
Dumpling GNN: Hybrid GNN Enables Better ADC Payload Activity Prediction Based on the Chemical Structure


Abstract
:
1. Introduction
1.1. Research Background and Problem Statement
- Ability to learn complex, non-linear structure–activity relationships from large datasets [9].
- Capacity to handle high-dimensional feature spaces characteristic of molecular data [10].
- Potential for end-to-end learning, reducing the need for manual feature engineering [11]
- Scalability to screen large virtual libraries of compounds efficiently [12].
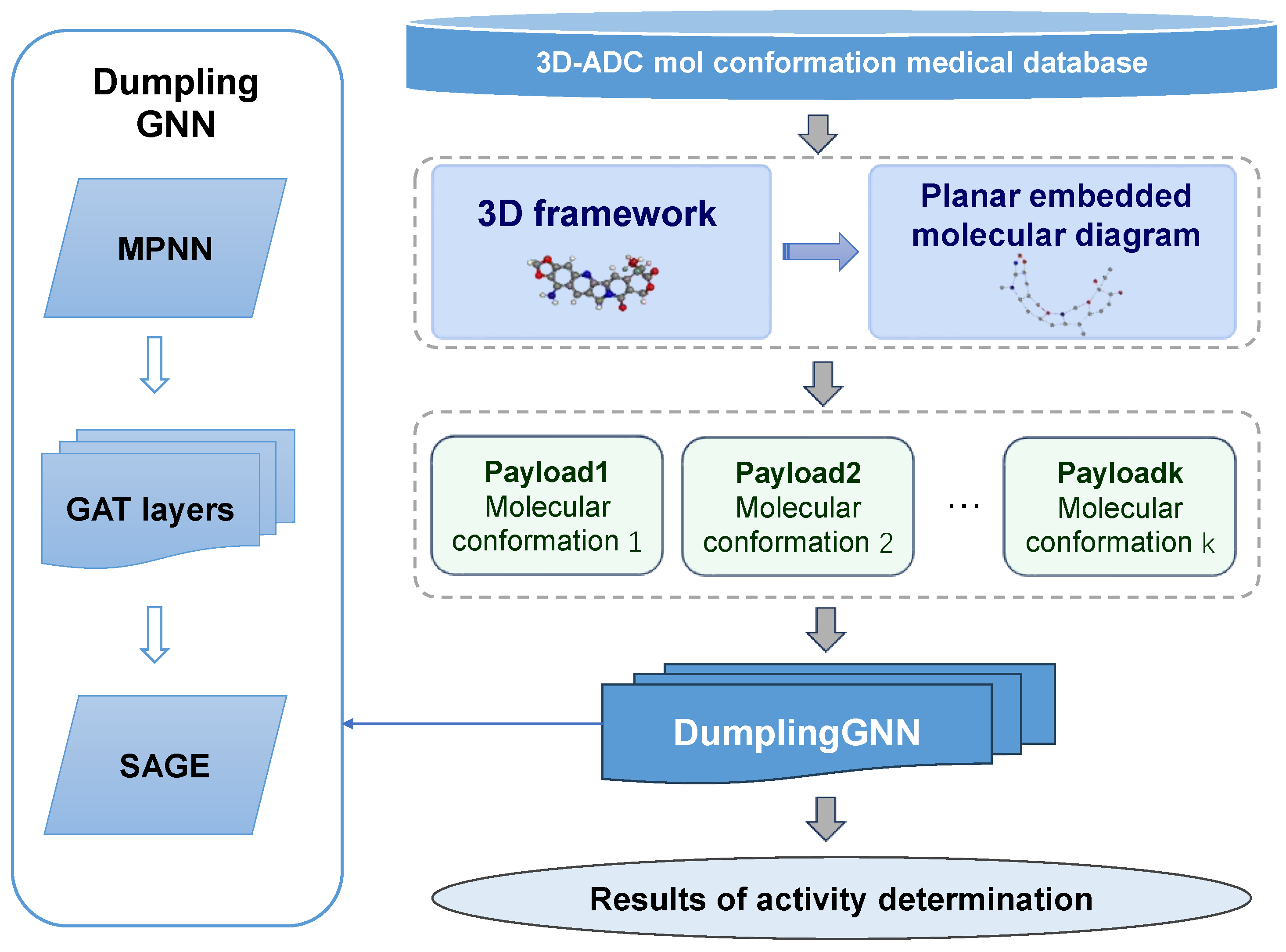
- A hybrid architecture combining Message Passing Neural Networks (MPNNs), Graph Attention Networks (GATs), and GraphSAGE layers, enabling comprehensive molecular feature extraction.
- An enhanced molecular graph construction algorithm incorporating both 2D topological and 3D structural information, providing a more nuanced representation of ADC payloads.
- A multi-task learning approach leveraging diverse molecular property prediction data to mitigate data scarcity issues in ADC payload datasets.
- An attention mechanism that enhances model interpretability, facilitating the identification of key substructures contributing to payload activity.
- A comprehensive interpretability framework that combines attention analysis with established cheminformatics techniques to provide biologically meaningful insights.
- A systematic approach to validating model predictions through their correlation with known pharmacophoric features and structure–activity relationships.
- We create a comprehensive ADC payload dataset, combining experimental data, high-quality computational predictions, and structures from recent patents. This dataset addresses the data scarcity issue and provides a valuable resource for the ADC research community.
- We develop an enhanced molecular graph construction algorithm that incorporates 3D structural information, improving the model’s ability to capture spatial features crucial for payload activity.
