
Examination of Anti-Inflammatory Effects After Propionate Supplementation in the R6/2 Mouse Model of Huntington’s Disease

 , ,
, , 
Abstract
:1. Introduction
1.1. Huntington’s Disease
1.2. Neuroinflammation in HD
1.3. Propionate
2. Results
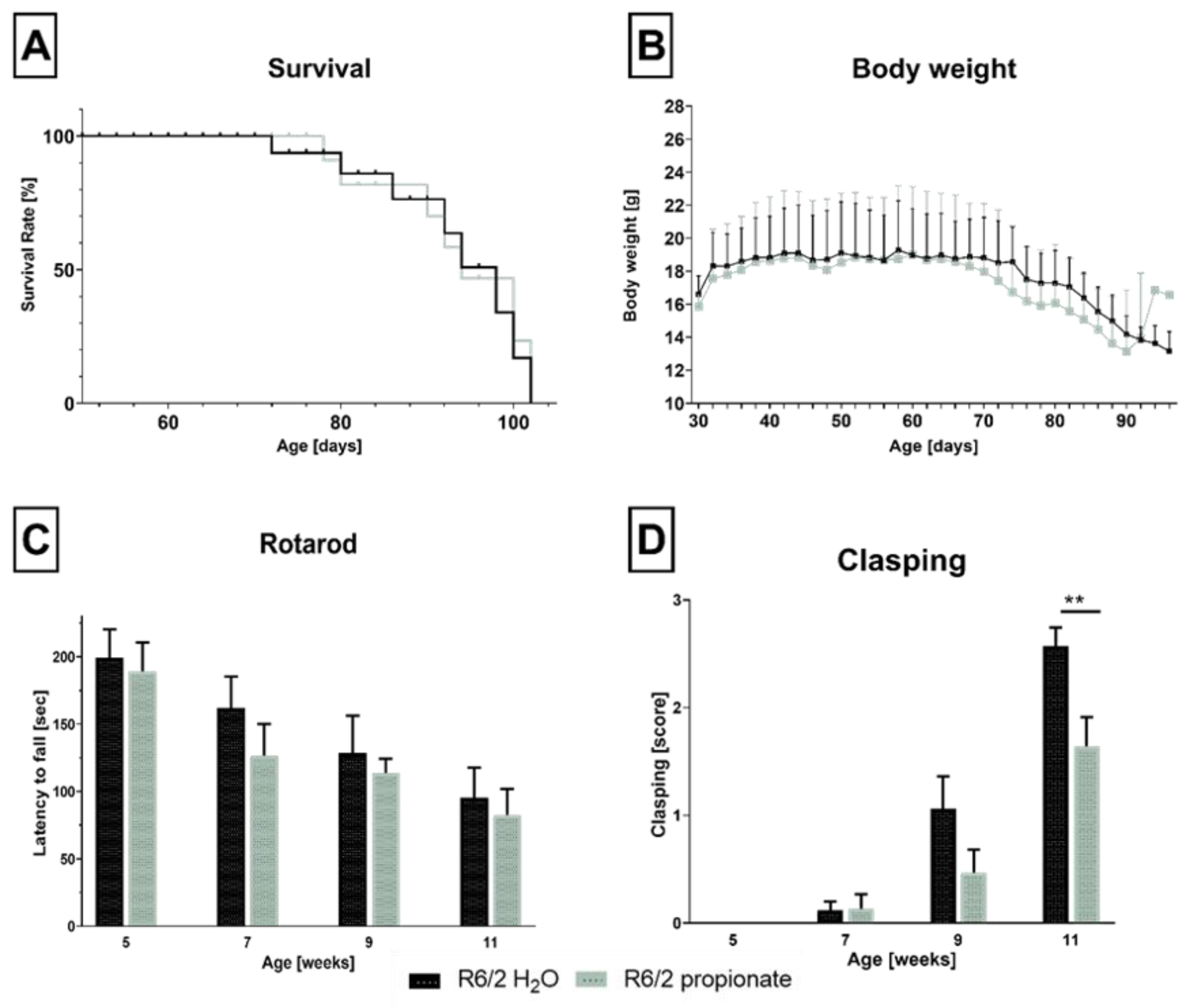
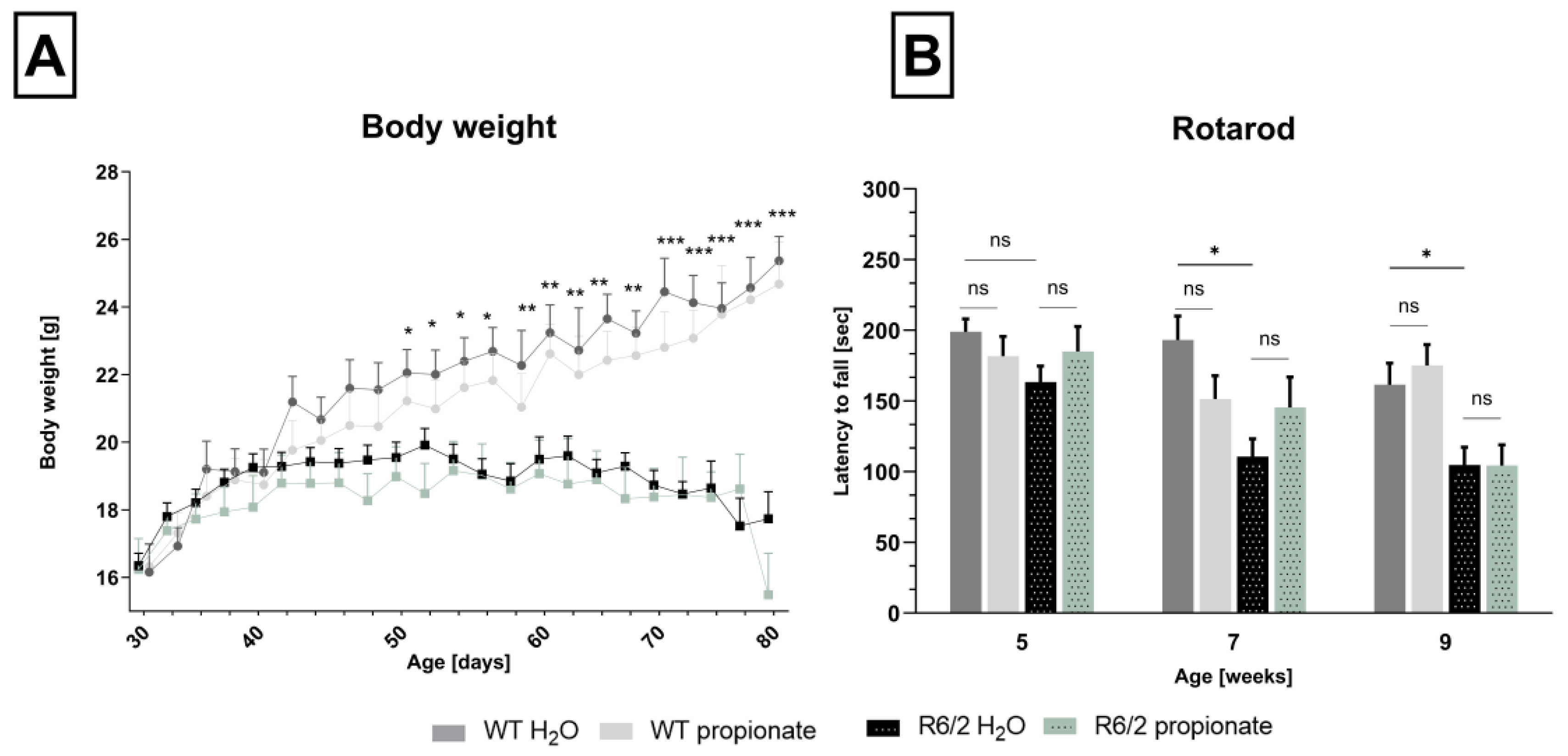
2.1. Propionate Treatment Did Not Affect Signs of Clinical Progression but Ameliorated Clasping Behavior in R6/2 Mice
2.2. Neuronal Decline Occurs in Propionate-Treated R6/2 Mice
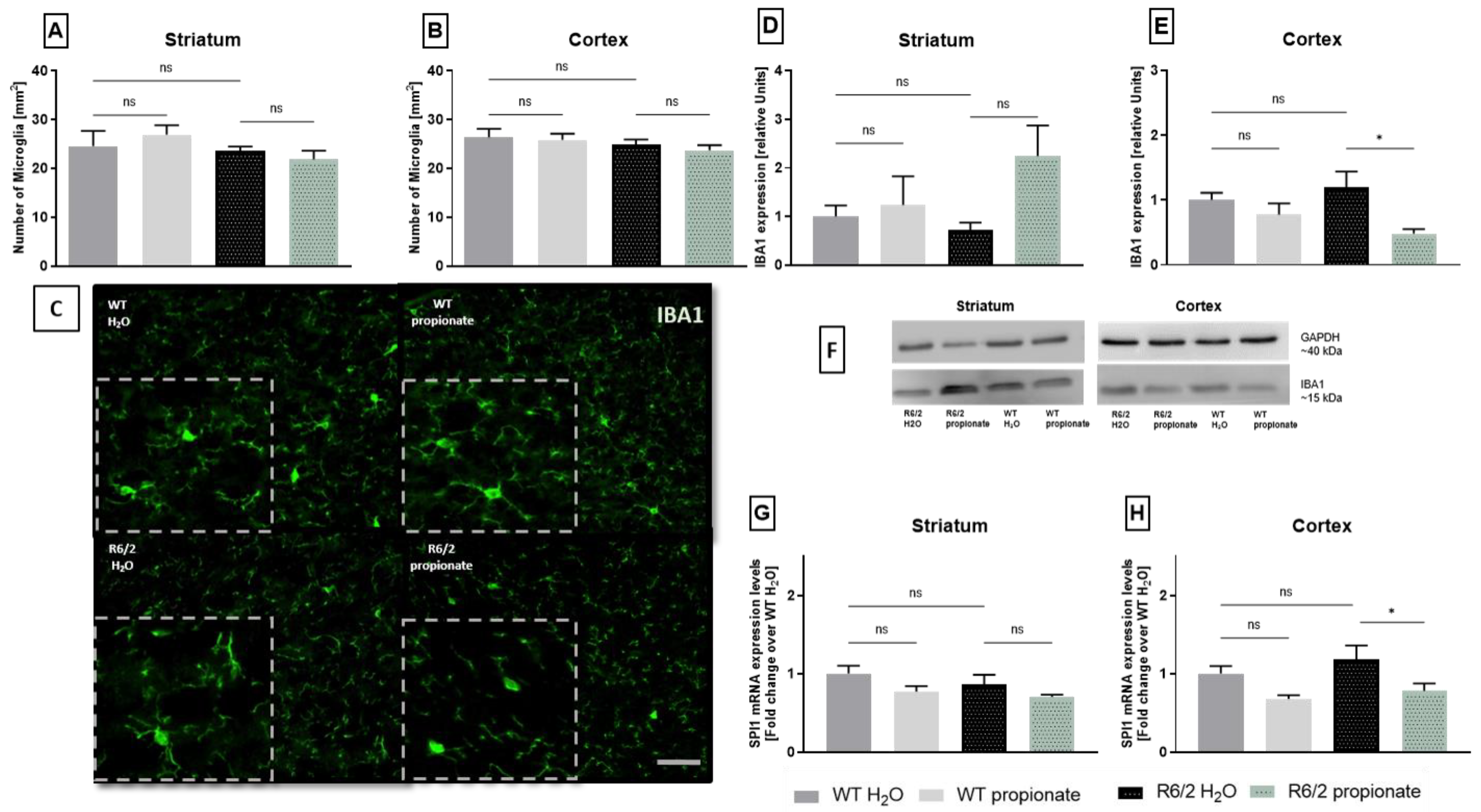
2.3. Examination of Microglia Activation
2.4. iNOS Expression and TNFα-mRNA Levels Are Altered in Propionate-Treated R6/2 Mice
3. Discussion
4. Materials and Methods
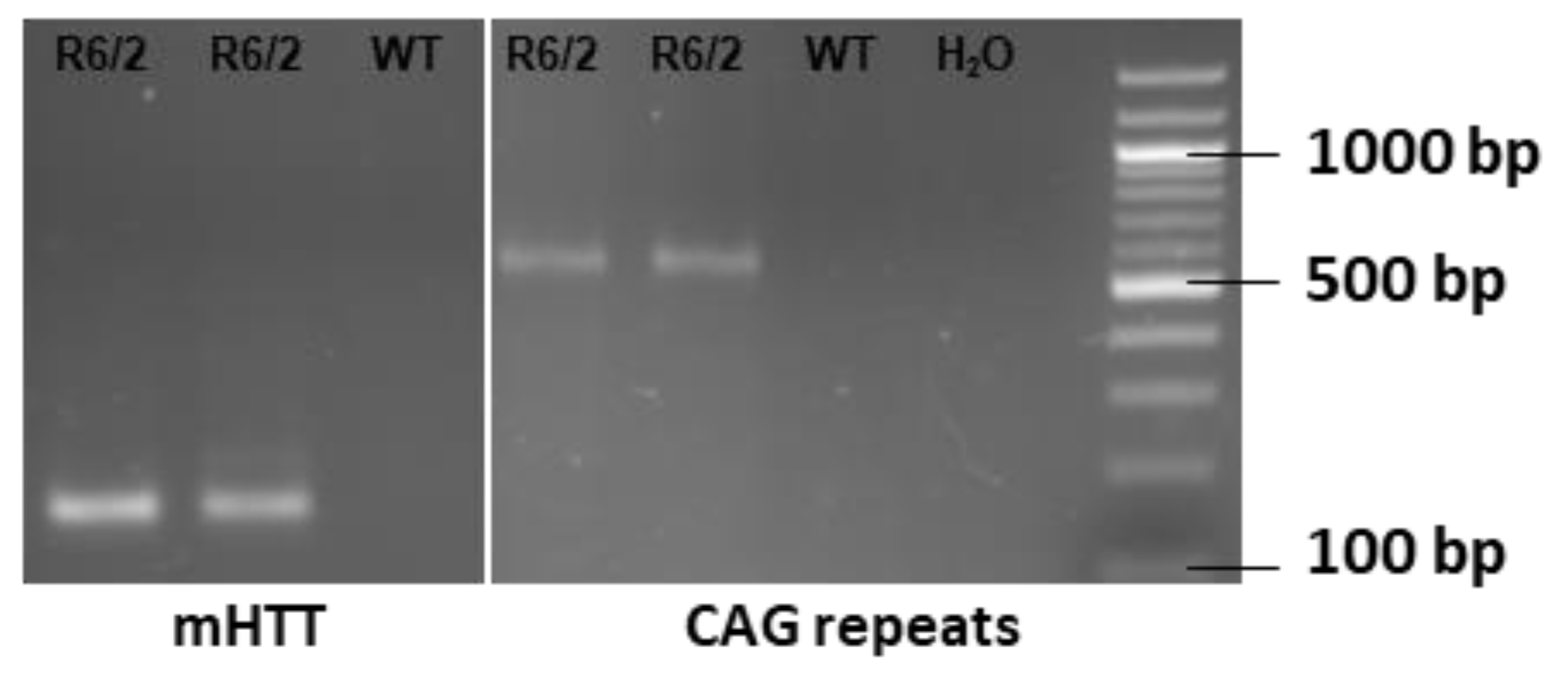
4.1. Mice Genotyping and Treatment
4.2. Behavioral Analysis
4.3. Tissue Preparation
4.4. Immunohistochemistry
4.5. Realtime-PCR
4.6. Western Blot
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neueder, A.; Landles, C.; Ghosh, R.; Howland, D.; Myers, R.H.; Faull, R.L.M.; Tabrizi, S.J.; Bates, G.P. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep. 2017, 7, 1307. [Google Scholar] [CrossRef]
- Han, I.; You, Y.; Kordower, J.H.; Brady, S.T.; Morfini, G.A. Differential vulnerability of neurons in Huntington’s disease: The role of cell type-specific features. J. Neurochem. 2010, 113, 1073–1091. [Google Scholar] [CrossRef] [PubMed]
- Dayalu, P.; Albin, R.L. Huntington disease: Pathogenesis and treatment. Neurol. Clin. 2015, 33, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Baake, V.; Reijntjes, R.H.A.M.; Dumas, E.M.; Thompson, J.C.; Roos, R.A.C. Cognitive decline in Huntington’s disease expansion gene carriers. Cortex 2017, 95, 51–62. [Google Scholar] [CrossRef]
- Lipe, H.; Bird, T. Late onset Huntington Disease: Clinical and genetic characteristics of 34 cases. J. Neurol. Sci. 2009, 276, 159–162. [Google Scholar] [CrossRef]
- Oosterloo, M.; Bijlsma, E.K.; van Kuijk, S.M.; Minkels, F.; Die-Smulders, C.E.d. Clinical and genetic characteristics of late-onset Huntington’s disease. Parkinsonism Relat. Disord. 2019, 61, 101–105. [Google Scholar] [CrossRef]
- Schultz, J.L.; Moser, A.D.; Nopoulos, P.C. The Association between CAG Repeat Length and Age of Onset of Juvenile-Onset Huntington’s Disease. Brain Sci. 2020, 10, 575. [Google Scholar] [CrossRef]
