
idopNetwork Analysis of Salt-Responsive Transcriptomes Reveals Hub Regulatory Modules and Genes in Populus euphratica


Abstract
1. Introduction
2. Results
2.1. Transcriptome Sequencing and Assembly
2.2. Analysis of Temporal DEGs
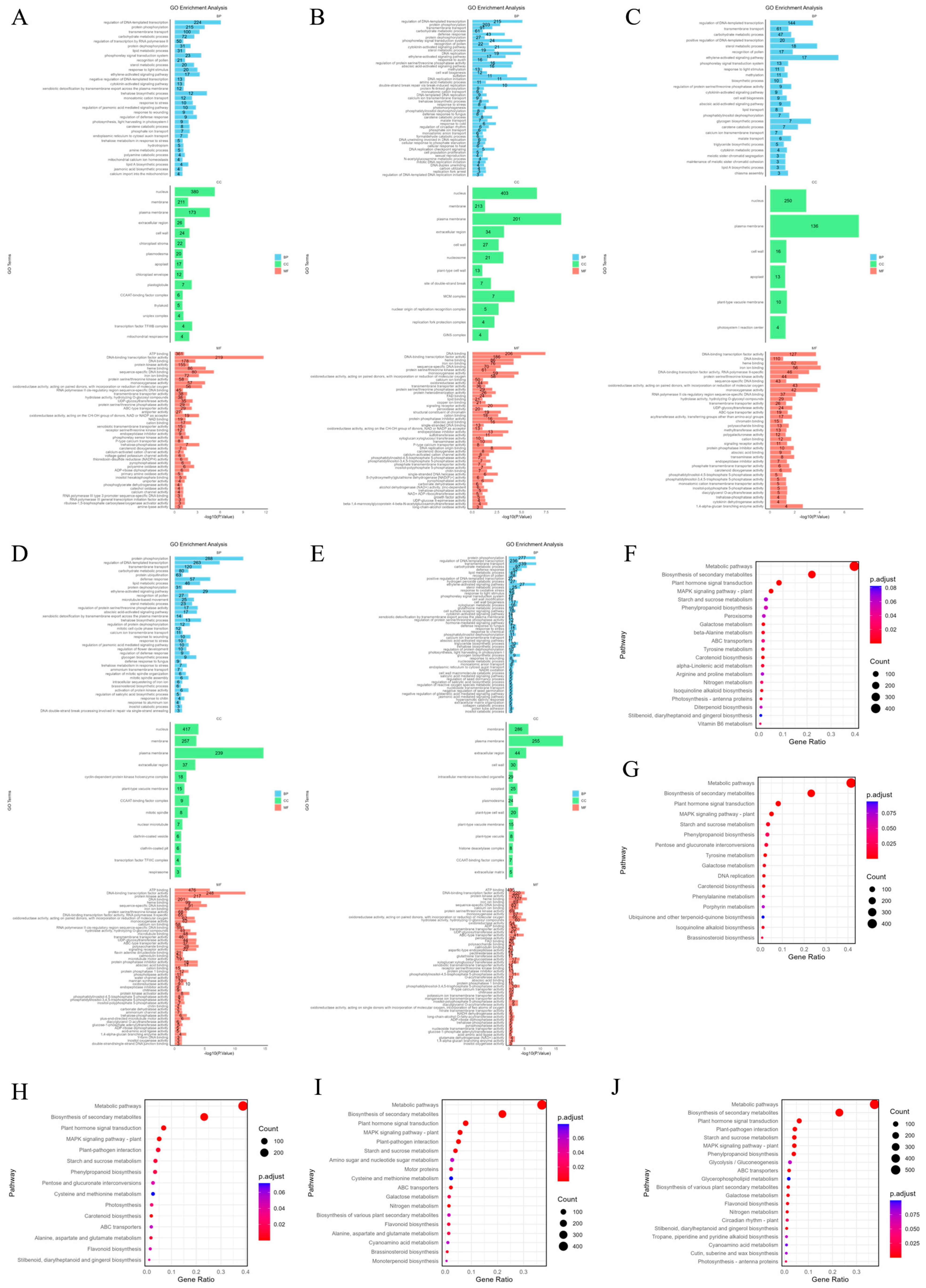
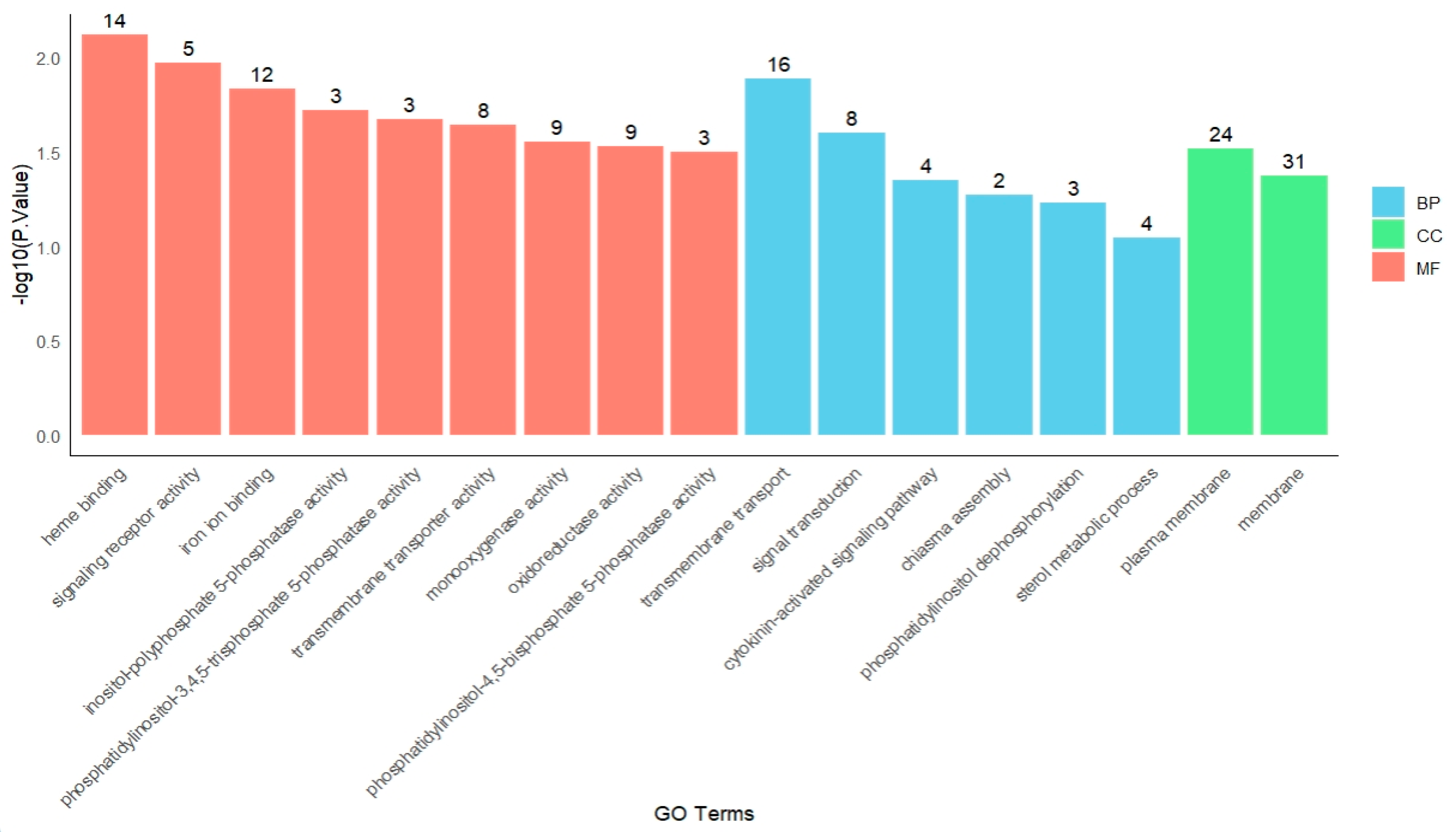
2.3. Gene Ontology (GO) Enrichment and KEGG Analysis of DEGs
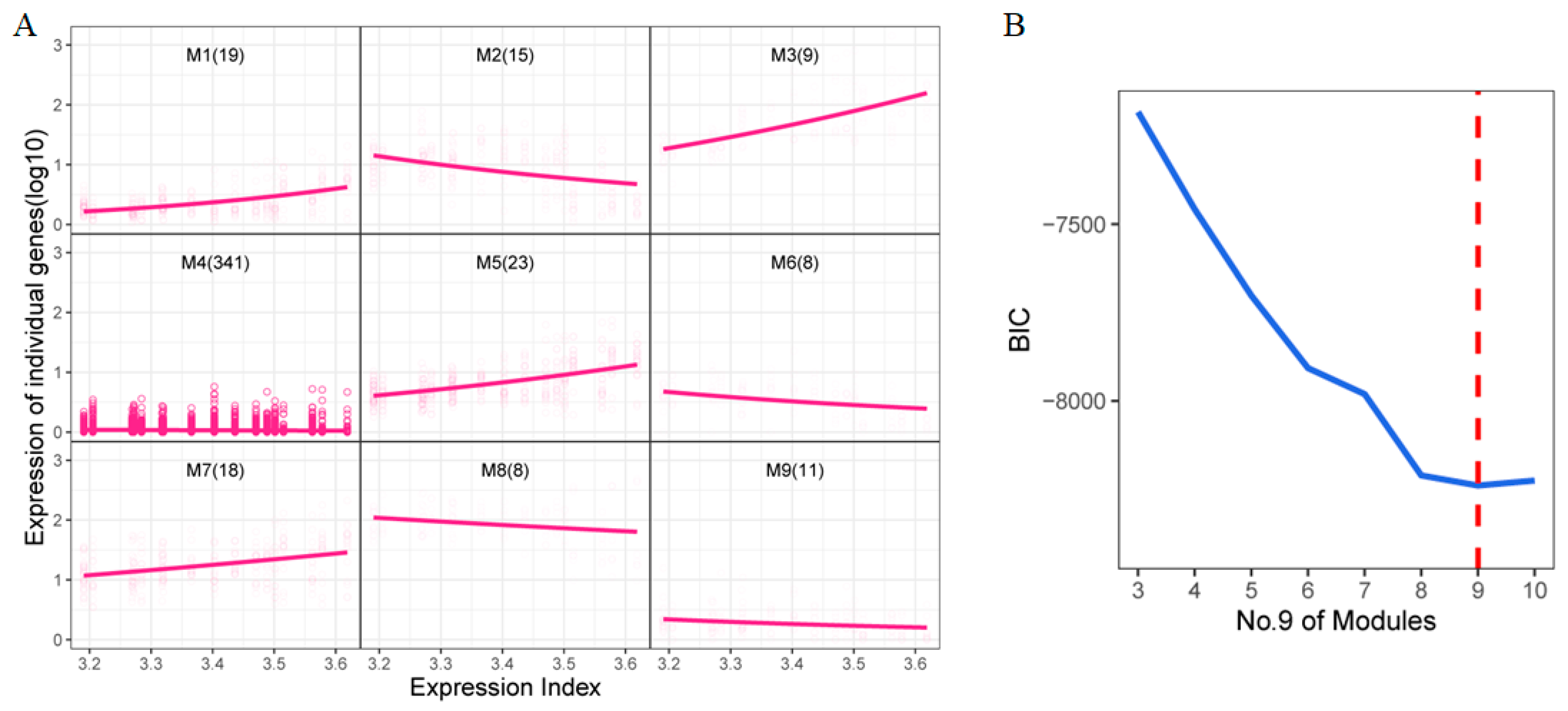
2.4. Functional Cluster of Temporal DEGs
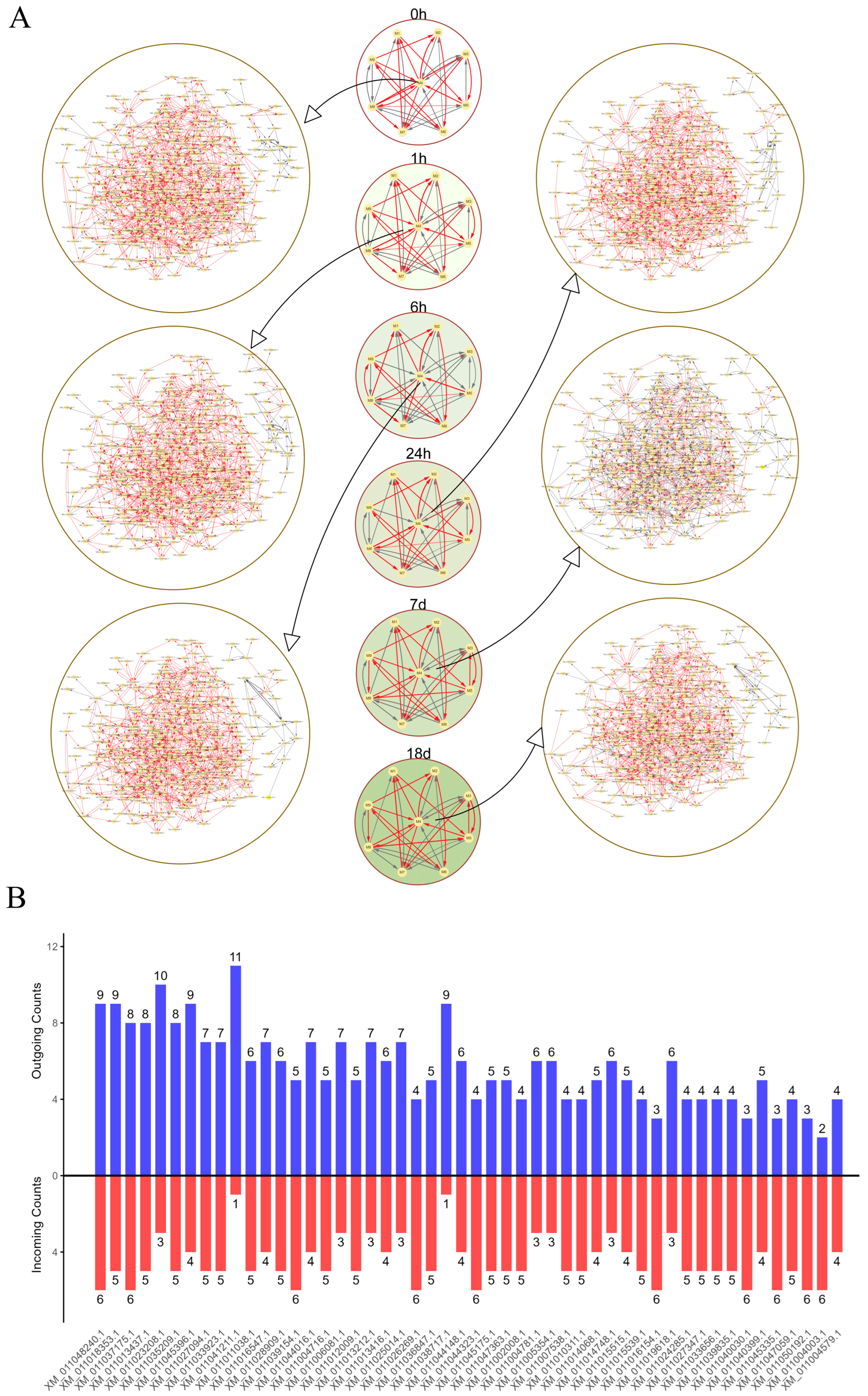
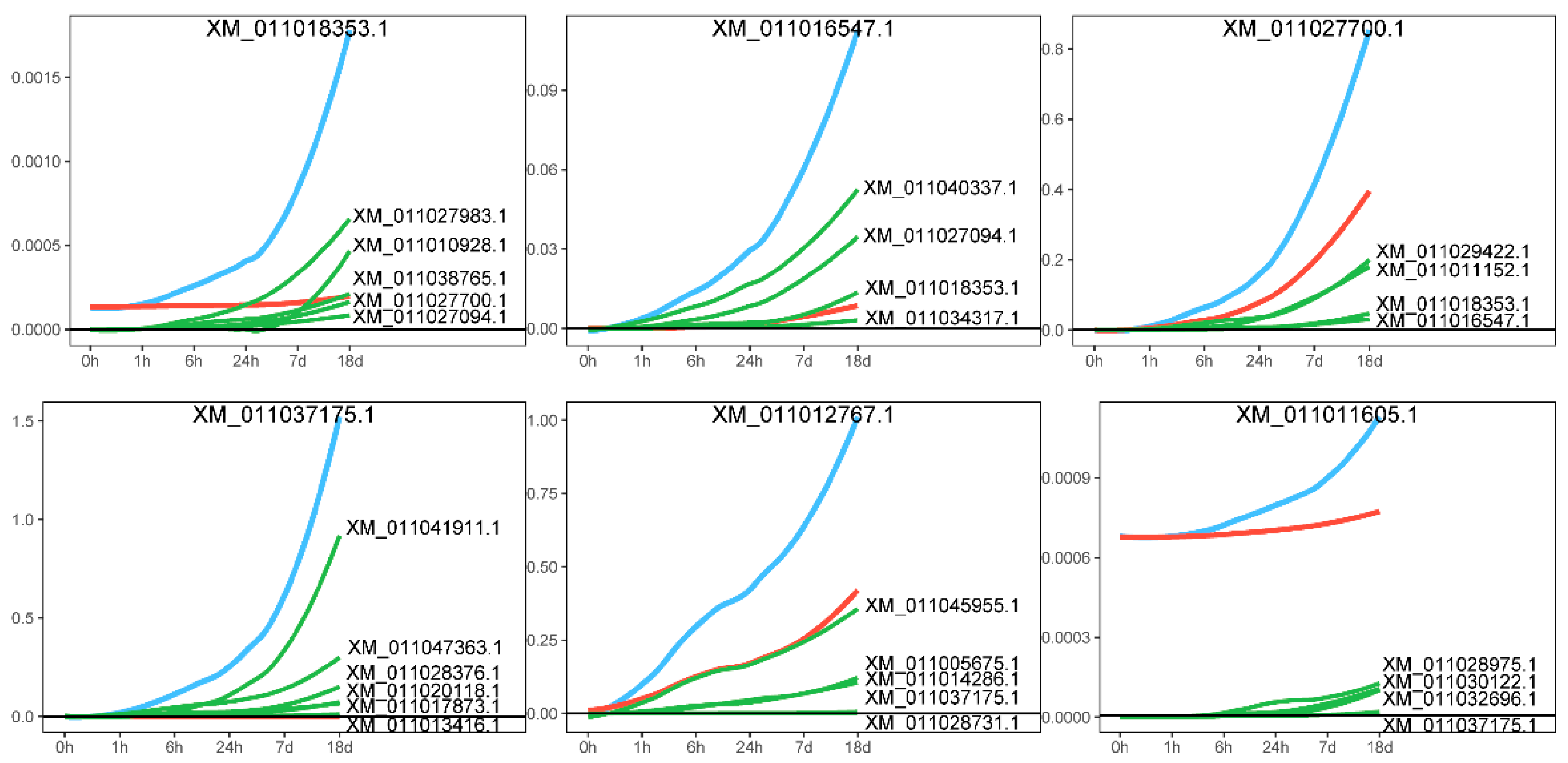
2.5. idopNetwork Reconstruction for DEGs
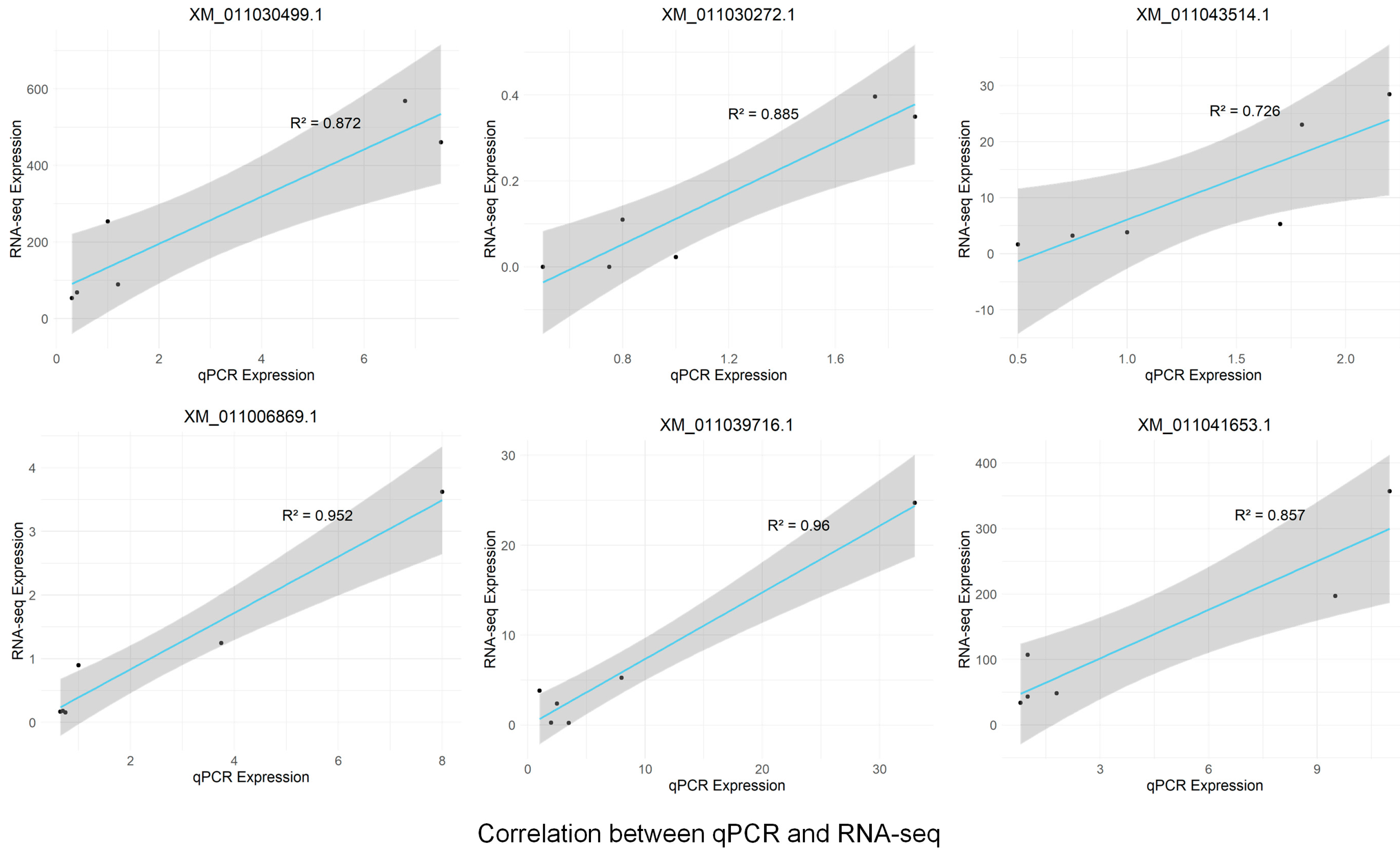
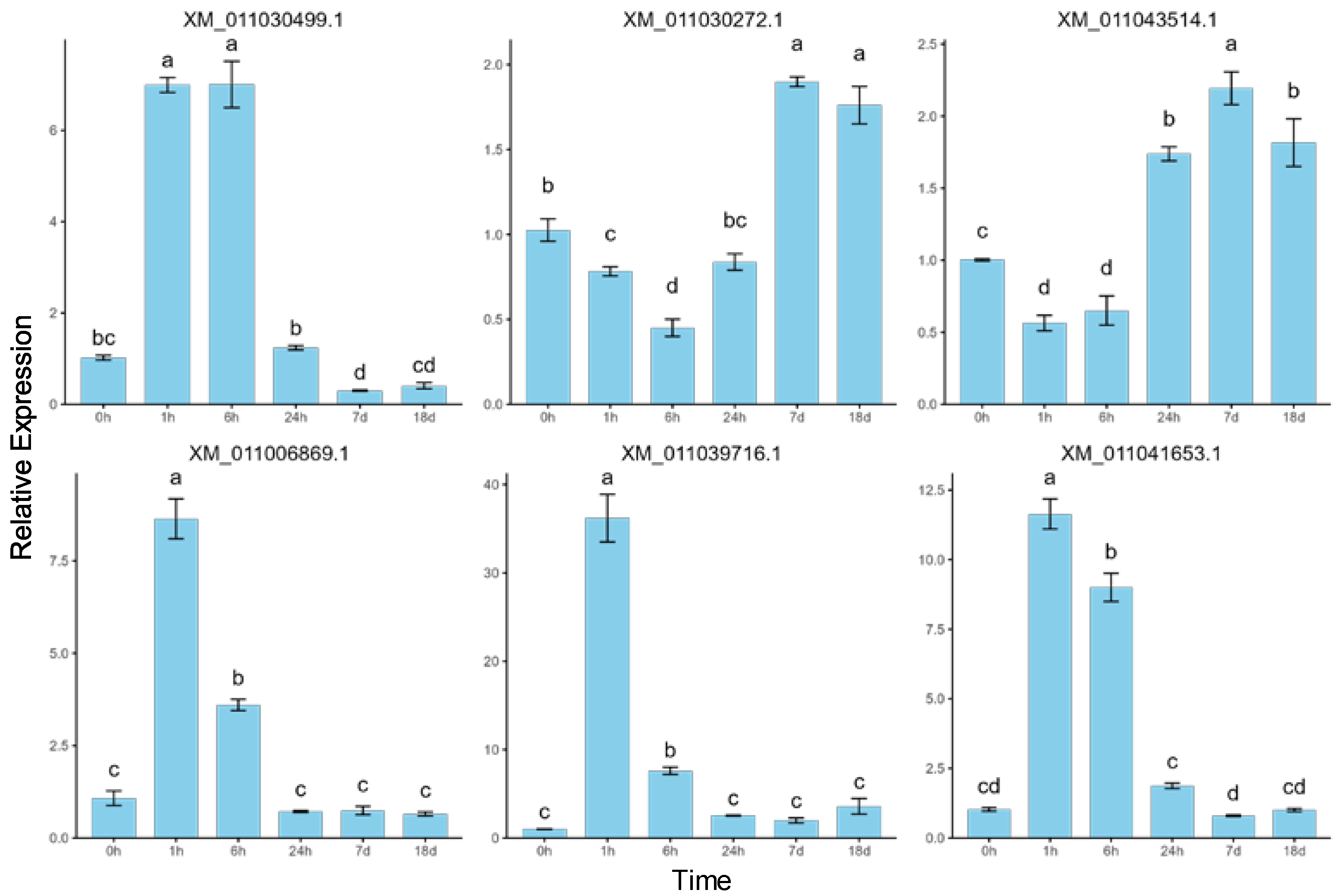
2.6. Validation of RNA-Seq Data by RT-qPCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Salt Stress Treatment
4.2. RNA Extraction and High-Throughput Sequencing
4.3. Reads Mapping and Analysis
4.4. Identification of Differentially Expressed Genes and GO Analysis
4.5. Clustering Analysis
4.6. Gene Regulatory Network Construction
4.7. Quantitative Real-Time PCR (RT-qPCR)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GRNs | gene regulatory networks |
| ST | short-term |
| LT | long-term |
| GO | Gene Ontology |
| P. euphratica | Populus euphratica |
| P. trichocarpa | Populus trichocarpa |
| DEGs | differentially expressed genes |
| PSGs | positive selective genes |
| BP | biological process |
| CC | cellular components |
| MF | molecular function |
| BIC | Bayesian Information Criterion |
| RT-qPCR | quantitative real-time PCR |
| qdODE | quasi-dynamic ordinary differential equations |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Functional Annotation | Primer Sequence (5′ to 3′) |
|---|---|---|
| XM_011030272.1 | transcription factor WER | Upstream: CGGGCGACGAAGTCACAGGT Downstream: GCCCCCAGGCTTAAACTAGG |
| XM_011043514.1 | transcription factor bHLH061 | Upstream: TCCTATTTAAAAGAGCGACCTCCACACACA Downstream: CGTTGTGGTGCATGAGGTAGGTGTT |
| XM_011041653.1 | TIFY10a | Upstream: GGGGCTCCTCGGAATTTTCCAT Downstream: AGCCGGAAGAATCGGTTGAAGACA |
| XM_011006869.1 | ERF022 | Upstream: CGCGCCGAGTATAAGAGTGGTACG Downstream: TTGGGCCCCCAGTCTCTTAAG |