- We introduce DumplingGNN, an innovative hybrid GNN architecture tailored for ADC payload activity prediction. This model demonstrates state-of-the-art performance across multiple benchmarks, achieving remarkable results on datasets such as BBBP (96.4% ROC-AUC), ToxCast (78.2% ROC-AUC), and PCBA (88.87% ROC-AUC), surpassing existing methods on various molecular property prediction tasks.
- We conduct extensive evaluations on multiple datasets, including our newly created ADC payload dataset and several public benchmarks from MoleculeNet. These comprehensive evaluations demonstrate the versatility and robustness of DumplingGNN across diverse molecular property prediction tasks.
1.2. Related Work
1.2.1. Graph Neural Networks in Drug Discovery
1.2.2. ADC Payload Activity Prediction
1.2.3. Incorporation of 3D Structural Information
1.2.4. Interpretability in Molecular GNNs
2. Results
2.1. Datasets
2.2. Experimental Setup
2.3. Results and Comparison with State-of-the-Art Models
2.4. Performance on ADC Payload Dataset
2.5. Ablation Studies
2.5.1. Significance of 3D Structural Information
2.5.2. Hierarchical Architecture Analysis
MPNN Layer
GAT Layers
GraphSAGE Layer
2.6. Model Interpretability and Biological Insights
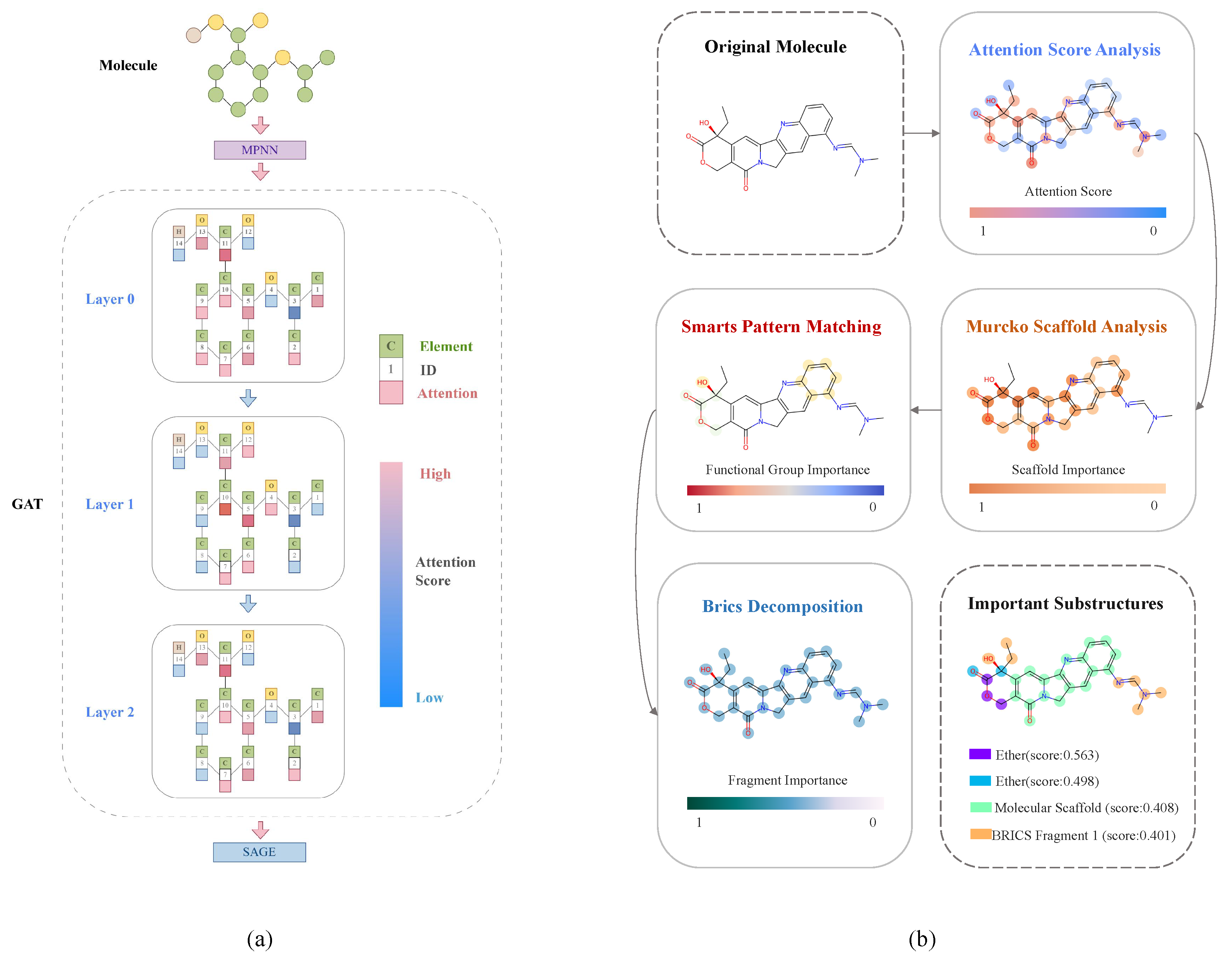
2.6.1. Attention Mechanism Analysis
- Layer 0: Shows broad feature detection with 6.61 effective attention heads and high diversity (), reflecting the initial exploration of the feature space.
- Layer 1: Demonstrates increased focus with 7.93 effective heads and stabilizing attention patterns (), indicating convergence on the relevant chemical patterns.
- Layer 2: Achieves refined feature selection with 7.97 effective heads and consistent attention distribution (), suggesting mature feature recognition.
