

Clinical Disorders in Cystic Fibrosis That Affect Emergency Procedures—A Case Report and Review

 , , , , and
, , , , and
Abstract

1. Introduction
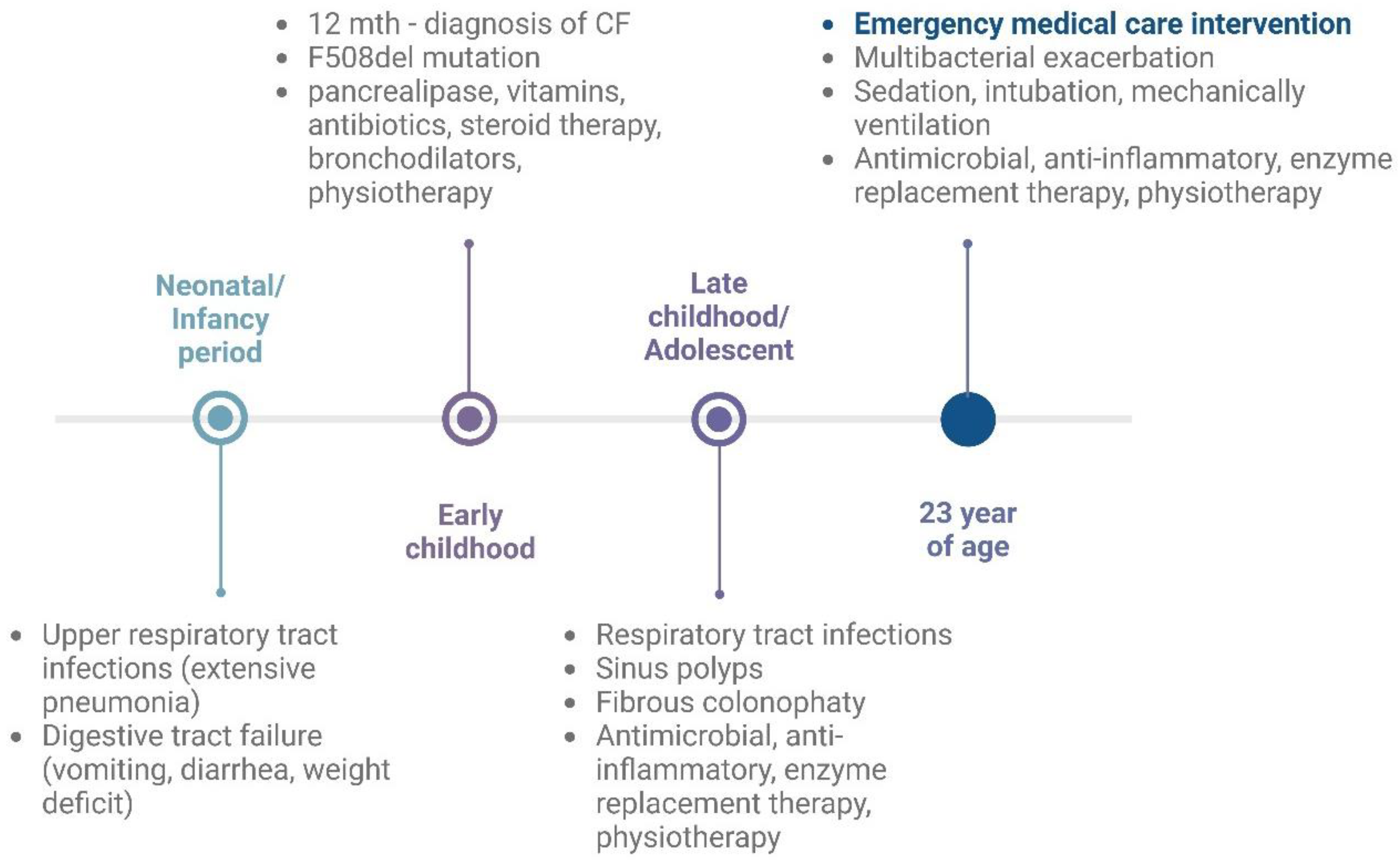
2. Cystic Fibrosis Case Report
2.1. Emergency Intervention in Critical Condition
2.2. Previous Medical History of a Patient
3. Narrative Review
3.1. Clinical Manifestation of Cystic Fibrosis
3.2. Cardiovascular Impact
3.3. Emergency Evaluation and Procedures for Patients with Cystic Fibrosis
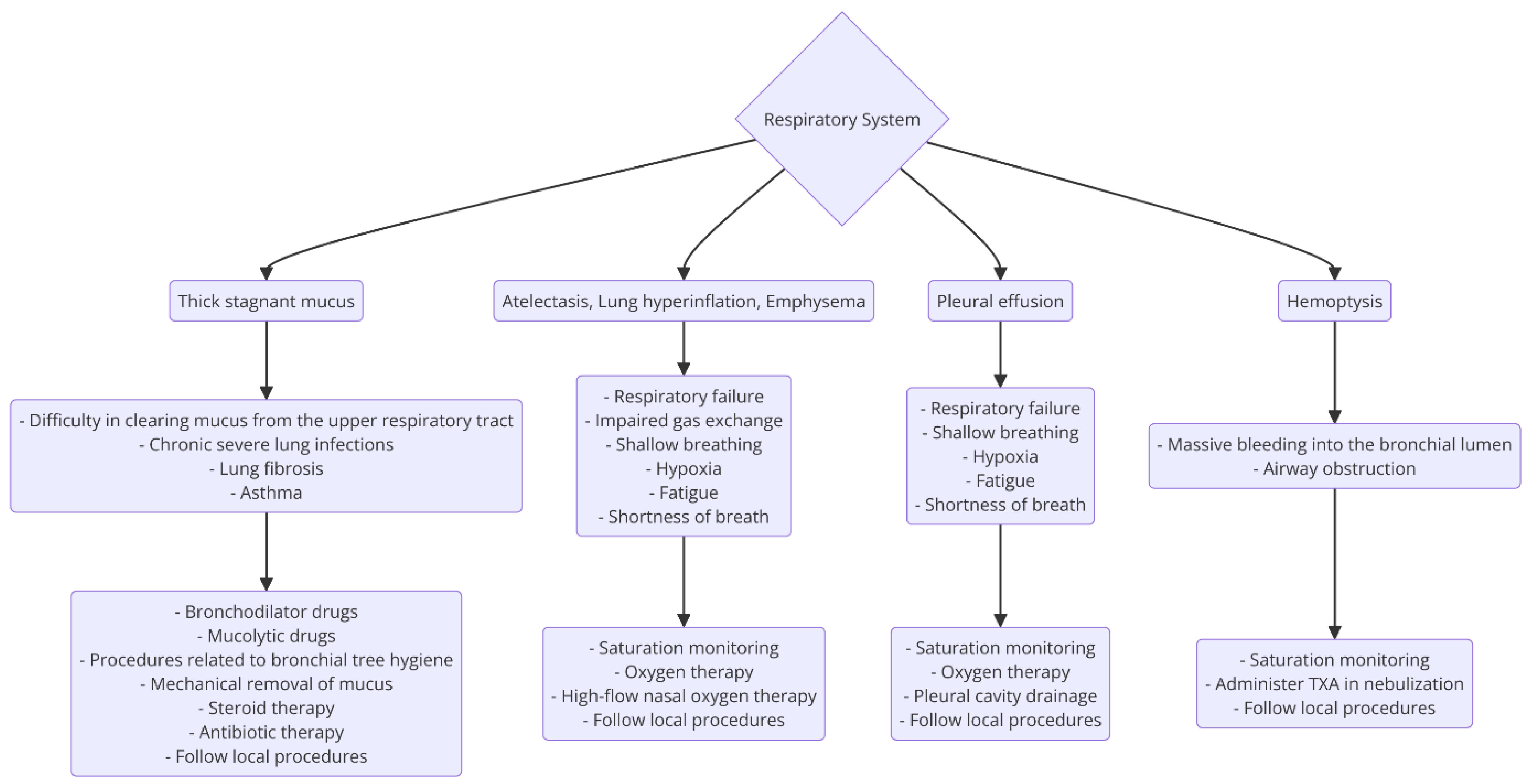
3.3.1. Respiratory Tract
- Cyanosis is a change in the color of the skin due to an increased concentration of deoxyhemoglobin >5 g/dL. Central cyanosis is observed around the mouth, mucous membranes of the oral cavity and tongue. Peripheral cyanosis is observed in the distal parts of the limbs. Differential cyanosis presents as a bluish discoloration between the upper and lower extremities [55];
- Pulmonary hypertension—hypoxia can lead to increased pulmonary artery pressure, contributing to pulmonary hypertension, which is a common complication in CF patients [56];
- Reduced lung function—hypoxia leads to a decreased forced expiratory volume and forced vital capacity in CF patients [56];
- Inflammation and infection—hypoxia worsens inflammation and promotes the growth of pathogens like Pseudomonas aeruginosa in CF lung infections [57];
- Anxiety, agitation, fear, or a loss of consciousness are signs of central nervous system hypoxia;
- Metabolic acidosis caused by hypercapnia is a sign of anaerobic metabolism in the tissues. It occurs in type II respiratory failure.