| XM_011039716.1 | hyoscyamine 6-dioxygenase | Upstream: CTTCCGACCACGCCAACTCTCTTC Downstream: GTCCGAACAAACCTCCCTCAATCAGAAA |
| XM_011030499.1 | zinc finger protein Zat10 | Upstream: GGGGCTGGAATCTTTCATCATC Downstream: CGTTGTGGTGCATGAGGTAGGTGTT |
| XM_011003616.1 | transcription faction WRKY53 | Upstream: GGGTTAACCTACTTGGTTTGTCCATCT Downstream: CCCAAGATAGTGGTGTGGAACGAGG |
| XM_011006625.1 | early responsive to dehydration 15 | Upstream: AGGCCAAAGCTACATTCCCATTATTA Downstream: AGGCCCATCTTTTTTGTTTGTTGAGGAAAAAAC |
| XM_011008621.1 | calcium-binding protein CML48 | Upstream: CGGGGCAAGCGTGTGAGACAGAGAGA Downstream: CGGCGCCTTTGTGTATGAGAGGTGA |
| XM_011020267.1 | transcription factor MYC2 | Upstream: CGGCGACCGTTTATCCGATCAT Downstream: CCCCTCTAGCAGTGGGTGGTGA |
| XM_011034907.1 | actin | Upstream: CGTGGACATCAGGAAGGACT Downstream: ACGCTCTGCTGTGGTGTGTGA |
| XM_011030272.1 | transcription factor WER | Upstream: CGGGCGACGAAGTCACAGGT Downstream: GCCCCCAGGCTTAAACTAGG |
| Time | Hub Genes |
|---|---|
| 0 h | XM_011018353.1 XM_011048240.1 XM_011037175.1 |
| 1 h | XM_011045396.1 XM_011041211.1 XM_011013437.1 |
| 6 h | XM_011035209.1 XM_011048240.1 XM_011027094.1 |
| 24 h | XM_011048240.1 XM_011018353.1 XM_011013437.1 |
| 7 d | XM_011048240.1 XM_011037175.1 XM_011023208.1 |
| 18 d | XM_011041211.1 XM_011048240.1 XM_011038717.1 |
Appendix B
References
- Zhu, J.-K. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef]
- Brosché, M.; Vinocur, B.; Alatalo, E.R.; Lamminmäki, A.; Teichmann, T.; Ottow, E.A.; Djilianov, D.; Afif, D.; Bogeat-Triboulot, M.-B.; Altman, A.; et al. Gene expression and metabolite profiling of Populus euphratica growing in the Negev desert. Genome Biol. 2005, 6, R101. [Google Scholar] [CrossRef]
- Gu, R.; Fonseca, S.; Puskás, L.G.; Hackler, L.; Zvara, Á.; Dudits, D.; Pais, M.S. Transcript identification and profiling during salt stress and recovery of Populus euphratica. Tree Physiol. 2004, 24, 265–276. [Google Scholar] [CrossRef]
- Chen, S.; Polle, A. Salinity tolerance of Populus. Plant Biol. 2010, 12, 317–333. [Google Scholar] [CrossRef]