2.6.2. Pharmacophore Recognition and Biological Validation
- Ester group (COC(=O)): As highlighted in Figure 1B(a), this moiety showed consistently high attention scores () with a remarkably low standard deviation. This group serves as a crucial linking element that stabilizes the positioning of Topoisomerase I inhibitors in the binding site, aligning with binding modes observed in the crystallographic studies of clinically approved DNA Topoisomerase I inhibitors.
- Hydroxyl group (C[OH]): Shown in Figure 1B(b), this group received equivalent attention scores for the ester group () and functions primarily as a hydrogen bond donor/acceptor, facilitating critical interactions with specific amino acid residues in the binding pocket of DNA Topoisomerase I.
- Five-membered ring system: Depicted in Figure 1B(c), this structural element was identified across all 177 analyzed molecules with a significant attention score (). Such a ring structure provides a rigid core scaffold that is often associated with planar aromatic systems for the enzyme inhibiting mechanism observed in DNA Topoisomerases. It provides the core scaffold essential for DNA intercalation and is a defining feature of camptothecin derivatives and other Topoisomerase I inhibitors currently used in clinical practice.
3. Discussion
3.1. Clinical and Drug Discovery Implications
- Structure–activity patterns: The consistent attention scores for key pharmacophoric elements ( for essential groups) provide quantitative guidance for structural optimization [24,52]. At the local scale, individual atom interactions and bond connectivity determine immediate activity, similar to how an enzyme recognizes functional groups. At a global scale, the overall molecular properties affect cellular uptake and distribution which aligns our molecular-level understanding with the phenotypes. The hierarchical approach adopted here not only improves prediction accuracy but also resonates with the way medicinal chemists think about structure–activity relationships.
- Safety-related features: The identification of potentially toxic substructures helps in the early risk assessment and modification of problematic molecular features.
- Design guidelines: The hierarchical attention patterns across layers (diversity from 0.057 to 0.007) suggest a systematic approach to molecular optimization. The integration of interpretability (via attention mechanism analysis) provides quantitative guidance on which molecular features are most critical for activity. This is valuable for the future drug design process, as it translates the abstract numbers of a machine learning model into tangible design principles, such as optimizing the positioning of ester or hydroxyl groups to enhance binding affinity and reduce toxicity. With the correlation of the observed attention patterns with known pharmacophoric requirements, our model validates its predictions through established biological principles, thus strengthening the case for its practical use in early drug discovery.
3.2. Limitations and Future Directions
- Attention pattern complexity: The evolution of attention scores through multiple layers (entropy from 0.248 to 0.260) can be challenging to interpret comprehensively.
- Validation challenges: While the model identifies known pharmacophoric elements, validating novel structural insights requires extensive experimental confirmation.
- Scale limitations: The current framework’s effectiveness in analyzing very large molecules or complex protein–ligand interactions needs further investigation.
4. Materials and Methods
4.1. Data: A Novel ADC Payload Dataset
4.2. Molecular Graph Construction
- –
- Atomic number: Indicates the type of element.
- –
- Degree: The number of bonds attached to the atom
- –
- Number of hydrogen atoms: Number of hydrogen atoms attached to the atom.
- –
- Implicit valence: The atomic valence level at which all chemical bonds are considered.
- –
- Aromaticity judgement: Whether the atom belongs to an aromatic ring or not.
- –
- Atomic coordinates: The position of the atom in three-dimensional space.
4.3. Network Architecture Design
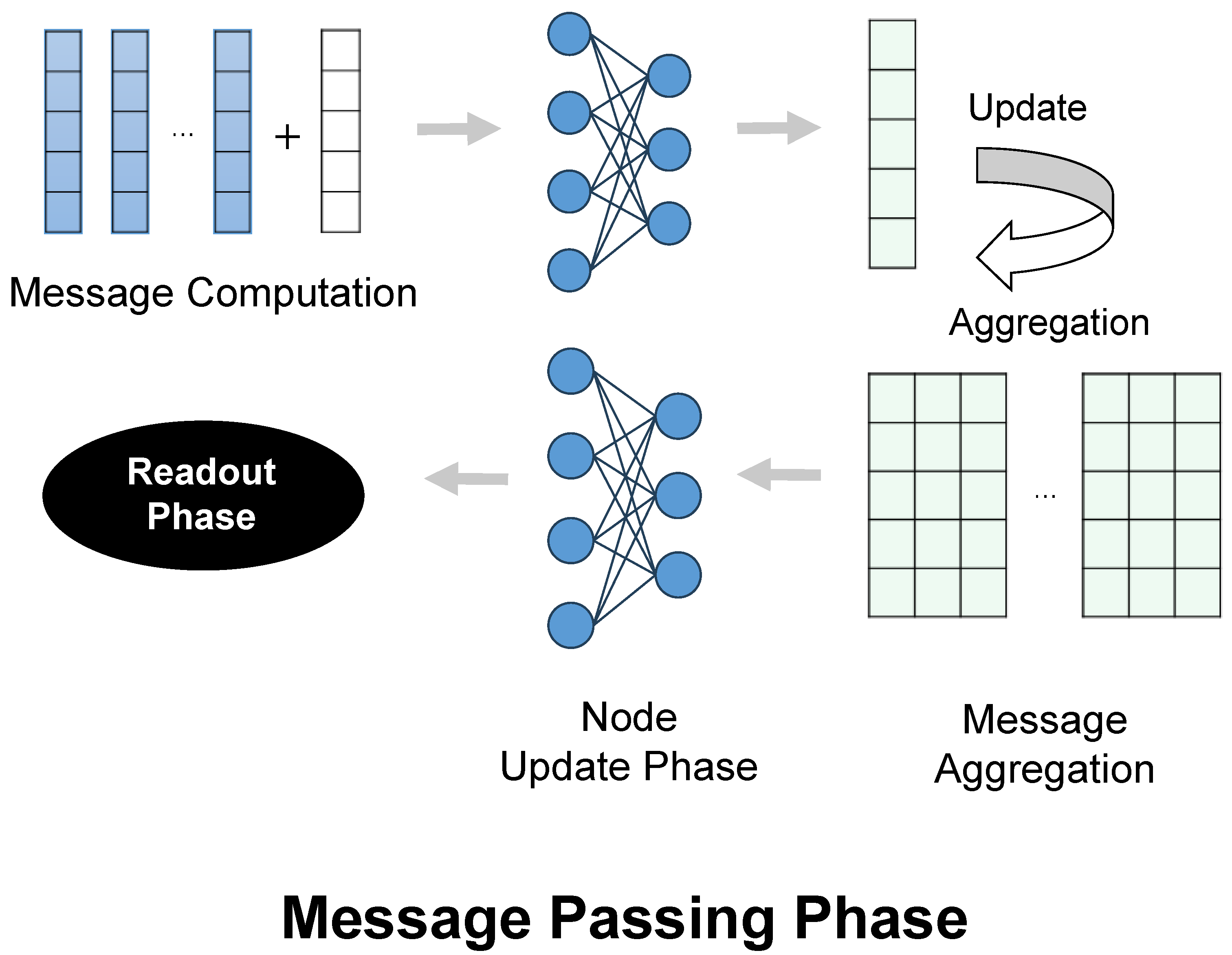
4.3.1. Message Passing Neural Network (MPNN) Layer
4.3.2. Graph Attention Network (GAT) Layers
- Diverse feature capture: Each head can specialize in different chemical patterns.