| Type of Oxygen Mask/Tube | The Obtained Values of FiO2 |
|---|---|
| Standard nasal cannula | It is widely accepted that 1 L/min = 24% (each increase by 1 L/min, in the range of 1–6 L/min, increases the concentration by about 4%). The precise measurement of FiO2 is difficult due to the variation in breathing patterns, including both tidal volume and respiratory rate. |
| Simple face masks | It is commonly accepted that 40–60% is used at a flow rate of 5–10 L/min, depending on the oxygen flow rate and the patient’s breathing pattern. A low rate of less than 5 L/min is not recommended due to the risk of CO2 rebreathing and increasing resistance during inhalation. |
| Venturi masks | Venturi masks allow for the precise administration of a specific oxygen concentration at the rate specified by the manufacturer (24%, 25%, 28%, 35%, 40%, 50%, 60%). If the patient is breathing at a rate of >30 breaths/min, the oxygen flow should be increased by 50% above the manufacturer’s recommendations. |
| High-concentration reservoir mask (non-rebreathing mask) | It is generally acknowledged that 60–90% is used at a flow rate of 15 L/min, but FiO2 depends on the patient’s breathing pattern and the proper fit of the mask to the face. |
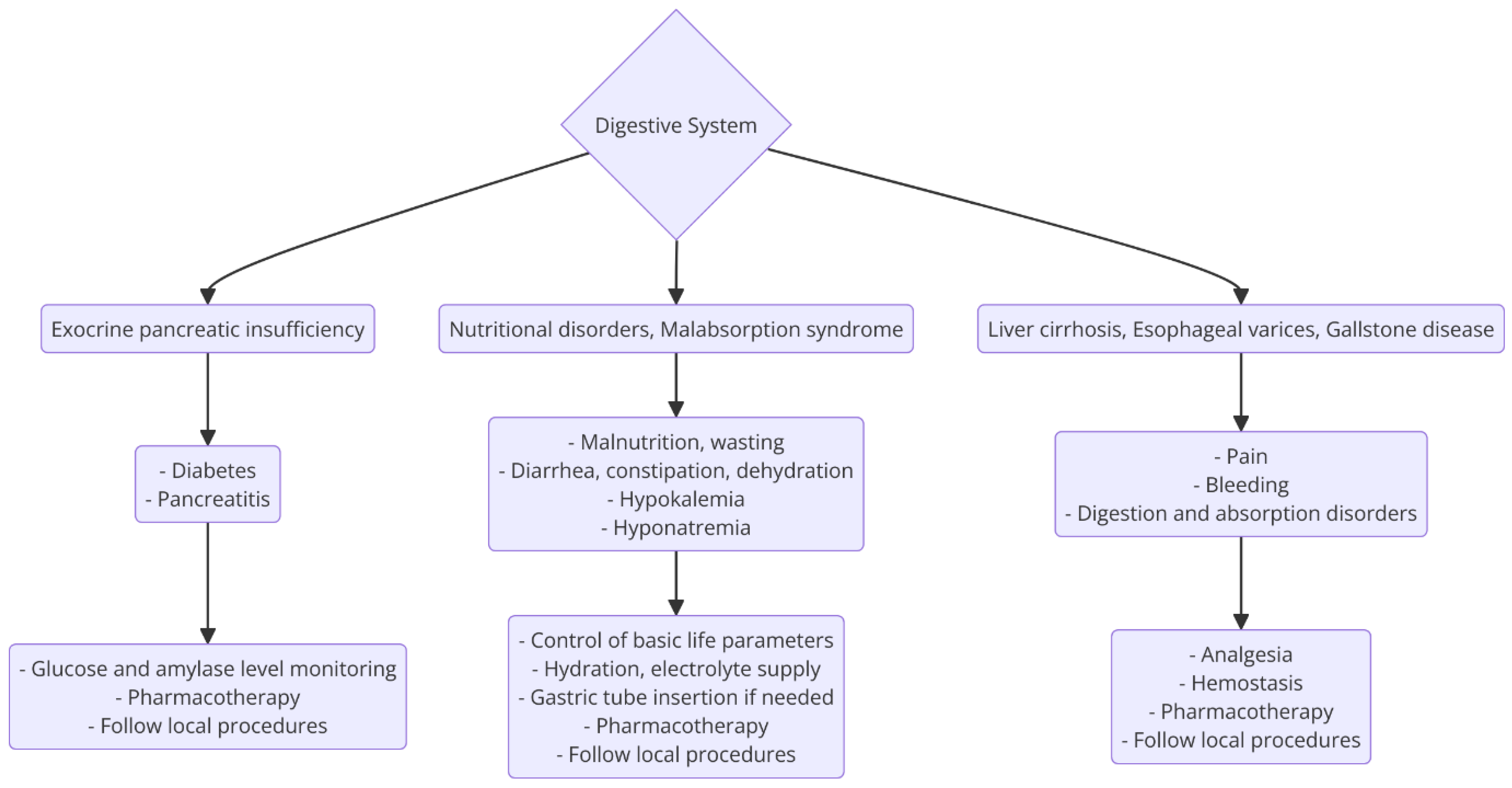
3.3.2. Digestive System
3.3.3. Sudden Cardiac Arrest
- ○
- Basic Life Support (BLS), which consists of chest compressions and rescue breaths (cardiopulmonary resuscitation, CPR), with the use of an automated external defibrillator (AED).
- ○
- Advanced Life Support (ALS), which includes CPR, defibrillation, maintaining airway patency, pharmacotherapy, and all other procedures aimed at restoring spontaneous cardiac action.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Kasam, R.K.; Gajjala, P.R.; Jegga, A.G.; Courtney, J.A.; Randell, S.H.; Kramer, E.L.; Clancy, J.P.; Madala, S.K. Fibrocyte accumulation in the lungs of cystic fibrosis patients. J. Cyst. Fibros. 2020, 19, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Mutation Database. Available online: http://www.genet.sickkids.on.ca/StatisticsPage.html (accessed on 25 January 2025).
- An Online Catalog of Human Genes and Genetic Disorders. Available online: https://omim.org/entry/602421 (accessed on 25 January 2025).
- Cystic Fibrosis Foundation. Available online: https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf (accessed on 26 January 2025).
