
Microbiota of Healthy Dental Pulp Under the Omics Loupe
 , ,
, ,
Abstract
:1. Introduction
2. Results
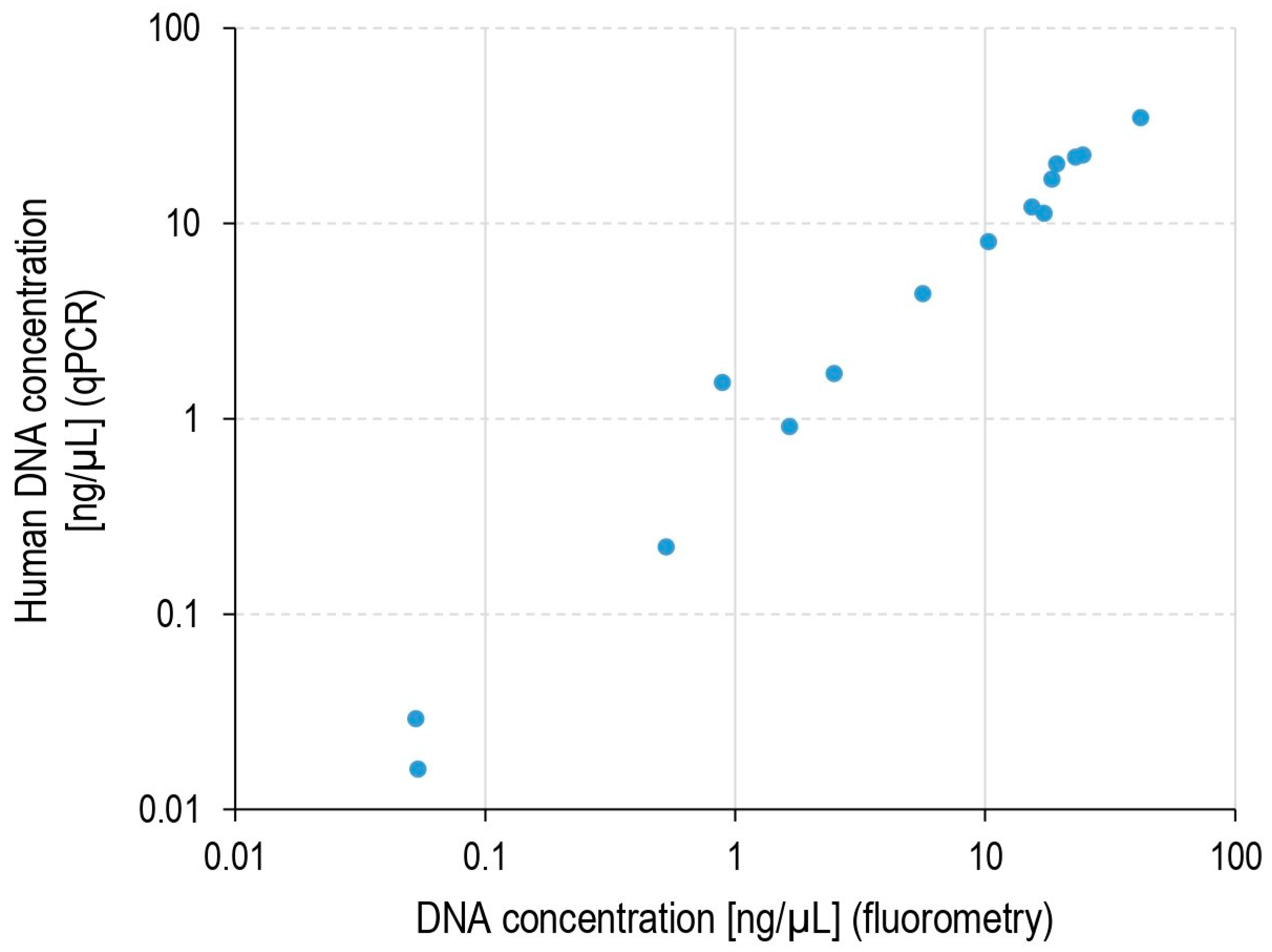
2.1. DNA Load in Pulp Samples
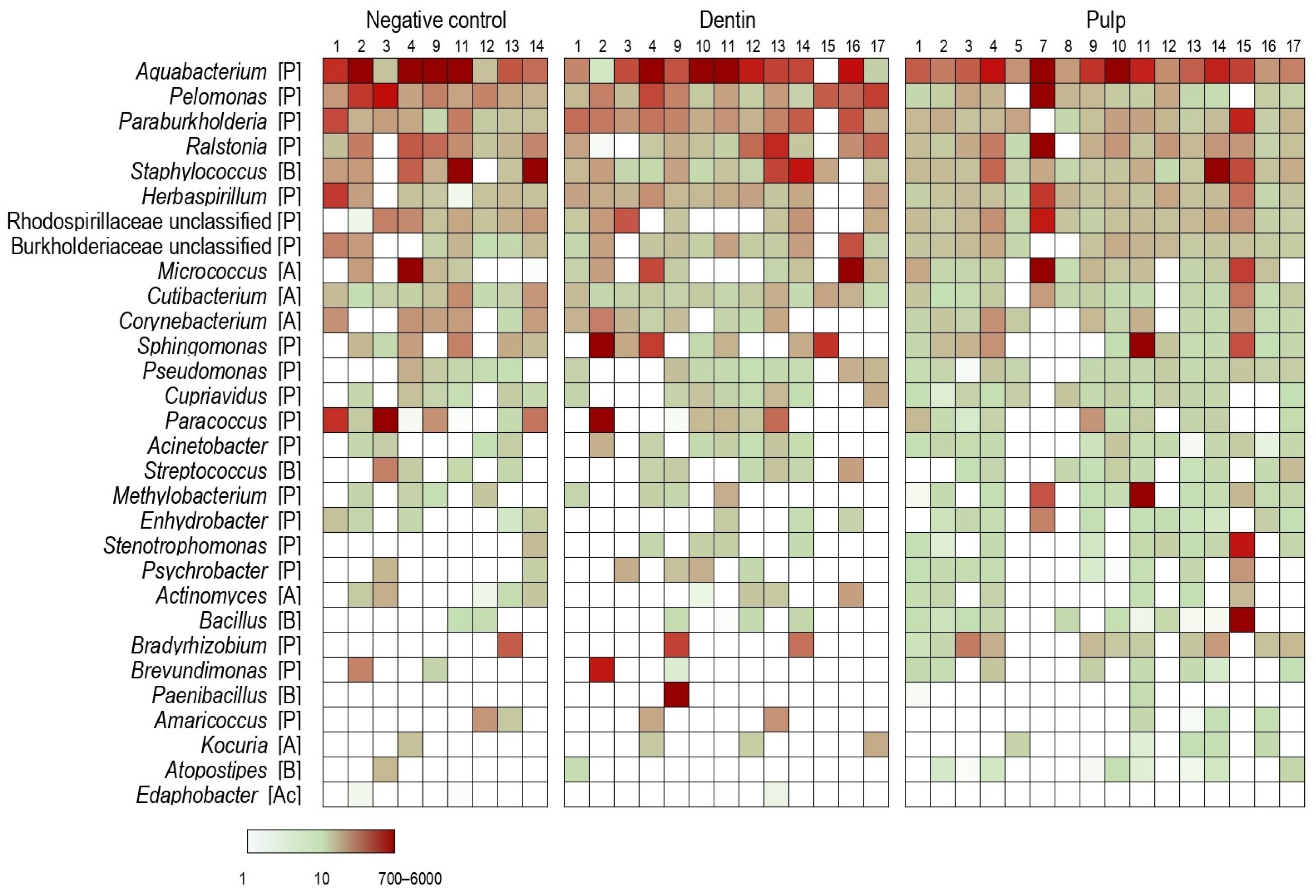
2.2. Bacteria in Dental Pulp, Root Dentin and Negative Controls
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Processing
4.2. DNA Extraction and Quantification
4.3. qPCR Assay
4.4. Amplicon Sequencing
4.5. Bioinformatics Analysis
4.6. Assessment of the Bacterial Load
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- European Society of Endodontology (ESE); Duncan, H.F.; Galler, K.M.; Tomson, P.L.; Simon, S.; El-Karim, I.; Kundzina, R.; Krastl, G.; Dammaschke, T.; Fransson, H.; et al. European Society of Endodontology position statement: Management of deep caries and the exposed pulp. Int. Endod. J. 2019, 52, 923–934. [Google Scholar] [PubMed]
- Widmer, C.; Skutas, J.; Easson, C.; Lopez, J.V.; Torneck, C.; Flax, M.; Sayin, T.C. Culture-independent characterization of the microbiome of healthy pulp. J. Endod. 2018, 44, 1132–1139.e2. [Google Scholar] [PubMed]
- Rocas, I.N.; Alves, F.R.; Rachid, C.T.; Lima, K.C.; Assuncao, I.V.; Gomes, P.N.; Siqueira, J.F., Jr. Microbiome of deep dentinal caries lesions in teeth with symptomatic irreversible pulpitis. PLoS ONE 2016, 11, e0154653. [Google Scholar]
- Bouillaguet, S.; Manoil, D.; Girard, M.; Louis, J.; Gaia, N.; Leo, S.; Schrenzel, J.; Lazarevic, V. Root microbiota in primary and secondary apical periodontitis. Front. Microbiol. 2018, 9, 2374. [Google Scholar]
- Amaral, R.R.; Braga, T.; Siqueira, J.F., Jr.; Rocas, I.N.; da Costa Rachid, C.T.C.; Oliveira, A.G.G.; de Souza Cortes, M.I.; Love, R.M. Root canal microbiome associated with asymptomatic apical periodontitis as determined by high-throughput sequencing. J. Endod. 2022, 48, 487–495. [Google Scholar]
- Siqueira, J.F., Jr.; Rocas, I.N. A critical analysis of research methods and experimental models to study the root canal microbiome. Int. Endod. J. 2022, 55 (Suppl. S1), 46–71. [Google Scholar]
- Grahn, N.; Olofsson, M.; Ellnebo-Svedlund, K.; Monstein, H.J.; Jonasson, J. Identification of mixed bacterial DNA contamination in broad-range PCR amplification of 16S rDNA V1 and V3 variable regions by pyrosequencing of cloned amplicons. FEMS Microbiol. Lett. 2003, 219, 87–91. [Google Scholar]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar]