- Janz, D.; Lautner, S.; Wildhagen, H.; Behnke, K.; Schnitzler, J.-P.; Rennenberg, H.; Fromm, J.; Polle, A. Salt stress induces the formation of a novel type of ‘pressure wood’ in two Populus species. New Phytol. 2012, 194, 129–141. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [PubMed]
- Ottow, E.A.; Brinker, M.; Teichmann, T.; Fritz, E.; Kaiser, W.; Brosché, M.; Kangasjärvi, J.; Jiang, X.; Polle, A. Populus euphratica Displays Apoplastic Sodium Accumulation, Osmotic Adjustment by Decreases in Calcium and Soluble Carbohydrates, and Develops Leaf Succulence under Salt Stress. Plant Physiol. 2005, 139, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ding, N.; Zhao, X.; Zhao, M.; Chang, Z.; Liu, J.; Zhang, L. Molecular characterization of PeSOS1: The putative Na+/H+ antiporter of Populus euphratica. Plant Mol. Biol. 2007, 65, 1–11. [Google Scholar] [CrossRef]
- Ottow, E.A.; Polle, A.; Brosché, M.; Kangasjärvi, J.; Dibrov, P.; Zörb, C.; Teichmann, T. Molecular characterization of PeNhaD1: The first member of the NhaD Na+/H+ antiporter family of plant origin. Plant Mol. Biol. 2005, 58, 75–88. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Wang, J.-P.; He, Y. A Populus euphratica NAC protein regulating Na+/K+ homeostasis improves salt tolerance in Arabidopsis thaliana. Gene 2013, 521, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, F.; Zhao, M.; An, L.; Zhang, L.; Chen, N. Properties of plasma membrane H+-ATPase in salt-treated Populus euphratica callus. Plant Cell Rep. 2006, 26, 229–235. [Google Scholar] [CrossRef]
- Ma, T.; Liu, Q.; Li, Z.; Zhang, X. Tonoplast H+-ATPase in response to salt stress in Populus euphratica cell suspensions. Plant Sci. 2002, 163, 499–505. [Google Scholar] [CrossRef]
- Shen, Z.; Yao, J.; Sun, J.; Chang, L.; Wang, S.; Ding, M.; Qian, Z.; Zhang, H.; Zhao, N.; Sa, G.; et al. Populus euphratica HSF binds the promoter of WRKY1 to enhance salt tolerance. Plant Sci. 2015, 235, 89–100. [Google Scholar] [CrossRef]
- Han, Y.; Wang, W.; Sun, J.; Ding, M.; Zhao, R.; Deng, S.; Wang, F.; Hu, Y.; Wang, Y.; Lu, Y.; et al. Populus euphratica XTH overexpression enhances salinity tolerance by the development of leaf succulence in transgenic tobacco plants. J. Exp. Bot. 2013, 64, 4225–4238. [Google Scholar] [CrossRef]