- Zolin, A.; Orenti, A.; Gambazza, S.; Adamoli, A.; van Rens, J.; Prasad, V.; Naehrlich, L.; Kirwan, L.; Krivec, U.; Zomer, D.; et al. ECFSPR Annual Report 2022; European Cystic Fibrosis Society Patient Registry: Karup, Denmark, 2024. [Google Scholar]
- Sands, D.; Skoczylas-Ligocka, A.; Cofta, S.; Walicka-Serzysko, K.; Woźniacki, Ł.; Natkaniec, M.; Megas, B.; Gilewski, D. Opieka nad Chorymi na Mukowiscydozę w Polsce. Stan Obecny i Rekomendacje Poprawy; Polskiego Towarzystwa Mukowiscydozy: Warsaw–Krakow, Poland, 2019. [Google Scholar]
- Ma, J.T.; Tang, C.; Kang, L.; Voynow, J.A.; Rubin, B.K. Cystic Fibrosis Sputum Rheology Correlates With Both Acute and Longitudinal Changes in Lung Function. Chest 2018, 154, 370–377. [Google Scholar] [CrossRef]
- Abrami, M.; Maschio, M.; Conese, M.; Confalonieri, M.; Gerin, F.; Dapas, B.; Farra, R.; Adrover, A.; Torelli, L.; Ruaro, B.; et al. Combined use of rheology and portable low-field NMR in cystic fibrosis patients. Respir. Med. 2021, 189, 106623. [Google Scholar] [CrossRef]
- Freeman Grossheim, L.; Gates, K.S. Emergency Department Care of Adult Cystic Fibrosis Patients; University of Texas Medical School at Houston: Houston, TX, USA, 2008. [Google Scholar]
- Walicka-Serzysko, K.; Orlik, T.; Sands, D.; Jeneralska, N.; Popiel, A.; Skorupa, W.; Pogorzelski, A.; Cofta, S. Nebulisation therapy in patients with cystic fibrosis—consensus of the Polish Cystic Fibrosis Society. Adv. Respir. Med. 2021, 89, 570–580. [Google Scholar] [CrossRef]
- Butzon, F.; Criddle, L.M. A 23-year-old woman with cystic fibrosis, acute hypotension, and cardiac arrest. J. Emerg. Nurs. 2006, 32, 506–508. [Google Scholar] [CrossRef]
- Sirhan, M.A.; Kalinin, M.; Cohen, L.; Gurevich, E. Uncommon Presentation of Cystic Fibrosis: A Case Report and Literature Review. Cureus 2023, 15, e45186. [Google Scholar] [CrossRef]
- Galante, G.; Freeman, A.J. Freeman, Gastrointestinal, Pancreatic, and Hepatic Manifestations of Cystic Fibrosis in the Newborn. Neoreviews 2019, 20, e12–e24. [Google Scholar] [CrossRef]
- Muyulema Muyulema, L.E.; Olivo Torres, R.E.d.C.; Tobar Armendariz, K.A. Clinical manifestations of cystic fibrosis. Interam. J. Health Sci. 2024, 4, 68. [Google Scholar] [CrossRef]
- Welsner, M.; Dietz-Terjung, S.; Stehling, F.; Schulte, T.; Niehammer, U.; Gahbiche, F.E.; Taube, C.; Strassburg, S.; Schoebel, C.; Weinreich, G.; et al. Obstructive sleep apnea and nocturnal hypoxemia in adult patients with cystic fibrosis. BMC Pulm. Med. 2022, 22, 446. [Google Scholar] [CrossRef]
- Cooney, A.L.; McCray, P.B., Jr.; Sinn, P.L. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 2018, 9, 538. [Google Scholar] [CrossRef] [PubMed]
- Bierlaagh, M.C.; Muilwijk, D.; Beekman, J.M.; van der Ent, C.K. A new era for people with cystic fibrosis. Eur. J. Pediatr. 2021, 180, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Lurquin, F.; Buysschaert, M.; Preumont, V. Advances in cystic fibrosis-related diabetes: Current status and future directions. Diabetes Metab. Syndr. 2023, 17, 102899. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.P.; De Boeck, K. A new era in the treatment of cystic fibrosis: Correction of the underlying CFTR defect. Lancet Respir. Med. 2013, 1, 158–163. [Google Scholar] [CrossRef]
- Ong, T.; Ramsey, B.W. Cystic Fibrosis: A Review. Jama 2023, 329, 1859–1871. [Google Scholar] [CrossRef]
- Turcios, N.L. Cystic Fibrosis Lung Disease: An Overview. Respir. Care 2020, 65, 233–251. [Google Scholar] [CrossRef]