- Boers, S.A.; Jansen, R.; Hays, J.P. Understanding and overcoming the pitfalls and biases of next-generation sequencing (NGS) methods for use in the routine clinical microbiological diagnostic laboratory. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1059–1070. [Google Scholar]
- Zaura, E.; Pappalardo, V.Y.; Buijs, M.J.; Volgenant, C.M.C.; Brandt, B.W. Optimizing the quality of clinical studies on oral microbiome: A practical guide for planning, performing, and reporting. Periodontol. 2000 2021, 85, 210–236. [Google Scholar]
- Hauser, S.; Lazarevic, V.; Tournoud, M.; Ruppé, E.; Santiago Allexant, E.; Guigon, G.; Schicklin, S.; Lanet, V.; Girard, M.; Mirande, C.; et al. A metagenomics method for the quantitative detection of bacterial pathogens causing hospital-associated and ventilator-associated pneumonia. Microbiol. Spectr. 2023, 11, e01294-23. [Google Scholar]
- Stammler, F.; Glasner, J.; Hiergeist, A.; Holler, E.; Weber, D.; Oefner, P.J.; Gessner, A.; Spang, R. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome 2016, 4, 28. [Google Scholar]
- Venkataraman, A.; Parlov, M.; Hu, P.; Schnell, D.; Wei, X.; Tiesman, J.P. Spike-in genomic DNA for validating performance of metagenomics workflows. Biotechniques 2018, 65, 315–321. [Google Scholar] [PubMed]
- Escapa, I.F.; Chen, T.; Huang, Y.; Gajare, P.; Dewhirst, F.E.; Lemon, K.P. New insights into human nostril microbiome from the expanded Human Oral Microbiome Database (eHOMD): A resource for the microbiome of the human aerodigestive tract. mSystems 2018, 3, e00187-18. [Google Scholar]
- Moossavi, S.; Fehr, K.; Khafipour, E.; Azad, M.B. Repeatability and reproducibility assessment in a large-scale population-based microbiota study: Case study on human milk microbiota. Microbiome 2021, 9, 41. [Google Scholar]
- Tsuzukibashi, O.; Uchibori, S.; Kobayashi, T.; Saito, M.; Umezawa, K.; Ohta, M.; Shinozaki-Kuwahara, N. A selective medium for the isolation of Microbacterium species in oral cavities. J. Microbiol. Methods 2015, 116, 60–65. [Google Scholar]