- Li, B.; Duan, H.; Li, J.; Deng, X.W.; Yin, W.; Xia, X. Global identification of miRNAs and targets in Populus euphratica under salt stress. Plant Mol. Biol. 2013, 81, 525–539. [Google Scholar] [CrossRef]
- Qiu, Q.; Ma, T.; Hu, Q.; Liu, B.; Wu, Y.; Zhou, H.; Wang, Q.; Wang, J.; Liu, J. Genome-scale transcriptome analysis of the desert poplar, Populus euphratica. Tree Physiol. 2011, 31, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Wang, J.; Zhou, G.; Yue, Z.; Hu, Q.; Chen, Y.; Liu, B.; Qiu, Q.; Wang, Z.; Zhang, J.; et al. Genomic insights into salt adaptation in a desert poplar. Nat. Commun. 2013, 4, 2797. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K. The Stress Concept in Plants: An Introduction. Ann. N. Y. Acad. Sci. 1998, 851, 187–198. [Google Scholar] [CrossRef]
- Sun, J.; Chen, S.; Dai, S.; Wang, R.; Li, N.; Shen, X.; Zhou, X.; Lu, C.; Zheng, X.; Hu, Z.; et al. NaCl-Induced Alternations of Cellular and Tissue Ion Fluxes in Roots of Salt-Resistant and Salt-Sensitive Poplar Species. Plant Physiol. 2009, 149, 1141–1153. [Google Scholar] [CrossRef]
- Dondelinger, F.; Husmeier, D.; Lèbre, S. Dynamic Bayesian networks in molecular plant science: Inferring gene regulatory networks from multiple gene expression time series. Euphytica 2012, 183, 361–377. [Google Scholar] [CrossRef]
- Kleyer, M.; Trinogga, J.; Cebrián-Piqueras, M.A.; Trenkamp, A.; Fløjgaard, C.; Ejrnæs, R.; Bouma, T.J.; Minden, V.; Maier, M.; Mantilla-Contreras, J. Trait correlation network analysis identifies biomass allocation traits and stem specific length as hub traits in herbaceous perennial plants. J. Ecol. 2019, 107, 829–842. [Google Scholar] [CrossRef]
- Cao, X.; Dong, A.; Kang, G.; Wang, X.; Duan, L.; Hou, H.; Zhao, T.; Wu, S.; Liu, X.; Huang, H. Modeling spatial interaction networks of the gut microbiota. Gut Microbes 2022, 14, 2106103. [Google Scholar] [CrossRef]
- Wang, Q.; Dong, A.; Zhao, J.; Wang, C.; Griffin, C.; Gragnoli, C.; Xue, F.; Wu, R. Vaginal microbiota networks as a mechanistic predictor of aerobic vaginitis. Front. Microbiol. 2022, 13, 998813. [Google Scholar] [CrossRef]