- Cardines, R.; Giufre, M.; Pompilio, A.; Fiscarelli, E.; Ricciotti, G.; Di Bonaventura, G.; Cerquetti, M. Haemophilus influenzae in children with cystic fibrosis: Antimicrobial susceptibility, molecular epidemiology, distribution of adhesins and biofilm formation. Int. J. Med. Microbiol. 2012, 302, 45–52. [Google Scholar] [CrossRef]
- Allen, P.; Borick, J.; Borick, J. Acute and Chronic Infection Management in CF. In Cystic Fibrosis in Primary Care: An Essential Guide to a Complex, Multi-System Disease; Lewis, M.D.F.D., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 69–87. [Google Scholar]
- Blanchard, A.C.; Waters, V.J. Microbiology of Cystic Fibrosis Airway Disease. Semin. Respir. Crit. Care Med. 2019, 40, 727–736. [Google Scholar] [CrossRef]
- Tunney, M.M.; Field, T.R.; Moriarty, T.F.; Patrick, S.; Doering, G.; Muhlebach, M.S.; Wolfgang, M.C.; Boucher, R.; Gilpin, D.F.; McDowell, A.; et al. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 995–1001. [Google Scholar] [CrossRef]
- Dasenbrook, E.C.; Checkley, W.; Merlo, C.A.; Konstan, M.W.; Lechtzin, N.; Boyle, M.P. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA 2010, 303, 2386–2392. [Google Scholar] [CrossRef]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 15010. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.R.; Saus, E.; Iraola-Guzman, S.; Cabello-Yeves, E.; Ksiezopolska, E.; Cozzuto, L.; Bejarano, L.A.; Andreu-Somavilla, N.; Alloza-Trabado, M.; Blanco, A.; et al. Citizen-science based study of the oral microbiome in Cystic fibrosis and matched controls reveals major differences in diversity and abundance of bacterial and fungal species. J. Oral Microbiol. 2021, 13, 1897328. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S. Gastrointestinal Manifestations of Cystic Fibrosis. Gastroenterol. Hepatol. 2016, 12, 43–47. [Google Scholar]
- Gibson-Corley, K.N.; Meyerholz, D.K.; Engelhardt, J.F. Pancreatic pathophysiology in cystic fibrosis. J. Pathol. 2016, 238, 311–320. [Google Scholar] [CrossRef]
- Hull, S.C.; Kass, N.E. Adults with cystic fibrosis and (in)fertility: How has the health care system responded? J. Androl. 2000, 21, 809–813. [Google Scholar] [CrossRef]
- Rahman, F.; Campbell, K.; Deebel, N.; Ghomeshi, A.; Zarli, M.; Domaradzki, L.; Tupayachi Ortiz, M.G.; Ramasamy, R. Assessing Infertility Literacy and Knowledge Gaps Among Patients with Cystic Fibrosis. Urol. Res. Pract. 2023, 49, 312–315. [Google Scholar] [CrossRef]
- Ratjen, F. Changes in strategies for optimal antibacterial therapy in cystic fibrosis. Int. J. Antimicrob. Agents 2001, 17, 93–96. [Google Scholar] [CrossRef]
- Bateman, R.M.; Sharpe, M.D.; Jagger, J.E.; Ellis, C.G.; Solé-Violán, J.; López-Rodríguez, M.; Herrera-Ramos, E.; Ruíz-Hernández, J.; Borderías, L.; Horcajada, J.; et al. 36th International Symposium on Intensive Care and Emergency Medicine: Brussels, Belgium. 15–18 March 2016. Crit. Care 2016, 20, 94. [Google Scholar] [CrossRef]
- Morrell, M.R.; Kiel, S.C.; Pilewski, J.M. Organ Transplantation for Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 842–856. [Google Scholar] [CrossRef]
- Huang, W.; Smith, A.T.; Korotun, M.; Iacono, A.; Wang, J. Lung Transplantation in a New Era in the Field of Cystic Fibrosis. Life 2023, 13, 1600. [Google Scholar] [CrossRef]
- Lung Transplantation in Patients with Cystic Fibrosis (Przeszczep Płuc u Chorych na Mukowiscydozę). Available online: https://ptwm.org.pl/pl/przeszczepy-mukowiscydoza (accessed on 15 April 2025).