- Myers, R.H. Huntington’s disease genetics. NeuroRx 2004, 1, 255–262. [Google Scholar] [CrossRef]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Goh, A.M.; Wibawa, P.; Loi, S.M.; Walterfang, M.; Velakoulis, D.; Looi, J.C. Huntington’s disease: Neuropsychiatric manifestations of Huntington’s disease. Australas. Psychiatry 2018, 26, 366–375. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [PubMed]
- Bachoud-Lévi, A.-C.; Ferreira, J.; Massart, R.; Youssov, K.; Rosser, A.; Busse, M.; Craufurd, D.; Reilmann, R.; de Michele, G.; Rae, D.; et al. International Guidelines for the Treatment of Huntington’s Disease. Front. Neurol. 2019, 10, 710. [Google Scholar] [CrossRef]
- Frank, S.; Testa, C.M.; Stamler, D.; Kayson, E.; Davis, C.; Edmondson, M.C.; Kinel, S.; Leavitt, B.; Oakes, D.; O’Neill, C.; et al. Effect of Deutetrabenazine on Chorea Among Patients with Huntington Disease: A Randomized Clinical Trial. JAMA 2016, 316, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease: A randomized controlled trial. Neurology 2006, 66, 366–372. [Google Scholar] [CrossRef]
- Saft, C.; Burgunder, J.-M.; Dose, M.; Jung, H.H.; Katzenschlager, R.; Priller, J.; Nguyen, H.P.; Reetz, K.; Reilmann, R.; Seppi, K.; et al. Differential diagnosis of chorea (guidelines of the German Neurological Society). Neurol. Res. Pract. 2023, 5, 63. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Kam, T.-I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Lavisse, S.; Goutal, S.; Wimberley, C.; Tonietto, M.; Bottlaender, M.; Gervais, P.; Kuhnast, B.; Peyronneau, M.-A.; Barret, O.; Lagarde, J.; et al. Increased microglial activation in patients with Parkinson disease using 18F-DPA714 TSPO PET imaging. Parkinsonism Relat. Disord. 2021, 82, 29–36. [Google Scholar] [CrossRef]
- Lois, C.; González, I.; Izquierdo-García, D.; Zürcher, N.R.; Wilkens, P.; Loggia, M.L.; Hooker, J.M.; Rosas, H.D. Neuroinflammation in Huntington’s Disease: New Insights with 11C-PBR28 PET/MRI. ACS Chem. Neurosci. 2018, 9, 2563–2571. [Google Scholar] [CrossRef]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef]
- von Essen, M.R.; Hellem, M.N.N.; Vinther-Jensen, T.; Ammitzbøll, C.; Hansen, R.H.; Hjermind, L.E.; Nielsen, T.T.; Nielsen, J.E.; Sellebjerg, F. Early Intrathecal T Helper 17.1 Cell Activity in Huntington Disease. Ann. Neurol. 2020, 87, 246–255. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Belarbi, K.; Jopson, T.; Tweedie, D.; Arellano, C.; Luo, W.; Greig, N.H.; Rosi, S. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J Neuroinflammation 2012, 9, 23. [Google Scholar] [CrossRef]