- Sun, L.; Jiang, L.; Grant, C.N.; Wang, H.-G.; Gragnoli, C.; Liu, Z.; Wu, R. Computational identification of gene networks as a biomarker of neuroblastoma risk. Cancers 2020, 12, 2086. [Google Scholar] [CrossRef]
- Sang, M.; Feng, L.; Dong, A.; Gragnoli, C.; Griffin, C.; Wu, R. The genomic physics of tumor–microenvironment crosstalk. Phys. Rep. 2023, 1029, 1–51. [Google Scholar] [CrossRef]
- Sang, M.; Dong, A.; Wu, S.; Li, F.; Wang, J.; Griffin, C.; Wu, R. A graph model of combination therapies. Drug Discov. Today 2022, 27, 1210–1217. [Google Scholar] [CrossRef]
- Dong, A.; Wu, S.; Che, J.; Wang, Y.; Wu, R. idopNetwork: A network tool to dissect spatial community ecology. Methods Ecol. Evol. 2023, 14, 2272–2283. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, L.; Fu, G.; Wang, M.; Wang, Y.; Shen, B.; Liu, Z.; Wang, Z.; Hou, W.; Berceli, S.; et al. An omnidirectional visualization model of personalized gene regulatory networks. Npj Syst. Biol. Appl. 2019, 5, 38. [Google Scholar] [CrossRef]
- Wu, R.; Jiang, L. Recovering dynamic networks in big static datasets. Phys. Rep. 2021, 912, 1–57. [Google Scholar] [CrossRef]
- Gries, D.; Zeng, F.; Foetzki, A.; Arndt, S.K.; Bruelheide, H.; Thomas, F.M.; Zhang, X.; Runge, M. Growth and water relations of Tamarix ramosissima and Populus euphratica on Taklamakan desert dunes in relation to depth to a permanent water table. Plant Cell Environ. 2003, 26, 725–736. [Google Scholar] [CrossRef]
- Ferreira, S.; Hjernø, K.; Larsen, M.; Wingsle, G.; Larsen, P.; Fey, S.; Roepstorff, P.; Pais, M.S. Proteome profiling of Populus euphratica Oliv. upon heat stress. Ann. Bot. 2006, 98, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, H.; Jamil, M.; Haq, A.; Ali, S.; Ahmad, R.; Malik, Z.; Parveen, Z. Salt stress manifestation on plants, mechanism of salt tolerance and potassium role in alleviating it: A review. Zemdirb.-Agric. 2016, 103, 229–238. [Google Scholar] [CrossRef]