- Pena, T.A.; Wright, B.; Parekh, K.R.; Kleney-Tait, J. Impact of elexacaftor-tezacaftor-ivacaftor in lung transplantation for cystic fibrosis in the United States. JHLT Open 2025, 7, 100171. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Taylor-Cousar, J.L. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur. Respir. Rev. 2020, 29, 190112. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2019, 10, 1662. [Google Scholar] [CrossRef]
- Martin, C.; Legeai, C.; Regard, L.; Cantrelle, C.; Dorent, R.; Carlier, N.; Kerbaul, F.; Burgel, P.R. Major Decrease in Lung Transplantation for Patients with Cystic Fibrosis in France. Am. J. Respir. Crit. Care Med. 2022, 205, 584–586. [Google Scholar] [CrossRef]
- King, C.S.; Brown, A.W.; Aryal, S.; Ahmad, K.; Donaldson, S. Critical Care of the Adult Patient With Cystic Fibrosis. Chest 2019, 155, 202–214. [Google Scholar] [CrossRef]
- Smith, D.; Bowden, T. Using the ABCDE approach to assess the deteriorating patient. Nurs. Stand. 2017, 32, 51–63. [Google Scholar] [CrossRef]
- Main, E.; Rand, S. Conventional chest physiotherapy compared to other airway clearance techniques for cystic fibrosis. Cochrane Database Syst. Rev. 2023, 5, CD002011. [Google Scholar] [CrossRef]
- Radtke, T. Role of physical activity and airway clearance therapy in cystic fibrosis: Moving forward in a rapidly changing landscape. Thorax 2023, 78, 3–4. [Google Scholar] [CrossRef]
- Nicholson, T.T.; Smith, A.; McKone, E.F.; Gallagher, C.G. Duration of intravenous antibiotic treatment for acute exacerbations of cystic fibrosis: A systematic review. J. Cyst. Fibros. 2022, 21, 562–573. [Google Scholar] [CrossRef]
- Schwarz, C.; Wimmer, E.; Holz, F.; Grehn, C.; Staab, D.; Eschenhagen, P.N. Antibiotic Therapy for Pulmonary Exacerbations in Cystic Fibrosis-A Single-Centre Prospective Observational Study. Antibiotics 2023, 12, 734. [Google Scholar] [CrossRef]
- Sands, D.; Walicka-Serzysko, K.; Doniec, Z.; Mastalerz-Migas, A.; Batura-Gabryel, H.; Pierzchała, W. ReCOMmendations for management in cystic fibrosis for General PrActitionerS—COMPAS CF—part 1. Pediatr. Pol. 2017, 92, 602–614. [Google Scholar]
- Ciuca, I.M.; Dediu, M.; Popin, D.; Pop, L.L.; Tamas, L.A.; Pilut, C.N.; Almajan Guta, B.; Popa, Z.L. Antibiotherapy in Children with Cystic Fibrosis-An Extensive Review. Children 2022, 9, 1258. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.; Flume, P.A. Pulmonary Complications of Cystic Fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 804–809. [Google Scholar] [CrossRef]
- Mirabile, V.S.; Shebl, E.; Sankari, A.; Burns, B. Respiratory Failure in Adults. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Bush, A.; Bilton, D.; Hodson, M. Hodson and Geddes’ Cystic Fibrosis, Fourth Edition; CRC Press: Boca Raton, FL, USA, 2015; pp. 236–260. [Google Scholar]
- Sands, D.; Walicka-Serzysko, K.; Doniec, Z.; Mastalerz-Migas, A.; Batura-Gabryel, H.; Pierzchała, W. ReCOMmendations for management in cystic fibrosis for General PrActitionerS—COMPAS CF—part 2. Pediatr. Pol. 2017, 92, 602–614. [Google Scholar] [CrossRef]
- Pahal, P.; Goyal, A. Central and Peripheral Cyanosis. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2025. [Google Scholar]
- Urquhart, D.S. Exploration of the Relationship Between Hypoxia and Measures of Clinical Status and Inflammation in Children with Cystic Fibrosis. Ph.D. Thesis, UCL (University College London), London, UK, 2010. [Google Scholar]
- Urquhart, D.; Montgomery, H.; Jaffé, A. Assessment of hypoxia in children with cystic fibrosis. Arch. Dis. Child. 2005, 90, 1138–1143. [Google Scholar] [CrossRef]
- O’Driscoll, B.; Howard, L.; Earis, J.; Mak, V.; Bajwah, S.; Beasley, R.; Curtis, K.; Davison, A.; Dorward, A.; Dyer, C. British Thoracic Society Guideline for oxygen use in adults in healthcare and emergency settings. BMJ Open Respir. Res. 2017, 4, i27. [Google Scholar] [CrossRef]
- Singer, M.; Young, P.J.; Laffey, J.G.; Asfar, P.; Taccone, F.S.; Skrifvars, M.B.; Meyhoff, C.S.; Radermacher, P. Dangers of hyperoxia. Crit. Care 2021, 25, 440. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, P.; Jaeschke, R.; Budaj, A.; Leśniak, W.; Krenke, R.; Mejza, F.; Niżankowska-Mogilnicka, E.; Dąbrowski, A.; Hartleb, M.; Nowakowska-Duława, E. Interna Szczeklika 2023; Medycyna Praktyczna: Kraków, Poland, 2023. [Google Scholar]
- Monsieurs, K.G.; Nolan, J.P.; Bossaert, L.L.; Greif, R.; Maconochie, I.K.; Nikolaou, N.I.; Perkins, G.D.; Soar, J.; Truhlář, A.; Wyllie, J.; et al. European Resuscitation Council Guidelines for Resuscitation 2015: Section 1. Executive summary. Resuscitation 2015, 95, 1–80. [Google Scholar] [CrossRef]
- Regulation of the Minister of Health of 28 February 2017 on the Type and Scope of Preventive, Diagnostic, Therapeutic, and Rehabilitative Services Provided Independently by Nurses or Midwives Without a Medical Order. Journal of Laws of the Republic of Poland, 2017, item 497. Available online: https://isap.sejm.gov.pl/isap.nsf/DocDetails.xsp?id=WDU20170000497 (accessed on 3 May 2025).
- Regulation of the Minister of Health of 22 June 2023 on Medical Emergency Procedures and Health Services Other Than Medical Emergency Procedures That Can Be Provided by Emergency Medical Technicians. Journal of Laws of the Republic of Poland, 2023, item 1180. Available online: https://isap.sejm.gov.pl/isap.nsf/DocDetails.xsp?id=WDU20230001180 (accessed on 3 May 2025).