- Yang, S.H.; Gangidine, M.; Pritts, T.A.; Goodman, M.D.; Lentsch, A.B. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock 2013, 40, 471–475. [Google Scholar] [CrossRef]
- Tassoni, A.; Mathur, V.; Vereen, J.; Cahir-McFarland, E.; Sankaranarayanan, S.; Yednock, T.; Andrews-Zwilling, Y. Inhibition of C1q Reduces Nerve Damage as Measured by Neurofilament Light Chain in the HD R6/2 Mouse Model (S36.004). Neurology 2022, 98, 23. [Google Scholar] [CrossRef]
- Chen, K.-P.; Hua, K.-F.; Tsai, F.-T.; Lin, T.-Y.; Cheng, C.-Y.; Yang, D.-I.; Hsu, H.-T.; Ju, T.-C. A selective inhibitor of the NLRP3 inflammasome as a potential therapeutic approach for neuroprotection in a transgenic mouse model of Huntington’s disease. J. Neuroinflamm. 2022, 19, 56. [Google Scholar] [CrossRef]
- Di Pardo, A.; Amico, E.; Favellato, M.; Castrataro, R.; Fucile, S.; Squitieri, F.; Maglione, V. FTY720 (fingolimod) is a neuroprotective and disease-modifying agent in cellular and mouse models of Huntington disease. Hum. Mol. Genet. 2014, 23, 2251–2265. [Google Scholar] [CrossRef]
- Labadorf, A.; Hoss, A.G.; Lagomarsino, V.; Latourelle, J.C.; Hadzi, T.C.; Bregu, J.; MacDonald, M.E.; Gusella, J.F.; Chen, J.-F.; Akbarian, S.; et al. Correction: RNA Sequence Analysis of Human Huntington Disease Brain Reveals an Extensive Increase in Inflammatory and Developmental Gene Expression. PLoS ONE 2016, 11, e0160295. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Blusch, A.; Fatoba, O.; Brunner, J.; Reick, C.; Hayardeny, L.; Hayden, M.; Sehr, D.; Winklhofer, K.F.; Saft, C.; et al. Laquinimod treatment in the R6/2 mouse model. Sci. Rep. 2017, 7, 4947. [Google Scholar] [CrossRef] [PubMed]
- Reilmann, R.; Anderson, K.E.; Feigin, A.; Tabrizi, S.J.; Leavitt, B.R.; Stout, J.C.; Piccini, P.; Schubert, R.; Loupe, P.; Wickenberg, A.; et al. Safety and efficacy of laquinimod for Huntington’s disease (LEGATO-HD): A multicentre, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Neurol. 2024, 23, 243–255. [Google Scholar] [CrossRef]