- Robust feature extraction: Multiple perspectives reduce the risk of missing important features.
- Enhanced stability: Averaging across heads provides more stable attention scores.
- Improved interpretability: Different heads can reveal various aspects of molecular recognition.
- First layer: Focuses on atomic-level features (mean attention = 0.246, std = 0.283).
- Second layer: Recognizes functional groups and local patterns (mean attention = 0.284, std = 0.412).
- Third layer: Captures global structural features (mean attention = 0.316, std = 0.430).
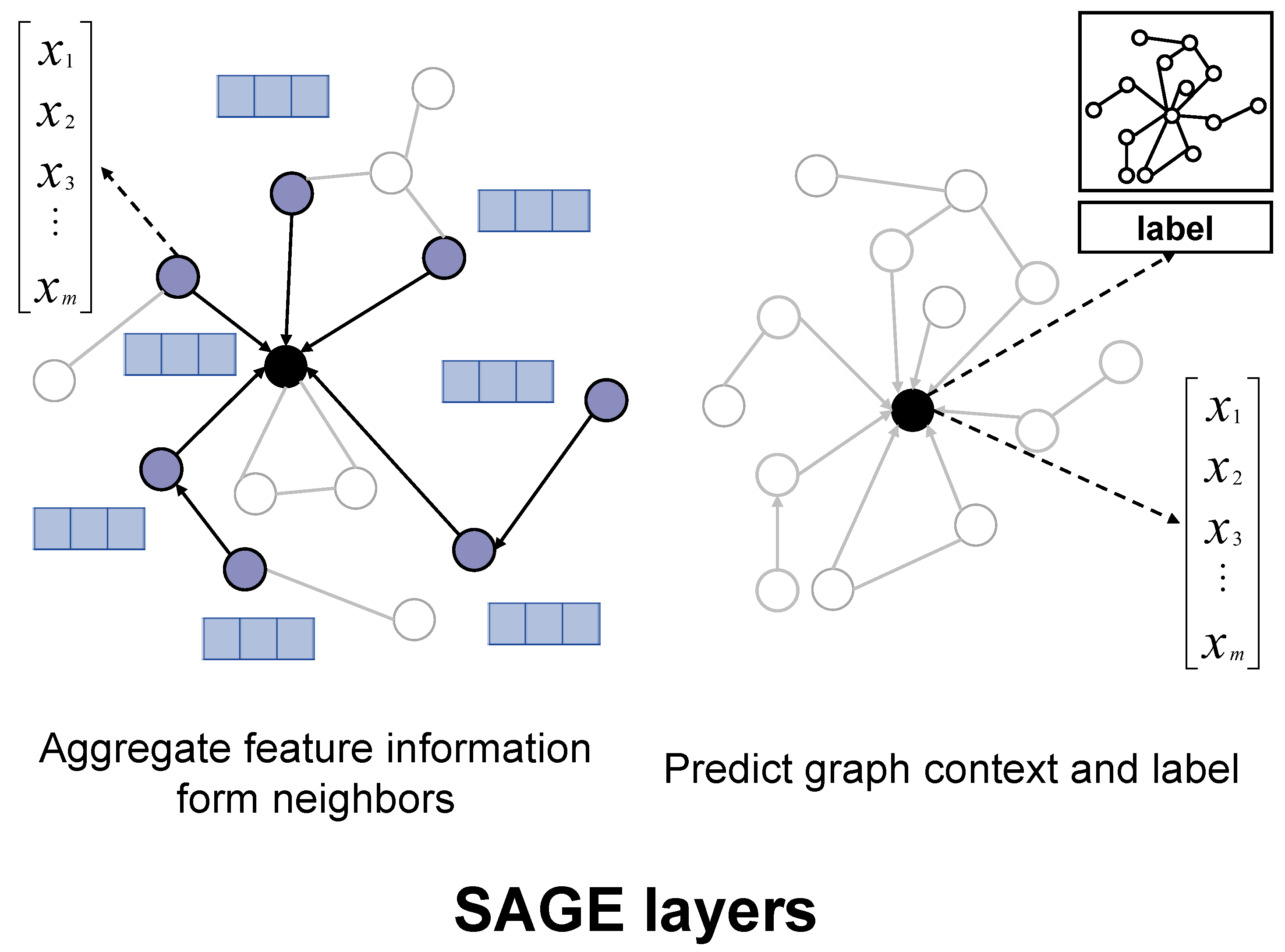
4.3.3. GraphSAGE Layer
4.4. Synergistic Effects and Biological Interpretability
- The MPNN layer captures local chemical interactions, modeling the reactivity and functional group behavior of the ADC payload.