- Paciorek, P.; Patrzała, A. Medyczne Czynności Ratunkowe; Wydawnictwo Lekarskie PZWL: Warszawa, Poland, 2015; ISBN 978-83-200-4891-9. [Google Scholar]
- Wrigley-Carr, H.E.; van Dorst, J.M.; Ooi, C.Y. Intestinal dysbiosis and inflammation in cystic fibrosis impacts gut and multi-organ axes. Med. Microecol. 2022, 13, 100057. [Google Scholar] [CrossRef]
- The European Resuscitation Council Guidelines for Resuscitation. Available online: https://cprguidelines.eu/ (accessed on 29 January 2025).
- Herrman, J.W.; Attia, M.W. Early use of oxygen in pediatric emergency patients: A standard but underrated therapy. J. Emerg. Nurs. 2002, 28, 440–443. [Google Scholar] [CrossRef]
- Pulmozyme, Characteristics of the Medicinal Product. Available online: http://chpl.com.pl/data_files/2011-07-05_110616_Pulmozyme_ChPL_C.pdf (accessed on 27 January 2025).
- Brodier, E.A.; Raithatha, M.; Kannan, S.; Karunasekara, N. Use of nebulised N-acetylcysteine as a life-saving mucolytic in intensive care: A case report. J. Intensive Care Soc. 2019, 21, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Beta2-mimetics Inhalation (pl. Beta2-mimetyki wziewne). Available online: https://www.mp.pl/pacjent/pochp/lekiileczenie/54235,beta2-mimetyki-wziewne/ (accessed on 27 January 2025).
- Atchinson, P.R.A.; Hatton, C.J.; Roginski, M.A.; Backer, E.D.; Long, B.; Lentz, S.A. The emergency department evaluation and management of massive hemoptysis. Am. J. Emerg. Med. 2021, 50, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.-Y.; Yang, S.-H.; Chiang, H.-S. Impact of Oxygen Concentration Delivered via Nasal Cannula on Different Lung Conditions: A Bench Study. Healthcare 2021, 9, 1235. [Google Scholar] [CrossRef]
- O’Driscoll, B.R.; Howard, L.S.; Earis, J.; Mak, V. BTS guideline for oxygen use in adults in healthcare and emergency settings. Thorax 2017, 72, ii1–ii90. [Google Scholar] [CrossRef]
- Jones, H.A.; Turner, S.L.; Hughes, J.M. Performance of the large-reservoir oxygen mask (Ventimask). Lancet 1984, 1, 1427–1431. [Google Scholar] [CrossRef]
- Assis, D.N.; Freedman, S.D. Gastrointestinal Disorders in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 109–118. [Google Scholar] [CrossRef]
- Collins, S. Nutritional management of cystic fibrosis—An update for the 21st century. Paediatr. Respir. Rev. 2018, 26, 4–6. [Google Scholar] [CrossRef]
- Scaparrotta, A.; Di Pillo, S.; Attanasi, M.; Consilvio, N.P.; Cingolani, A.; Rapino, D.; Mohn, A.; Chiarelli, F. Growth failure in children with cystic fibrosis. J. Pediatr. Endocrinol. Metab. 2012, 25, 393–405. [Google Scholar] [CrossRef]
- Sutherland, R.; Katz, T.; Liu, V.; Quintano, J.; Brunner, R.; Tong, C.W.; Collins, C.E.; Ooi, C.Y. Dietary intake of energy-dense, nutrient-poor and nutrient-dense food sources in children with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 804–810. [Google Scholar] [CrossRef]
- Escobedo-Monge, M.F.; Barrado, E.; Parodi-Roman, J.; Escobedo-Monge, M.A.; Marcos-Temprano, M.; Marugan-Miguelsanz, J.M. Magnesium Status and Calcium/Magnesium Ratios in a Series of Cystic Fibrosis Patients. Nutrients 2022, 14, 1793. [Google Scholar] [CrossRef]
- Shape, J.M.; Sala, M.A. Nutrition management in adults with cystic fibrosis. Nutr. Clin. Pract. 2022, 37, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Altman, K.; McDonald, C.M.; Michel, S.H.; Maguiness, K. Nutrition in cystic fibrosis: From the past to the present and into the future. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S56–S73. [Google Scholar] [CrossRef] [PubMed]
- Ha, A.C.T.; Doumouras, B.S.; Wang, C.N.; Tranmer, J.; Lee, D.S. Prediction of Sudden Cardiac Arrest in the General Population: Review of Traditional and Emerging Risk Factors. Can. J. Cardiol. 2022, 38, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Dykowska, G.; Śmigrocka, E.; Borawska-Kowalczyk, U.; Sands, D.; Sienkiewicz, Z.; Leńczuk-Gruba, A.; Gorczyca, D.; Głowacka, M. Parents’ Knowledge of the Impact of Cystic Fibrosis on the Quality of Life of Children and Adolescents Suffering from This Disease as an Element of Patient Safety. J. Clin. Med. 2023, 12, 5214. [Google Scholar] [CrossRef]