- Achenbach, J.; Saft, C.; Faissner, S.; Ellrichmann, G. Positive effect of immunomodulatory therapies on disease progression in Huntington’s disease? Data from a real-world cohort. Ther. Adv. Neurol. Disord. 2022, 15, 17562864221109750. [Google Scholar] [CrossRef]
- Pascale, A.; Marchesi, N.; Marelli, C.; Coppola, A.; Luzi, L.; Govoni, S.; Giustina, A.; Gazzaruso, C. Microbiota and metabolic diseases. Endocrine 2018, 61, 357–371. [Google Scholar] [CrossRef]
- Tsukahara, T.; Matsukawa, N.; Tomonaga, S.; Inoue, R.; Ushida, K.; Ochiai, K. High-sensitivity detection of short-chain fatty acids in porcine ileal, cecal, portal and abdominal blood by gas chromatography-mass spectrometry. Anim. Sci. J. 2014, 85, 494–498. [Google Scholar] [CrossRef]
- Breuer, R.I.; Soergel, K.H.; Lashner, B.A.; Christ, M.L.; Hanauer, S.B.; Vanagunas, A.; Harig, J.M.; Keshavarzian, A.; Robinson, M.; Sellin, J.H.; et al. Short chain fatty acid rectal irrigation for left-sided ulcerative colitis: A randomised, placebo controlled trial. Gut 1997, 40, 485–491. [Google Scholar] [CrossRef]
- D’Argenio, G.; Mazzacca, G. Short-chain fatty acid in the human colon. Relation to inflammatory bowel diseases and colon cancer. Adv. Exp. Med. Biol. 1999, 472, 149–158. [Google Scholar] [CrossRef]
- Vernia, P.; Marcheggiano, A.; Caprilli, R.; Frieri, G.; Corrao, G.; Valpiani, D.; Di Paolo, M.C.; Paoluzi, P.; Torsoli, A. Short-chain fatty acid topical treatment in distal ulcerative colitis. Aliment. Pharmacol. Ther. 1995, 9, 309–313. [Google Scholar] [CrossRef]
- Duscha, A.; Gisevius, B.; Hirschberg, S.; Yissachar, N.; Stangl, G.I.; Eilers, E.; Bader, V.; Haase, S.; Kaisler, J.; David, C.; et al. Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067–1080.e16. [Google Scholar] [CrossRef]
- Wasser, C.I.; Mercieca, E.-C.; Kong, G.; Hannan, A.J.; McKeown, S.J.; Glikmann-Johnston, Y.; Stout, J.C. Gut dysbiosis in Huntington’s disease: Associations among gut microbiota, cognitive performance and clinical outcomes. Brain Commun. 2020, 2, fcaa110. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.-H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.J.; Lione, L.A.; Humby, T.; Mangiarini, L.; Mahal, A.; Bates, G.P.; Dunnett, S.B.; Morton, A.J. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J. Neurosci. 1999, 19, 3248–3257. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Filippone, A.; Lanza, M.; Campolo, M.; Casili, G.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Protective effect of sodium propionate in Aβ1-42 -induced neurotoxicity and spinal cord trauma. Neuropharmacology 2020, 166, 107977. [Google Scholar] [CrossRef]