- Ran, X.; Wang, X.; Gao, X.; Liang, H.; Liu, B.; Huang, X. Effects of salt stress on the photosynthetic physiology and mineral ion absorption and distribution in white willow (Salix alba L.). PLoS ONE 2021, 16, e0260086. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Wang, W.; Zhou, T.; Zhang, Y.; Chen, J.; Xiao, B.; Yang, Y.; Yu, Y. Transcriptomic analysis reveals the molecular mechanisms of Camellia sinensis in response to salt stress. Plant Growth Regul. 2018, 84, 481–492. [Google Scholar] [CrossRef]
- Duan, L.; Sebastian, J.; Dinneny, J.R. Salt-stress regulation of root system growth and architecture in Arabidopsis seedlings. In Plant Cell Expansion: Methods and Protocols; Humana Press: New York, NY, USA, 2015; pp. 105–122. [Google Scholar]
- Rewald, B.; Shelef, O.; Ephrath, J.E.; Rachmilevitch, S. Adaptive plasticity of salt-stressed root systems. In Ecophysiology and Responses of Plants Under Salt Stress; Springer: New York, NY, USA, 2013; pp. 169–201. [Google Scholar]
- Jiang, L.; Griffin, C.H.; Wu, R. SEGN: Inferring real-time gene networks mediating phenotypic plasticity. Comput. Struct. Biotechnol. J. 2020, 18, 2510–2521. [Google Scholar] [CrossRef]
- Li, X.; Nair, A.; Wang, S.; Wang, L. Quality Control of RNA-Seq Experiments. In RNA Bioinformatics; Picardi, E., Ed.; Springer: New York, NY, USA, 2015; pp. 137–146. [Google Scholar]
- Hu, L.; Xie, Y.; Fan, S.; Wang, Z.; Wang, F.; Zhang, B.; Li, H.; Song, J.; Kong, L. Comparative analysis of root transcriptome profiles between drought-tolerant and susceptible wheat genotypes in response to water stress. Plant Sci. 2018, 272, 276–293. [Google Scholar] [CrossRef]
- Cui, W.; Long, Q.; Xiao, M.; Wang, X.; Feng, G.; Li, X.; Wang, P.; Zhou, Y. Refining computational inference of gene regulatory networks: Integrating knockout data within a multi-task framework. Brief. Bioinform. 2024, 25, bbae361. [Google Scholar] [CrossRef]
- Wu, S.; Liu, X.; Dong, A.; Gragnoli, C.; Griffin, C.; Wu, J.; Yau, S.-T.; Wu, R. The metabolomic physics of complex diseases. Proc. Natl. Acad. Sci. USA 2023, 120, e2308496120. [Google Scholar] [CrossRef]