- The GAT layers identify key substructures and atomic relationships, mimicking the concept of pharmacophores in drug discovery.
- The GraphSAGE layer aggregates information to model global molecular properties, which are crucial for predicting the payload’s behavior in biological systems.
- Layer-wise attention collection: Each layer’s attention weights are collected and normalized, providing insights into the model’s focus at different abstraction levels.
- Multi-scale feature analysis: Attention patterns reveal the hierarchical recognition of molecular features, from atomic properties to global structural characteristics.
- Temporal evolution tracking: The progression of attention scores through layers demonstrates how the model builds its understanding of molecular properties.
- Attention score analysis: Identifies atoms and bonds crucial for activity prediction, highlighting potential pharmacophoric elements.
- SMARTS pattern matching [61]: Recognizes functional groups and their relative importance based on attention scores.
- Murcko scaffold analysis [62]: Evaluates the contribution of core molecular frameworks to predicted activity.
- BRICS decomposition [63]: Identifies key fragments and their hierarchical relationships in molecular recognition.
- Structure–activity relationships: Understanding which molecular features contribute most significantly to the activity.
- Design guidelines: Identifying preferred scaffolds and functional groups for optimization.
- Mechanism insights: Revealing potential binding modes and interaction patterns.
- Safety assessment: Highlighting structural features that might contribute to toxicity.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | Antibody–Drug Conjugate |
| GNN | Graph Neural Network |
| MPNN | Message Passing Neural Network |
| GAT | Graph Attention Network |
| ROC-AUC | Receiver Operating Characteristic-Area Under Curve |
| BBBP | Blood–Brain Barrier Penetration |
| PCBA | PubChem Bioassay |
References
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaía, N. Next-generation antibody–drug conjugates for cancer therapy. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 195–209. [Google Scholar] [CrossRef]
- Peters, I.F.; Senter, P.D. Advances in antibody-drug conjugate drug development. Expert Opin. Drug Deliv. 2018, 15, 775–788. [Google Scholar]
- Lambert, J.M. Antibody-drug conjugates: Design and selection of/linker-payload. Cancer Chemother. Pharmacol. 2017, 79, 1019–1028. [Google Scholar]
- Zhao, X.D.; Li, J.Q.; Wang, Y.X.; Wang, L.X.; Li, J.; Zhang, X. Anti-cancer payload development: Current status and future perspectives. J. Control. Release 2020, 321, 138–149. [Google Scholar]
- Carter, P.J.; Senter, P.D. Site-specific antibody-drug conjugates: The next generation. Drug Discov. Today 2018, 23, 1534–1542. [Google Scholar]
- Schneider, G.; Fechner, N.; Fischer, B.; Neumann, M. Rethinking molecular similarity: Towards a machine learning approach. J. Chem. Inf. Model. 2020, 60, 5155–5165. [Google Scholar]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Yang, K.; Li, Y.; Zhou, J.; Tu, Z.; Chen, L.; Cui, P.; Wang, S. Analyzed and Improved Graph Neural Networks for Molecular Property Prediction. arXiv 2019, arXiv:1903.01307. [Google Scholar]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A Benchmark for Molecular Machine Learning. Chem. Sci. 2020, 9, 513–530. [Google Scholar] [CrossRef]
- Duvenaud, D.K.; Maclaurin, D.; Iparraguirre, J.; Bombarell, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional networks on graphs for learning molecular fingerprints. Adv. Neural Inf. Process. Syst. 2015, 28, 2224–2232. [Google Scholar]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Philip, S.Y. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2020, 32, 4–24. [Google Scholar] [CrossRef]
- Goh, G.; Li, Y.; Tan, J.; Ong, C.; Tan, A.; Sung, W.; Tan, P. Machine Learning with Tensor Flow in Medicine and Biology. arXiv 2017, arXiv:1706.05274. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph neural networks: A review of methods and applications. AI Open 2020, 1, 57–81. [Google Scholar] [CrossRef]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural message passing for quantum chemistry. In Proceedings of the International Conference on Machine Learning, Sydney, NSW, Australia, 6–11 August 2017; PMLR: Breckenridge, CO, USA, 2017; pp. 1263–1272. [Google Scholar]