- Cabanas, M.; Piquemal, M.; Pistono, C.; Arnaud, S.; Rakesh, D.; Poinama, E.; Guillou, J.-L.; Garret, M.; Cho, Y.H. Correlations Between Mutant Huntingtin Aggregates and Behavioral Changes in R6/1 Mice. J. Huntingtons Dis. 2020, 9, 33–45. [Google Scholar] [CrossRef]
- Crotti, A.; Benner, C.; Kerman, B.E.; Gosselin, D.; Lagier-Tourenne, C.; Zuccato, C.; Cattaneo, E.; Gage, F.H.; Cleveland, D.W.; Glass, C.K. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 2014, 17, 513–521. [Google Scholar] [CrossRef]
- Heneka, M.T.; Feinstein, D.L. Expression and function of inducible nitric oxide synthase in neurons. J. Neuroimmunol. 2001, 114, 8–18. [Google Scholar] [CrossRef]
- Lee, D.-H.; Heidecke, H.; Schröder, A.; Paul, F.; Wachter, R.; Hoffmann, R.; Ellrichmann, G.; Dragun, D.; Waschbisch, A.; Stegbauer, J.; et al. Increase of angiotensin II type 1 receptor auto-antibodies in Huntington’s disease. Mol. Neurodegener. 2014, 9, 49. [Google Scholar] [CrossRef]
- Novati, A.; Nguyen, H.P.; Schulze-Hentrich, J. Environmental stimulation in Huntington disease patients and animal models. Neurobiol. Dis. 2022, 171, 105725. [Google Scholar] [CrossRef]
- Wronka, D.; Karlik, A.; Misiorek, J.O.; Przybyl, L. What the Gut Tells the Brain-Is There a Link between Microbiota and Huntington’s Disease? Int. J. Mol. Sci. 2023, 24, 4477. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Ellul, S.; Narayana, V.K.; Kanojia, K.; Ha, H.T.T.; Li, S.; Renoir, T.; Cao, K.-A.L.; Hannan, A.J. An integrated metagenomics and metabolomics approach implicates the microbiota-gut-brain axis in the pathogenesis of Huntington’s disease. Neurobiol. Dis. 2021, 148, 105199. [Google Scholar] [CrossRef]
- Guo, Z.; Rudow, G.; Pletnikova, O.; Codispoti, K.-E.; Orr, B.A.; Crain, B.J.; Duan, W.; Margolis, R.L.; Rosenblatt, A.; Ross, C.A.; et al. Striatal neuronal loss correlates with clinical motor impairment in Huntington’s disease. Mov. Disord. 2012, 27, 1379–1386. [Google Scholar] [CrossRef]
- Canals, J.M.; Pineda, J.R.; Torres-Peraza, J.F.; Bosch, M.; Martín-Ibañez, R.; Muñoz, M.T.; Mengod, G.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 2004, 24, 7727–7739. [Google Scholar] [CrossRef]