- O’Brien, M.J.; Ansari, A. Protein interaction network revealed by quantitative proteomic analysis links TFIIB to multiple aspects of the transcription cycle. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2024, 1872, 140968. [Google Scholar] [CrossRef]
- Yuan, C.; Hu, Y.; Liu, Q.; Xu, J.; Zhou, W.; Yu, H.; Shen, L.; Qin, C. MED8 regulates floral transition in Arabidopsis by interacting with FPA. Plant J. 2023, 116, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, Y.; Zhang, Y.; Li, C.; Gong, S.; Yan, S.; Li, G.; Hu, G.; Ren, H.; Yang, J.; et al. Comparative transcriptome analysis of salt-sensitive and salt-tolerant maize reveals potential mechanisms to enhance salt resistance. Genes Genom. 2019, 41, 781–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, S.; Dai, X.; Yang, Y.; Luo, Y.; Gao, Y.; Liu, X.; Wei, W.; Wang, H.; Xu, X. PSDX: A comprehensive multi-omics association database of Populus trichocarpa with a focus on the secondary growth in response to stresses. Front. Plant Sci. 2021, 12, 655565. [Google Scholar] [CrossRef]
- Fu, Y.; Li, F.; Mu, S.; Jiang, L.; Ye, M.; Wu, R. Heterophylly quantitative trait loci respond to salt stress in the desert tree Populus euphratica. Front. Plant Sci. 2021, 12, 692494. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality Control Tool for High Throughput Sequence Data. Babraham Bioinformatics. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 April 2025).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Hill, S.M. Network clustering: Probing biological heterogeneity by sparse graphical models. Bioinformatics 2011, 27, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Allesina, S.; Tang, S. Stability criteria for complex ecosystems. Nature 2012, 483, 205–208. [Google Scholar] [CrossRef]
- Kim, B.-R.; Zhang, L.; Berg, A.; Fan, J.; Wu, R. A computational approach to the functional clustering of periodic gene-expression profiles. Genetics 2008, 180, 821–834. [Google Scholar] [CrossRef] [PubMed]

| Samples | Raw Reads | Clean Reads | Clean Bases | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|
| eSR_0h1 | 120,061,832 | 117,037,376 | 17.56G | 93.42 | 43.91 |