- Chou, Y.J.; Sheu, S.Y.; Sheu, D.S.; Wang, J.T.; Chen, W.M. Schlegelella aquatica sp. nov., a novel thermophilic bacterium isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 2006, 56, 2793–2797. [Google Scholar]
- Cotta, M.A.; Whitehead, T.R.; Collins, M.D.; Lawson, P.A. Atopostipes suicloacale gen. nov., sp. nov., isolated from an underground swine manure storage pit. Anaerobe 2004, 10, 191–195. [Google Scholar]
- Rasmussen, M.; Mohlin, A.W.; Nilson, B. From contamination to infective endocarditis—A population-based retrospective study of Corynebacterium isolated from blood cultures. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 113–119. [Google Scholar]
- Arruda-Vasconcelos, R.; Chiarelli-Neto, V.M.; Louzada, L.M.; Aveiro, E.; Alves-Silva, E.G.; de-Jesus-Soares, A.; Ferraz, C.C.R.; Almeida, J.F.A.; Marciano, M.A.; Pecorari, V.G.A.; et al. Quantitative analysis of culturable bacteria, levels of endotoxins, inflammatory mediators and substance P in teeth with symptomatic irreversible pulpitis and in teeth with vital normal pulp tissues. Int. Endod. J. 2023, 56, 827–836. [Google Scholar]
- Martins, C.C.; Lockhart, P.B.; Firmino, R.T.; Kilmartin, C.; Cahill, T.J.; Dayer, M.; Occhi-Alexandre, I.G.P.; Lai, H.; Ge, L.; Thornhill, M.H. Bacteremia following different oral procedures: Systematic review and meta-analysis. Oral Dis. 2024, 30, 846–854. [Google Scholar] [PubMed]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome datasets are compositional: And this is not optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar]
- Siqueira, J.; Rôças, I. Microbiology of endodontic infections. In Cohen’s Pathways of the Pulp, 11th ed.; Hargreaves, K.M., Cohen, S.M., Berman, L.H., Eds.; Mosby Elsevier: St. Louis, MO, USA, 2016; pp. 599–629. [Google Scholar]
- Leo, S.; Lazarevic, V.; von Dach, E.; Kaiser, L.; Prendki, V.; Schrenzel, J.; Huttner, B.D.; Huttner, A. Effects of antibiotic duration on the intestinal microbiota and resistome: The PIRATE RESISTANCE project, a cohort study nested within a randomized trial. EBioMedicine 2021, 71, 103566. [Google Scholar]