- Torres-Cruz, F.M.; Mendoza, E.; Vivar-Cortés, I.C.; García-Sierra, F.; Hernández-Echeagaray, E. Do BDNF and NT-4/5 exert synergistic or occlusive effects on corticostriatal transmission in a male mouse model of Huntington’s disease? J. Neurosci. Res. 2019, 97, 1665–1677. [Google Scholar] [CrossRef]
- Yu, C.; Li, C.H.; Chen, S.; Yoo, H.; Qin, X.; Park, H. Decreased BDNF Release in Cortical Neurons of a Knock-in Mouse Model of Huntington’s Disease. Sci. Rep. 2018, 8, 16976. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- Etxeberria-Rekalde, E.; Alzola-Aldamizetxebarria, S.; Flunkert, S.; Hable, I.; Daurer, M.; Neddens, J.; Hutter-Paier, B. Quantification of Huntington’s Disease Related Markers in the R6/2 Mouse Model. Front. Mol. Neurosci. 2020, 13, 617229. [Google Scholar] [CrossRef]
- Paldino, E.; Balducci, C.; La Vitola, P.; Artioli, L.; D’Angelo, V.; Giampà, C.; Artuso, V.; Forloni, G.; Fusco, F.R. Neuroprotective Effects of Doxycycline in the R6/2 Mouse Model of Huntington’s Disease. Mol. Neurobiol. 2020, 57, 1889–1903. [Google Scholar] [CrossRef]
- Paldino, E.; D’Angelo, V.; Sancesario, G.; Fusco, F.R. Pyroptotic cell death in the R6/2 mouse model of Huntington’s disease: New insight on the inflammasome. Cell Death Discov. 2020, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; St-Pierre, M.-K.; Carrier, M.; El Hajj, H.; Novak, S.W.; Sanchez, M.G.; Cicchetti, F.; Tremblay, M.-È. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflam. 2020, 17, 98. [Google Scholar] [CrossRef] [PubMed]
- Yu-Taeger, L.; Stricker-Shaver, J.; Arnold, K.; Bambynek-Dziuk, P.; Novati, A.; Singer, E.; Lourhmati, A.; Fabian, C.; Magg, J.; Riess, O.; et al. Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease. Cells 2019, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Hopperton, K.E.; Mohammad, D.; Trépanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Smith, A.M.; Smyth, L.C.; Jansson, D.; Scotter, E.L.; Swanson, M.E.V.; Aalderink, M.; Coppieters, N.; Narayan, P.; Handley, R.; et al. PU.1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol. Neurodegener. 2018, 13, 44. [Google Scholar] [CrossRef]
- Gasser, J.; Gillet, G.; Valadas, J.S.; Rouvière, L.; Kotian, A.; Fan, W.; Keaney, J.; Kadiu, I. Innate immune activation and aberrant function in the R6/2 mouse model and Huntington’s disease iPSC-derived microglia. Front. Mol. Neurosci. 2023, 16, 1191324. [Google Scholar] [CrossRef]