| eSR_0h2 | 126,544,958 | 123,009,778 | 18.45G | 93.24 | 43.74 |
| eSR_0h3 | 127,970,524 | 124,812,530 | 18.72G | 93.59 | 43.41 |
| eSR_1h1 | 123,483,826 | 120,287,176 | 18.04G | 94.23 | 44.15 |
| eSR_1h2 | 108,341,308 | 105,816,148 | 15.87G | 94.21 | 44.66 |
| eSR_1h3 | 126,160,386 | 122,993,670 | 18.45G | 94.10 | 44.35 |
| eSR_6h1 | 131,378,764 | 124,558,814 | 18.68G | 94.36 | 45.28 |
| eSR_6h2 | 120,654,756 | 117,432,120 | 17.61G | 93.86 | 44.31 |
| eSR_6h3 | 144,094,448 | 139,794,392 | 20.97G | 94.19 | 45.05 |
| eSR_24h1 | 153,980,886 | 148,217,346 | 22.23G | 92.62 | 42.85 |
| eSR_24h2 | 140,975,854 | 135,396,226 | 20.31G | 92.60 | 43.29 |
| eSR_24h3 | 159,602,918 | 153,235,106 | 22.99G | 92.65 | 42.98 |
| eSR_7d1 | 115,600,962 | 111,504,632 | 16.73G | 91.49 | 43.63 |
| eSR_7d2 | 142,277,696 | 137,337,968 | 20.6G | 91.56 | 43.79 |
| eSR_7d3 | 146,228,598 | 141,376,682 | 21.21G | 91.80 | 43.45 |
| eSR_18d1 | 128,754,356 | 125,175,170 | 18.78G | 93.80 | 44.47 |
| eSR_18d2 | 131,503,632 | 127,914,480 | 19.19G | 93.65 | 43.96 |
| eSR_18d3 | 114,477,542 | 111,408,754 | 16.71G | 94.04 | 43.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.; Pan, W.; Dong, A. idopNetwork Analysis of Salt-Responsive Transcriptomes Reveals Hub Regulatory Modules and Genes in Populus euphratica. Int. J. Mol. Sci. 2025, 26, 4091. https://doi.org/10.3390/ijms26094091
Wu S, Pan W, Dong A. idopNetwork Analysis of Salt-Responsive Transcriptomes Reveals Hub Regulatory Modules and Genes in Populus euphratica. International Journal of Molecular Sciences. 2025; 26(9):4091. https://doi.org/10.3390/ijms26094091
Chicago/Turabian StyleWu, Shuang, Wenqi Pan, and Ang Dong. 2025. "idopNetwork Analysis of Salt-Responsive Transcriptomes Reveals Hub Regulatory Modules and Genes in Populus euphratica" International Journal of Molecular Sciences 26, no. 9: 4091. https://doi.org/10.3390/ijms26094091
APA StyleWu, S., Pan, W., & Dong, A. (2025). idopNetwork Analysis of Salt-Responsive Transcriptomes Reveals Hub Regulatory Modules and Genes in Populus euphratica. International Journal of Molecular Sciences, 26(9), 4091. https://doi.org/10.3390/ijms26094091