- Apostolopoulos, N.; Glaeser, S.P.; Bagwe, R.; Janssen, S.; Mayer, U.; Ewers, C.; Kampfer, P.; Neiger, R.; Thom, N. Description and comparison of the skin and ear canal microbiota of non-allergic and allergic German shepherd dogs using next generation sequencing. PLoS ONE 2021, 16, e0250695. [Google Scholar]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRXiv 2016. [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.; Schmidt, T.M. rrnDB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Taxon | Abundance [Median (Minimum; Q1; Q3; Maximum)] | p * | ||||
|---|---|---|---|---|---|---|
| Negative Control | Dentin | Pulp | Pulp vs. Negative Control | Pulp vs. Dentin | ||
| Species | Atopostipes unclassified | 0 (0;0;0;0.12) | 0 (0;0;0;0.36) | 2.1 (0;0;6.5;15) | 0.022 | 0.009 |
| Bacillus gottheilii | 0 (0;0;0;0) | 0 (0;0;0;0) | 0.014 (0;0;0.04;5995) | 0.022 | 0.006 | |
| Brevibacterium ammoniilyticum | 0 (0;0;0;0) | 0 (0;0;0;0) | 0 (0;0;5.9;32) | 0.059 | 0.036 | |
| Brevundimonas albigilva | 0 (0;0;0.13;295) | 0 (0;0;0.035;0.61) | 0 (0;0;17;65) | 0.402 | 0.035 | |
| Corynebacterium afermentans | 0 (0;0;0;215) | 0 (0;0;0;0) | 0 (0;0;1.7;119) | 0.402 | 0.036 | |
| Cutibacterium acnes | 60 (19;38;123;267) | 58.4 (24;45;101;154) | 30 (0;16;54;216) | 0.008 | 0.027 | |
| Edaphobacter unclassified | 0 (0;0;0.65;3) | 0.50 (0;0;0.83;4.2) | 0 (0;0;0;0.77) | 0.100 | 0.042 | |
| Finegoldia magna | 0 (0;0;0;0.085) | 0 (0;0;0;0) | 0 (0;0;5.4;26) | 0.093 | 0.022 | |
| Fretibacterium unclassified | 0 (0;0;0;0) | 0 (0;0;0;0) | 0 (0;0;0.039;304) | 0.036 | 0.022 | |
| Fusobacterium nucleatum | 0 (0;0;0;36) | 0 (0;0;0;0) | 0 (0;0;8.7;36) | 0.295 | 0.036 | |
| Corynebacterium KI515728_s | 0 (0;0;0;22) | 0 (0;0;0;0.018) | 0 (0;0;8.1;18) | 0.402 | 0.022 | |
| Microbacterium unclassified | 0 (0;0;0;46) | 0 (0;0;0;0.035) | 0 (0;0;16;36) | 0.106 | 0.022 | |
| Paraburkholderia unclassified | 150 (35;101;215;473) | 296 (0;171;345;446) | 132 (0;93;173;593) | 0.359 | 0.040 | |
| Paracoccus aestuarii | 2.5 (0;0.23;183;1321) | 0 (0;0;0.19;2.2) | 0 (0;0;12;27) | 0.076 | 0.032 | |
| Pelomonas unclassified | 230 (152;198;307;663) | 226 (50;122;385;507) | 71 (0;36;144;1656) | 0.004 | 0.006 | |
| Pseudomonas poae | 0 (0;0;0.14;51) | 0 (0;0;0.093;17) | 11 (0;1.8;21;50) | 0.098 | 0.003 | |
| Rothia amarae | 0 (0;0;0;111) | 0 (0;0;0;30) | 0 (0;0;3.5;157) | 0.100 | 0.035 | |
| Schlegelella aquatica | 0 (0;0;0;0) | 0 (0;0;0;0.21) | 0.030 (0;0;11;136) | 0.036 | 0.014 | |