- Saha, R.N.; Pahan, K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid. Redox Signal. 2006, 8, 929–947. [Google Scholar] [CrossRef]
- Madrigal, J.L.M.; Hurtado, O.; Moro, M.A.; Lizasoain, I.; Lorenzo, P.; Castrillo, A.; Boscá, L.; Leza, J.C. The increase in TNF-alpha levels is implicated in NF-kappaB activation and inducible nitric oxide synthase expression in brain cortex after immobilization stress. Neuropsychopharmacology 2002, 26, 155–163. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.-S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Liu, T.; Li, J.; Liu, Y.; Xiao, N.; Suo, H.; Xie, K.; Yang, C.; Wu, C. Short-chain fatty acids suppress lipopolysaccharide-induced production of nitric oxide and proinflammatory cytokines through inhibition of NF-κB pathway in RAW264.7 cells. Inflammation 2012, 35, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Kuno, R.; Wang, J.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Autocrine activation of microglia by tumor necrosis factor-alpha. J. Neuroimmunol. 2005, 162, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef]
- Eide, S.; Misztal, M.; Feng, Z.-P. Interleukin-6 as a marker of Huntington’s disease progression: Systematic review and meta-analysis. Brain Behav. Immun. Health 2023, 30, 100635. [Google Scholar] [CrossRef]
- Wertz, M.H.; Pineda, S.S.; Lee, H.; Kulicke, R.; Kellis, M.; Heiman, M. Interleukin-6 deficiency exacerbates Huntington’s disease model phenotypes. Mol. Neurodegener. 2020, 15, 29. [Google Scholar] [CrossRef]
- Hao, C.; Gao, Z.; Liu, X.; Rong, Z.; Jia, J.; Kang, K.; Guo, W.; Li, J. Intravenous administration of sodium propionate induces antidepressant or prodepressant effect in a dose dependent manner. Sci. Rep. 2020, 10, 19917. [Google Scholar] [CrossRef]
- Mondelli, V.; Vernon, A.C.; Turkheimer, F.; Dazzan, P.; Pariante, C.M. Brain microglia in psychiatric disorders. Lancet Psychiatry 2017, 4, 563–572. [Google Scholar] [CrossRef]
- Réus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef]