- Sands, D.; Pogorzelski, A.; Skoczylas-Ligocka, A. Epidemiologia i Organizacja Opieki Medycznej nad Chorymi na Mukowiscydozę. Mukowiscydoza, Choroba Wieloukładowa; Termedia sp. z o.o.: Poznań, Poland, 2018. [Google Scholar]
- Nash, E.F.; Choyce, J.; Carrolan, V.; Justice, E.; Shaw, K.L.; Sitch, A.; Mistry, H.; Whitehouse, J.L. A prospective randomised controlled mixed-methods pilot study of home monitoring in adults with cystic fibrosis. Ther. Adv. Respir. Dis. 2022, 16, 17534666211070133. [Google Scholar] [CrossRef]
- Becheva, M.; Atanasov, P. Therapeutic guidelines for the treatment of cystic fibrosis. Pharmacia 2021, 68, 151–154. [Google Scholar] [CrossRef]
- Bhowmick, T.; Jagpal, S.; Hussain, S. Antimicrobial stewardship in the treatment of infections among patients with cystic fibrosis. Curr. Treat. Options Infect. Dis. 2018, 10, 263–269. [Google Scholar] [CrossRef]
- Ramstrom, H.; Erwander, I.; Mared, L.; Kornfalt, R.; Seiving, B. Pharmaceutical intervention in the care of cystic fibrosis patients. J. Clin. Pharm. Ther. 2000, 25, 427–434. [Google Scholar] [CrossRef]
- Elborn, J.S. Current approaches to the management of infection in cystic fibrosis. Curr. Pediatr. Rep. 2013, 1, 141–148. [Google Scholar] [CrossRef]
- Sreenivasulu, H.; Muppalla, S.K.; Vuppalapati, S.; Shokrolahi, M.; Reddy Pulliahgaru, A. Hope in Every Breath: Navigating the Therapeutic Landscape of Cystic Fibrosis. Cureus 2023, 15, e43603. [Google Scholar] [CrossRef]
- Filipow, N.; Stanojevic, S.; Raywood, E.; Shannon, H.; Tanriver, G.; Kapoor, K.; Douglas, H.; Davies, G.; O’Connor, R.; Murray, N. Real-world effectiveness of airway clearance techniques in children with cystic fibrosis. Eur. Respir. J. 2023, 62, 2300522. [Google Scholar] [CrossRef] [PubMed]
- Oliwko, E.; Babuśka-Roczniak, M.; Wojtanowska-Kaczka, M.; Lyubinets, O.; Roczniak, W. Physiotherapeutic recommendations for patients with cystic fibrosis. J. Educ. Health Sport 2020, 10, 96–103. [Google Scholar] [CrossRef]
- Barry, P.J.; Plant, B.J. Editorial: The changing landscape of cystic fibrosis: New therapies, challenges and a global pandemic. Curr. Opin. Pulm. Med. 2020, 26, 668–670. [Google Scholar] [CrossRef]
- Somaraju, U.R.R.; Solis-Moya, A. Pancreatic enzyme replacement therapy for people with cystic fibrosis. Cochrane Database Syst. Rev. 2020, 8, CD008227. [Google Scholar] [CrossRef]
- Lipuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef]
- Juniper, M. NEWS2, patient safety and hypercapnic respiratory failure. Clin. Med. 2022, 22, 518–521. [Google Scholar] [CrossRef]
- Sofianopoulos, S.; Williams, B. Pre-hospital management of cystic fibrosis patients presenting with haemoptysis. J. Paramed. Pract. 2013, 5, 139–145. [Google Scholar] [CrossRef]
- Grossheim, L.F. Emergency Department Care of Adult Cystic Fibrosis Patients. Emerg. Med. Rep. 2008, 29, 109–120. [Google Scholar]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarzynka, S.; Dobrosz, M.; Jaworski, S.; Jóźwicki, K.; Wierzba, S.; Barbarska, O.; Minkiewicz-Zochniak, A. Clinical Disorders in Cystic Fibrosis That Affect Emergency Procedures—A Case Report and Review. J. Clin. Med. 2025, 14, 3187. https://doi.org/10.3390/jcm14093187
Jarzynka S, Dobrosz M, Jaworski S, Jóźwicki K, Wierzba S, Barbarska O, Minkiewicz-Zochniak A. Clinical Disorders in Cystic Fibrosis That Affect Emergency Procedures—A Case Report and Review. Journal of Clinical Medicine. 2025; 14(9):3187. https://doi.org/10.3390/jcm14093187
Chicago/Turabian StyleJarzynka, Sylwia, Mateusz Dobrosz, Sebastian Jaworski, Kamil Jóźwicki, Sebastian Wierzba, Olga Barbarska, and Anna Minkiewicz-Zochniak. 2025. "Clinical Disorders in Cystic Fibrosis That Affect Emergency Procedures—A Case Report and Review" Journal of Clinical Medicine 14, no. 9: 3187. https://doi.org/10.3390/jcm14093187
APA StyleJarzynka, S., Dobrosz, M., Jaworski, S., Jóźwicki, K., Wierzba, S., Barbarska, O., & Minkiewicz-Zochniak, A. (2025). Clinical Disorders in Cystic Fibrosis That Affect Emergency Procedures—A Case Report and Review. Journal of Clinical Medicine, 14(9), 3187. https://doi.org/10.3390/jcm14093187