by fasting and intercurrent illnesses | • Elevated long chain acylcarnitines (especially C16 and C18:1) • Low free carnitine | • Frequent feeding, avoidance of fasting • Low fat and high carbohydrates diet • MCT • Carnitine supplementation b | [56,64,65,66] | |
| CPT II deficiency | CPT2 | • Myopathic form: recurrent attacks of rhabdomyolysis triggered by prolonged exercise, infection, fasting, and cold • Neonatal form: hypoketotic hypoglycemia, liver failure, arrhythmias, cardiomyopathy, seizures, dysmorphic features, renal and brain malformations • Infantile form: hypoketotic hypoglycemia, hepatomegaly, liver failure, cardiomyopathy, and arrhythmias | • Elevated long chain acylcarnitines (especially C16 and C18:1) • Low free carnitine | • Frequent feeding, avoidance of fasting • Low fat and high carbohydrates diet • MCT • Carnitine supplementation b | [56,58,67,69,70,71,72,73,74,75] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almannai, M.; Alfadhel, M.; El-Hattab, A.W. Carnitine Inborn Errors of Metabolism. Molecules 2019, 24, 3251. https://doi.org/10.3390/molecules24183251
Almannai M, Alfadhel M, El-Hattab AW. Carnitine Inborn Errors of Metabolism. Molecules. 2019; 24(18):3251. https://doi.org/10.3390/molecules24183251
Chicago/Turabian StyleAlmannai, Mohammed, Majid Alfadhel, and Ayman W. El-Hattab. 2019. "Carnitine Inborn Errors of Metabolism" Molecules 24, no. 18: 3251. https://doi.org/10.3390/molecules24183251
APA StyleAlmannai, M., Alfadhel, M., & El-Hattab, A. W. (2019). Carnitine Inborn Errors of Metabolism. Molecules, 24(18), 3251. https://doi.org/10.3390/molecules24183251