| Genus | Treponema | 0 (0;0;0;0) | 0 (0;0;0;0) | 0 (0;0;0.12;1482) | 0.036 | 0.036 |
| Bacillus | 0 (0;0;0.36;21) | 0.039 (0;0;1.6;36) | 6.3 (0;0.058;20;5995) | 0.039 | 0.126 | |
| Cutibacterium | 60 (19;57;123;268) | 73 (24;45;121;193) | 37 (0;22;79;339) | 0.020 | 0.244 | |
| Edaphobacter | 0 (0;0;0.65;3) | 0.50 (0;0;0.83;4.2) | 0 (0;0;0;0.77) | 0.100 | 0.042 | |
| Finegoldia | 0 (0;0;0;0.085) | 0 (0;0;0;0) | 0 (0;0;5.4;26) | 0.093 | 0.022 | |
| Fretibacterium | 0 (0;0;0;0) | 0 (0;0;0;0) | 0.01 (0;0;0.068;447) | 0.036 | 0.014 | |
| Fusobacterium | 0 (0;0;0;36) | 0 (0;0;0;0) | 0 (0;0;8.7;36) | 0.295 | 0.036 | |
| Microbacterium | 0 (0;0;0;46) | 0 (0;0;0;0.035) | 0 (0;0;16;36) | 0.106 | 0.022 | |
| Paraburkholderia | 150 (35;101;215;473) | 296 (0;171;345;446) | 132 (0;93;173;593) | 0.359 | 0.040 | |
| Pelomonas | 230 (152;198;307;663) | 226 (50;122;385;507) | 71 (0;36;144;1656) | 0.004 | 0.006 | |
| Pseudoalteromonas | 0 (0;0;0.011;23) | 0 (0;0;0;0.0044) | 0 (0;0;0.012;11) | 0.554 | 0.036 | |
| Schlegelella | 0 (0;0;0;0) | 0 (0;0;0;0.21) | 0.030 (0;0;11;136) | 0.036 | 0.014 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bérard, A.; Mauffrey, F.; Gaïa, N.; Perez, A.; Chaabane, C.; Schrenzel, J.; Leprince, J.G.; Bouillaguet, S.; Lazarevic, V. Microbiota of Healthy Dental Pulp Under the Omics Loupe. Int. J. Mol. Sci. 2025, 26, 3232. https://doi.org/10.3390/ijms26073232
Bérard A, Mauffrey F, Gaïa N, Perez A, Chaabane C, Schrenzel J, Leprince JG, Bouillaguet S, Lazarevic V. Microbiota of Healthy Dental Pulp Under the Omics Loupe. International Journal of Molecular Sciences. 2025; 26(7):3232. https://doi.org/10.3390/ijms26073232
Chicago/Turabian StyleBérard, Alan, Florian Mauffrey, Nadia Gaïa, Alexandre Perez, Chiraz Chaabane, Jacques Schrenzel, Julian Grégoire Leprince, Serge Bouillaguet, and Vladimir Lazarevic. 2025. "Microbiota of Healthy Dental Pulp Under the Omics Loupe" International Journal of Molecular Sciences 26, no. 7: 3232. https://doi.org/10.3390/ijms26073232
APA StyleBérard, A., Mauffrey, F., Gaïa, N., Perez, A., Chaabane, C., Schrenzel, J., Leprince, J. G., Bouillaguet, S., & Lazarevic, V. (2025). Microbiota of Healthy Dental Pulp Under the Omics Loupe. International Journal of Molecular Sciences, 26(7), 3232. https://doi.org/10.3390/ijms26073232