- Fatoba, O.; Kloster, E.; Reick, C.; Saft, C.; Gold, R.; Epplen, J.T.; Arning, L.; Ellrichmann, G. Activation of NPY-Y2 receptors ameliorates disease pathology in the R6/2 mouse and PC12 cell models of Huntington’s disease. Exp. Neurol. 2018, 302, 112–128. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. Paxinos and Franklin’s The Mouse Brain in Stereotaxic Coordinates, 5th ed.; Academic Press an Imprint of Elsevier: London, UK, 2019; ISBN 0128161582. [Google Scholar]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer Sequences |
|---|---|
| mutated Htt | 5′-CCG CTC AGG TTC TGC TTT TA-3′ sen |
| 5′-TGG AAG GAC TTG AGG GAC TC-3′ ase | |
| CAG-repeat | 5′-GGC GGC TGA GGA AGC TGA GGA G-3′ sen |
| 5′-ATG AAG GCC TTC GAG TCC CTC AAG TCC TTC-3′ ase |
| Target Gene | Primer Sequences |
|---|---|
| β-Actin | 5′ CCT CTA TGC CAA CAC ACT GC sen |
| 5′ CAT CGT ACT CCT GCT TGC TG ase | |
| 18S ribosomal RNA | 5′ GTA ACC CGT TGA ACC CCA TT sen |
| 5′ CCA TCC AAT CGG TAG TAG ase | |
| IL-6 | 5′ CCG GAG AGG AGA CTT CAC AG sen |
| 5′ GGA AAT TGG GGT AGG AAG GA ase | |
| TNFα | 5′ GCC TCT TCT CAT TCC CGC TT sen |
| 5′ CTG ATG AGA GGG AGG CCA TT ase | |
| BDNF | 5′ CTT ATG AAT CGC CAG CCA ATT CTC sen |
| 5′ TGC AGG GGC ATA GAC AAA AGG ase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
König, J.; Blusch, A.; Fatoba, O.; Gold, R.; Saft, C.; Ellrichmann-Wilms, G. Examination of Anti-Inflammatory Effects After Propionate Supplementation in the R6/2 Mouse Model of Huntington’s Disease. Int. J. Mol. Sci. 2025, 26, 3318. https://doi.org/10.3390/ijms26073318
König J, Blusch A, Fatoba O, Gold R, Saft C, Ellrichmann-Wilms G. Examination of Anti-Inflammatory Effects After Propionate Supplementation in the R6/2 Mouse Model of Huntington’s Disease. International Journal of Molecular Sciences. 2025; 26(7):3318. https://doi.org/10.3390/ijms26073318
Chicago/Turabian StyleKönig, Jennifer, Alina Blusch, Oluwaseun Fatoba, Ralf Gold, Carsten Saft, and Gisa Ellrichmann-Wilms. 2025. "Examination of Anti-Inflammatory Effects After Propionate Supplementation in the R6/2 Mouse Model of Huntington’s Disease" International Journal of Molecular Sciences 26, no. 7: 3318. https://doi.org/10.3390/ijms26073318
APA StyleKönig, J., Blusch, A., Fatoba, O., Gold, R., Saft, C., & Ellrichmann-Wilms, G. (2025). Examination of Anti-Inflammatory Effects After Propionate Supplementation in the R6/2 Mouse Model of Huntington’s Disease. International Journal of Molecular Sciences, 26(7), 3318. https://doi.org/10.3390/ijms26073318