- Ying, R.; Bourgeois, D.; You, J.; Zitnik, M.; Leskovec, J. GNNExplainer: Generating Explanations for Graph Neural Networks. arXiv 2019, arXiv:1903.03894. [Google Scholar]
- Xu, K.; Hu, W.; Leskovec, J.; Jegelka, S. How powerful are graph neural networks? arXiv 2018, arXiv:1810.00826. [Google Scholar]
- Klicpera, J.; Gross, J. Directional Message Passing on Molecular Graphs via Synthetic Coordinates. arXiv 2021, arXiv:2111.04718. [Google Scholar]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J.Comput.-Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef]
- Velickovic, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. Stat 2017, 1050, 10–48550. [Google Scholar]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 2019, 63, 8749–8760. [Google Scholar] [CrossRef]
- Liu, J.; Gao, Z.; Liu, H.; Shen, W. Graph convolutional networks meet graph recurrent neural networks for molecular property prediction. J. Chem. Inf. Model. 2021, 61, 1695–1704. [Google Scholar]
- Rong, Z.H.; Zhang, Q.; Xu, J.; Zhu, X.M.; Li, H. Self-supervised Graph Structure Learning for Molecular Property Prediction. J. Chem. Inf. Model. 2020, 60, 3500–3509. [Google Scholar]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload diversification: A key step in the development of antibody–drug conjugates. J. Hematol. Oncol. 2023, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Shi, L.; Hu, Z.; Gao, T.; Liu, J.; Lv, X.; Liu, H.; Tang, H.; Liu, B.; Xu, L.; et al. Quantitative structure-activity relationship (QSAR) modeling of chemical mutagens and carcinogens by machine learning methods: A review. Chem. Res. Toxicol. 2018, 31, 585–603. [Google Scholar]
- Chen, L.; Li, B.; Chen, Y.; Lin, M.; Zhang, S.; Li, C.; Pang, Y.; Wang, L. ADCNet: A unified framework for predicting the activity of antibody-drug conjugates. arXiv 2024, arXiv:2401.09176. [Google Scholar]
- Schütt, K.T.; Arbabzadah, F.; Chmiela, S.; Müller, K.R.; Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 13890. [Google Scholar] [CrossRef]
- Townshend, R.J.L.; Vögele, M.; Suriana, P.; Derry, A.; Powers, A.; Laloudakis, Y.; Bedi, R.; Rangarajan, S.; Groban, E.; Mallet, V.; et al. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. ACS Cent. Sci. 2021, 7, 784–801. [Google Scholar] [CrossRef]
- Jing, B.; Eismann, S.; Soni, P.N.; Dror, R.O. Equivariant graph neural networks for 3D macroeconomic structure. arXiv 2021, arXiv:2106.03843v. [Google Scholar]
- Pope, P.E.; Kolouri, S.; Rostami, M.; Martin, C.E.; Hoffmann, H. Explainability methods for graph convolutional neural networks. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 15–20 June 2019; pp. 10772–10781. [Google Scholar]
- Schnake, T.; Eberle, O.; Lederer, J.; Nakajima, S.; Schütt, K.T.; Müller, K.R.; Montavon, G. Higher-order explanations of graph neural networks via relevant walks. IEEE Trans. Pattern Anal. Mach. Intell. 2021, 44, 9248–9261. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Demirel, M.F.; Liang, Y. N-Gram Graph: Simple Unsupervised Representation for Graphs, with Applications to Molecules. arXiv 2019, arXiv:1806.09206. [Google Scholar]
- Rong, Y.; Bian, Y.; Xu, T.; Xie, W.; Wei, Y.; Huang, W.; Huang, J. Self-supervised graph transformer on large-scale molecular data. In Proceedings of the 34th International Conference on Neural Information Processing Systems, NIPS ’20, Red Hook, NY, USA, 6–12 December 2020. [Google Scholar]
- Liu, S.; Wang, H.; Liu, W.; Lasenby, J.; Guo, H.; Tang, J. Pre-training Molecular Graph Representation with 3D Geometry. In Proceedings of the International Conference on Learning Representations, online, 25 April 2022. [Google Scholar]
- Liu, Z.; Zhang, W.; Xia, Y.; Wu, L.; Xie, S.; Qin, T.; Zhang, M.; Liu, T.Y. MolXPT: Wrapping Molecules with Text for Generative Pre-training. arXiv 2023, arXiv:2305.10688. [Google Scholar]
- Tao, N.; Abe, M. Bayesian Flow Network Framework for Chemistry Tasks. J. Chem. Inf. Model. 2025, 65, 1178–1187. [Google Scholar] [CrossRef]
- Li, J.; Cai, D.; He, X. Learning Graph-Level Representation for Drug Discovery. arXiv 2017, arXiv:1709.03741. [Google Scholar]
- Hu, W.; Liu, B.; Gomes, J.; Zitnik, M.; Liang, P.; Pande, V.; Leskovec, J. Strategies for Pre-training Graph Neural Networks. In Proceedings of the International Conference on Learning Representations, Addis Ababa, Ethiopia, 26–30 April 2020. [Google Scholar]
- Wang, Y.; Wang, J.; Cao, Z.; Barati Farimani, A. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 2022, 4, 279–287. [Google Scholar] [CrossRef]
- Fang, X.; Liu, L.; Lei, J.; He, D.; Zhang, S.; Zhou, J.; Wang, F.; Wu, H.; Wang, H. Geometry-enhanced molecular representation learning for property prediction. Nat. Mach. Intell. 2022, 4, 127–134. [Google Scholar] [CrossRef]
- Zhou, G.; Gao, Z.; Ding, Q.; Zheng, H.; Xu, H.; Wei, Z.; Zhang, L.; Ke, G. Uni-Mol: A Universal 3D Molecular Representation Learning Framework. In Proceedings of the The Eleventh International Conference on Learning Representations, Kigali, Rwanda, 1–5 May 2023. [Google Scholar]
- Paykan Heyrati, M.; Ghorbanali, Z.; Akbari, M.; Pishgahi, G.; Zare-Mirakabad, F. BioAct-Het: A Heterogeneous Siamese Neural Network for Bioactivity Prediction Using Novel Bioactivity Representation. ACS Omega 2023, 8, 44757–44772. [Google Scholar] [CrossRef]
- Fang, W.; Xu, M.; Xu, H. Geometry-Enhanced Molecular Representations Improve Graph Neural Networks for Molecular Property Prediction. J. Chem. Inf. Model. 2021, 61, 5077–5090. [Google Scholar] [CrossRef]
- Wang, X.; Girshick, R.; Gupta, A.; He, K. Attention Mechanisms in Deep Learning: A Comprehensive Survey. IEEE Trans. Pattern Anal. Mach. Intell. 2023, 45, 3–35. [Google Scholar]
- Chen, C.; Wang, S.; Xu, Y.; Zhang, W. Interpreting Attention Mechanisms in Graph Neural Networks for Molecular Property Prediction. J. Chem. Inf. Model. 2023, 63, 2234–2245. [Google Scholar]
- Li, Y.; Zhang, H.; Wang, J. Statistical Analysis of Attention Patterns in Deep Learning Models. Nat. Mach. Intell. 2023, 5, 456–468. [Google Scholar]
- Fout, A.R.; Byrd, J.N.; Smith, W.H.; Yang, Y.; Tan, C.; Li, B.; Pearlman, D.A.; Hanson, R.M.; Lin, S.; Cao, Y.; et al. Protein-ligand binding affinity prediction using an ensemble docking approach. PLoS Comput. Biol. 2017, 13, e1005409. [Google Scholar]
- Jiang, D.; Wu, Z.; Hsieh, C.Y.; Chen, G.; Liao, B.; Wang, Z.; Shen, C.; Cao, D.; Wu, J.; Hou, T. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminform. 2021, 13, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Wang, S.; Xu, Y.; Zhang, W.; Ouyang, D.; Gao, J. Graph neural networks for antibody-drug conjugate property prediction. J. Chem. Inf. Model. 2023, 63, 1276–1286. [Google Scholar]
- Watanabe, T.; Arashida, N.; Fujii, T.; Shikida, N.; Ito, K.; Shimbo, K.; Seki, T.; Iwai, Y.; Hirama, R.; Hatada, N.; et al. Exo-Cleavable Linkers: Enhanced Stability and Therapeutic Efficacy in Antibody–Drug Conjugates. J. Med. Chem. 2024, 67, 18124–18138. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Hutchison, G.R. Fast, efficient fragment-based coordinate generation for Open Babel. J. Cheminform. 2019, 11, 49. [Google Scholar] [CrossRef]
- Tosco, P.; Stiefl, N.; Landrum, G. Bringing the MMFF force field to the RDKit: Implementation and validation. J. Cheminform. 2014, 6, 37. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, K.; Wu, Q.; Huang, S.Y. NLDock: A Fast Nucleic Acid–Ligand Docking Algorithm for Modeling RNA/DNA–Ligand Complexes. J. Chem. Inf. Model. 2021, 61, 4771–4782. [Google Scholar] [CrossRef]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W.; Ying, Z.; Leskovec, J. Inductive representation learning on large graphs. Adv. Neural Inf. Process. Syst. 2017, 30, 1024–1034. [Google Scholar]
- Vaswani, A. Attention is all you need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Zhang, W.; Liu, Y.; Li, H.; Yang, J. Hierarchical Attention Networks for Molecular Property Prediction: From Atoms to Molecules. J. Chem. Theory Comput. 2023, 19, 4123–4135. [Google Scholar]
- DCI Systems. SMARTS—A Language for Describing Molecular Patterns; Technical Report; Daylight Chemical Information Systems Inc.: Laguna Niguel, CA, USA, 1997. [Google Scholar]
- Bemis, G.W.; Murcko, M.A. Properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Degen, J.; Wegscheid-Gerlach, C.; Bunke, A.; Hindle, S.; Guba, W.; Landrum, G. The art of splitting sets: BRICS decomposition and fragment recombination for 3D fragment-based lead generation. ChemMedChem 2008, 3, 1503–1507. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Molecules | Tasks | Description |
|---|---|---|---|
| BBBP | 2039 | 1 | Blood–Brain Barrier Penetration |
| BACE | 1513 | 1 | Inhibition of Beta-Secretase 1 |
| ClinTox | 1478 | 2 | Clinical Trial Toxicity |
| Tox21 | 7831 | 12 | Toxicology in the 21st Century |
| ToxCast | 8575 | 617 | EPA Toxicity Forecaster |
| SIDER | 1427 | 27 | Drug Side Effect Resource |
| HIV | 41,127 | 1 | HIV Replication Inhibition |
| PCBA | 437,929 | 128 | PubChem Bioassay Data |
| Model Category | Model | BBBP | BACE | ClinTox | Tox21 | ToxCast | SIDER | HIV | PCBA |
|---|---|---|---|---|---|---|---|---|---|
| Proposed Model | DumplingGNN | 96.4 (0.7) | 88.2 (0.5) | 95.9 (2.0) | 82.3 (0.4) | 78.2 (0.1) | 74.0 (0.6) | 79.4 (0.2) | 88.87 (0.2) |
| Single-Architecture GNNs | D-MPNN [34] | 71.0 (0.3) | 80.9 (0.6) | 90.6 (0.6) | 75.9 (0.7) | 65.5 (0.3) | 57.0 (0.7) | 77.1 (0.5) | 86.2 (0.1) |
| Attentive FP [23] | 85.5 | 78.4 (0.022) | 84.7 (0.3) | 76.1 (0.5) | 63.7 (0.2) | 60.6 (3.2) | 75.7 (1.4) | 80.1 (1.4) | |
| GraphConv + dummy super node [40] | – | – | – | 85.4 | 76.8 | – | 85.1 | 86.7 | |
| Traditional ML | N-Gram RF [35] | 69.7 (0.6) | 77.9 (1.5) | 77.5 (4.0) | 74.3 (0.4) | – | 66.8 (0.7) | 77.2 (0.1) | – |
| N-Gram xGB [35] | 69.1 (0.8) | 79.1 (1.3) | 87.5 (2.7) | 75.8 (0.9) | – | 65.5 (0.7) | 78.7 (0.4) | – | |
| Pre-trained Models | PretrainGNN [41] | 68.7 (1.3) | 84.5 (0.7) | 72.6 (1.5) | 78.1 (0.6) | 65.7 (0.6) | 62.7 (0.8) | 79.9 (0.7) | 86.0 (0.1) |
| GROVERlarge [36] | 69.5 (0.1) | 81.0 (1.4) | 76.2 (3.7) | 73.5 (0.1) | 65.3 (0.5) | 65.4 (0.1) | 68.2 (1.1) | 83.0 (0.4) | |
| GraphMVP [37] | 72.4 (1.6) | 81.2 (0.9) | 79.1 (2.8) | 75.9 (0.5) | 63.1 (0.4) | 63.9 (1.2) | 77.0 (1.2) | – | |
| MolCLR [42] | 72.2 (2.1) | 82.4 (0.9) | 91.2 (3.5) | 75.0 (0.2) | – | 58.9 (1.4) | 78.1 (0.5) | – | |
| GEM [43] | 72.4 (0.4) | 85.6 (1.1) | 90.1 (1.3) | 78.1 (0.1) | 69.2 (0.4) | 67.2 (0.4) | 80.6 (0.9) | 86.6 (0.1) | |
| Uni-Mol [44] | 72.9 (0.6) | 85.7 (0.2) | 91.9 (1.8) | 79.6 (0.5) | 69.6 (0.1) | 65.9 (1.3) | 80.8 (0.3) | 88.5 (0.1) | |
| Advanced Approaches | MolXPT [38] | 80.5 (0.5) | 88.4 | 95.3 (0.2) | 77.1 | – | 71.7 | 78.1 | – |
| ChemBFN [39] | 95.74 | 73.56 | 99.18 | – | – | – | 79.37 | – | |
| Previous SOTA | (Best reported results) | 95.74 [39] | 88.4 [38] | 99.18 [39] | 89.9 [45] | 77.7 [44] | 91.1 [45] | 80.8 [35] | 88.5 [44] |
| Architecture Type | Model | Accuracy | Sensitivity | Specificity | MCC | AUC-ROC | F1 Score | Balanced Accuracy | AUC-PR |
|---|---|---|---|---|---|---|---|---|---|
| Hybrid Architecture | DumplingGNN | 0.9148 | 0.9508 | 0.9754 | 0.8287 | 0.9547 | 0.9243 | 0.9111 | 0.9531 |
| Single-Architecture GNNs | FiveLayerMPNN | 0.8655 | 0.9262 | 0.7921 | 0.7301 | 0.9281 | 0.8828 | 0.8592 | 0.9342 |
| FiveLayerGAT | 0.8565 | 0.9180 | 0.7822 | 0.7117 | 0.8741 | 0.8750 | 0.8501 | 0.8653 | |
| FiveLayerSAGE | 0.7982 | 0.8525 | 0.7327 | 0.5917 | 0.8509 | 0.8221 | 0.7926 | 0.8668 | |
| FiveLayerGCN | 0.7623 | 0.8525 | 0.6535 | 0.5197 | 0.8301 | 0.7969 | 0.7530 | 0.8589 |
| Variant Category | Model Variant | Accuracy | Sensitivity | Specificity | MCC | AUC-ROC | F1 Score | Balanced Accuracy | AUC-PR |
|---|---|---|---|---|---|---|---|---|---|
| Complete Architecture | Full DumplingGNN | 0.915 | 0.951 | 0.975 | 0.829 | 0.955 | 0.924 | 0.911 | 0.953 |
| Input Representation | SMILES-Only | 0.734 | 0.768 | 0.949 | 0.461 | 0.782 | 0.775 | 0.721 | 0.786 |
| Component Ablation | No GraphSAGE | 0.870 | 0.902 | 0.832 | 0.737 | 0.940 | 0.884 | 0.867 | 0.939 |
| No GAT | 0.803 | 0.869 | 0.723 | 0.601 | 0.861 | 0.828 | 0.796 | 0.882 | |
| No MPNN | 0.812 | 0.869 | 0.743 | 0.619 | 0.878 | 0.835 | 0.806 | 0.840 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Xie, L.; Dai, R.; Lyu, Z. Dumpling GNN: Hybrid GNN Enables Better ADC Payload Activity Prediction Based on the Chemical Structure. Int. J. Mol. Sci. 2025, 26, 4859. https://doi.org/10.3390/ijms26104859
Xu S, Xie L, Dai R, Lyu Z. Dumpling GNN: Hybrid GNN Enables Better ADC Payload Activity Prediction Based on the Chemical Structure. International Journal of Molecular Sciences. 2025; 26(10):4859. https://doi.org/10.3390/ijms26104859
Chicago/Turabian StyleXu, Shengjie, Lingxi Xie, Rujie Dai, and Zehua Lyu. 2025. "Dumpling GNN: Hybrid GNN Enables Better ADC Payload Activity Prediction Based on the Chemical Structure" International Journal of Molecular Sciences 26, no. 10: 4859. https://doi.org/10.3390/ijms26104859
APA StyleXu, S., Xie, L., Dai, R., & Lyu, Z. (2025). Dumpling GNN: Hybrid GNN Enables Better ADC Payload Activity Prediction Based on the Chemical Structure. International Journal of Molecular Sciences, 26(10), 4859. https://doi.org/10.3390/ijms26104859