
Novel Compounds Target Aberrant Calcium Signaling in the Treatment of Relapsed High-Risk Neuroblastoma

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Screening a Library of Synthesized Compounds
2.2. Chemical Structures of Compound 248 and Compound 249
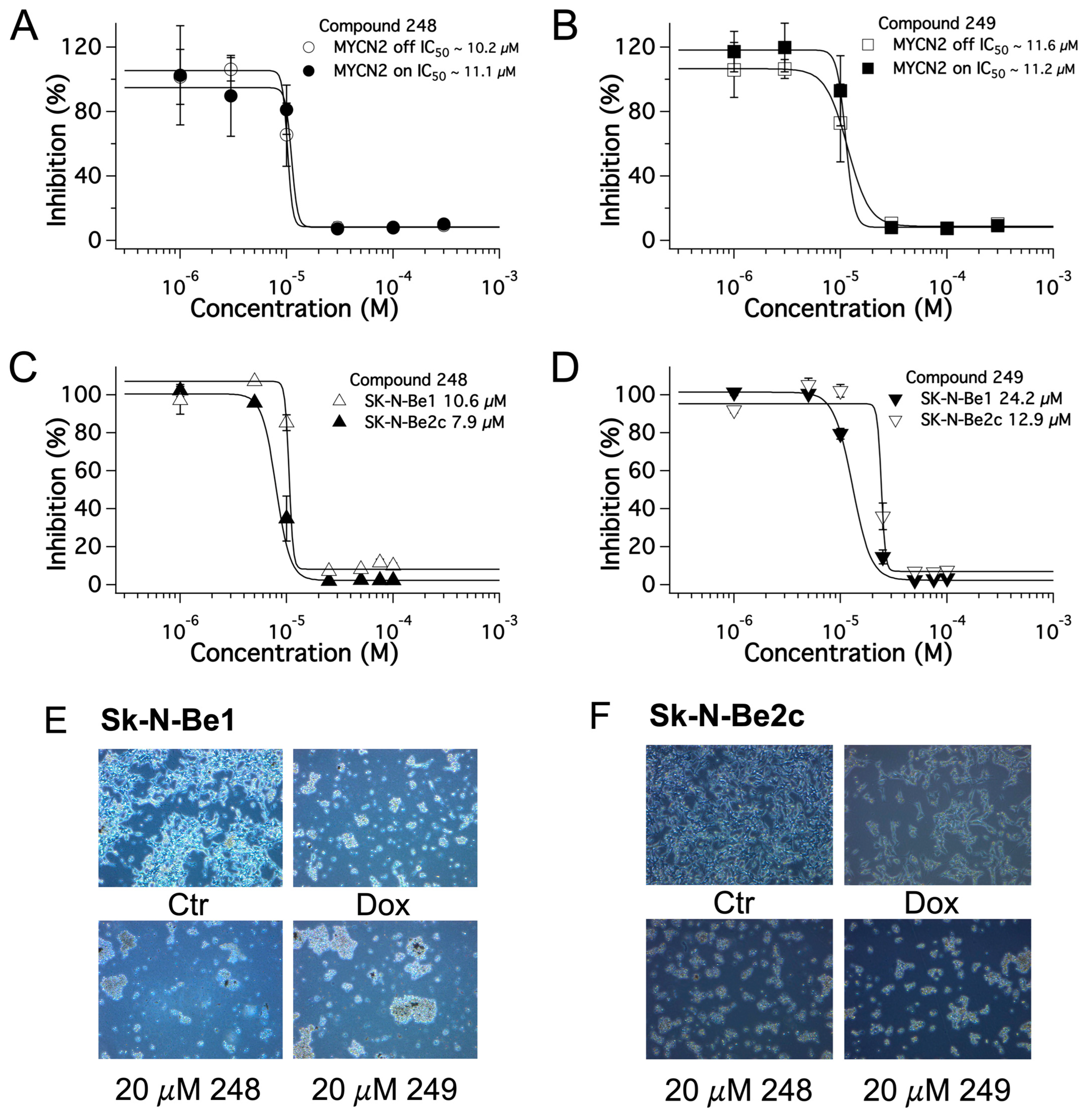
2.3. Dose-Dependent Effects of Compound 248 and Compound 249 in NB
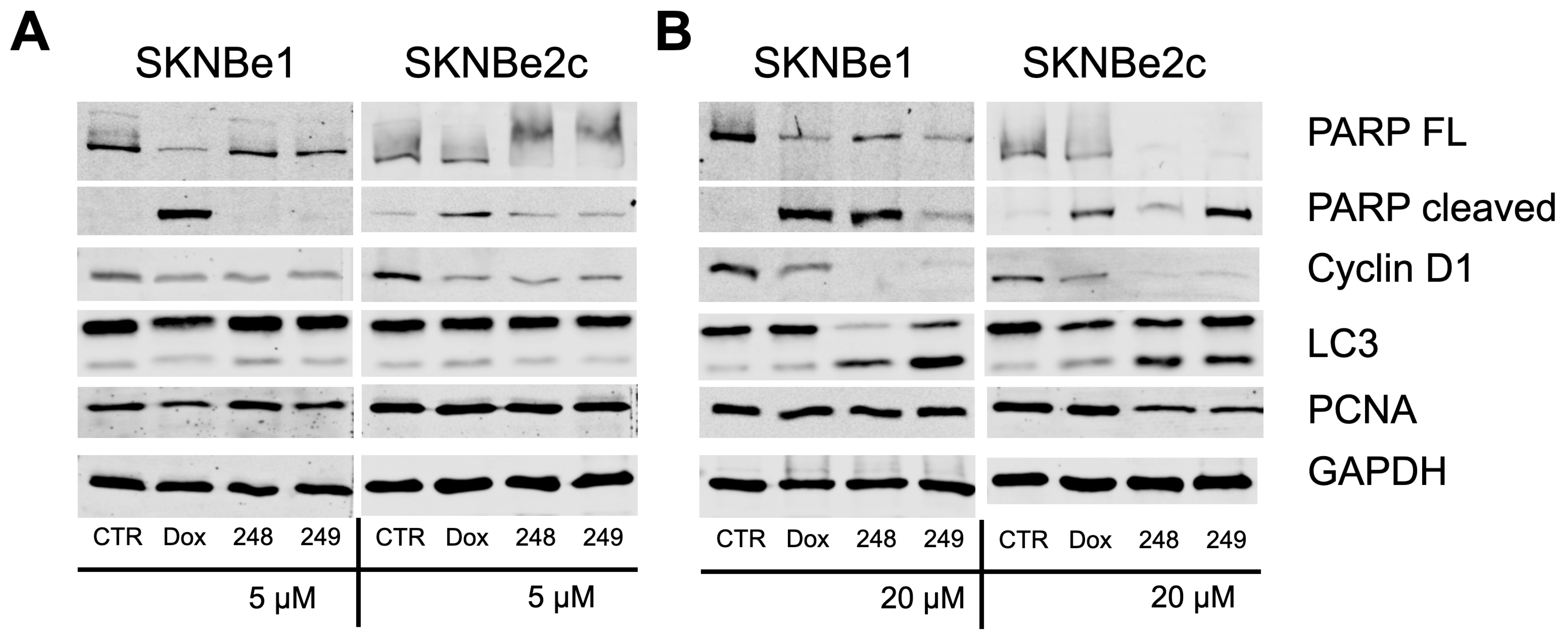
2.4. Compounds 248 and 249 Decrease Cell Cycle Proteins and Increase Apoptosis Markers
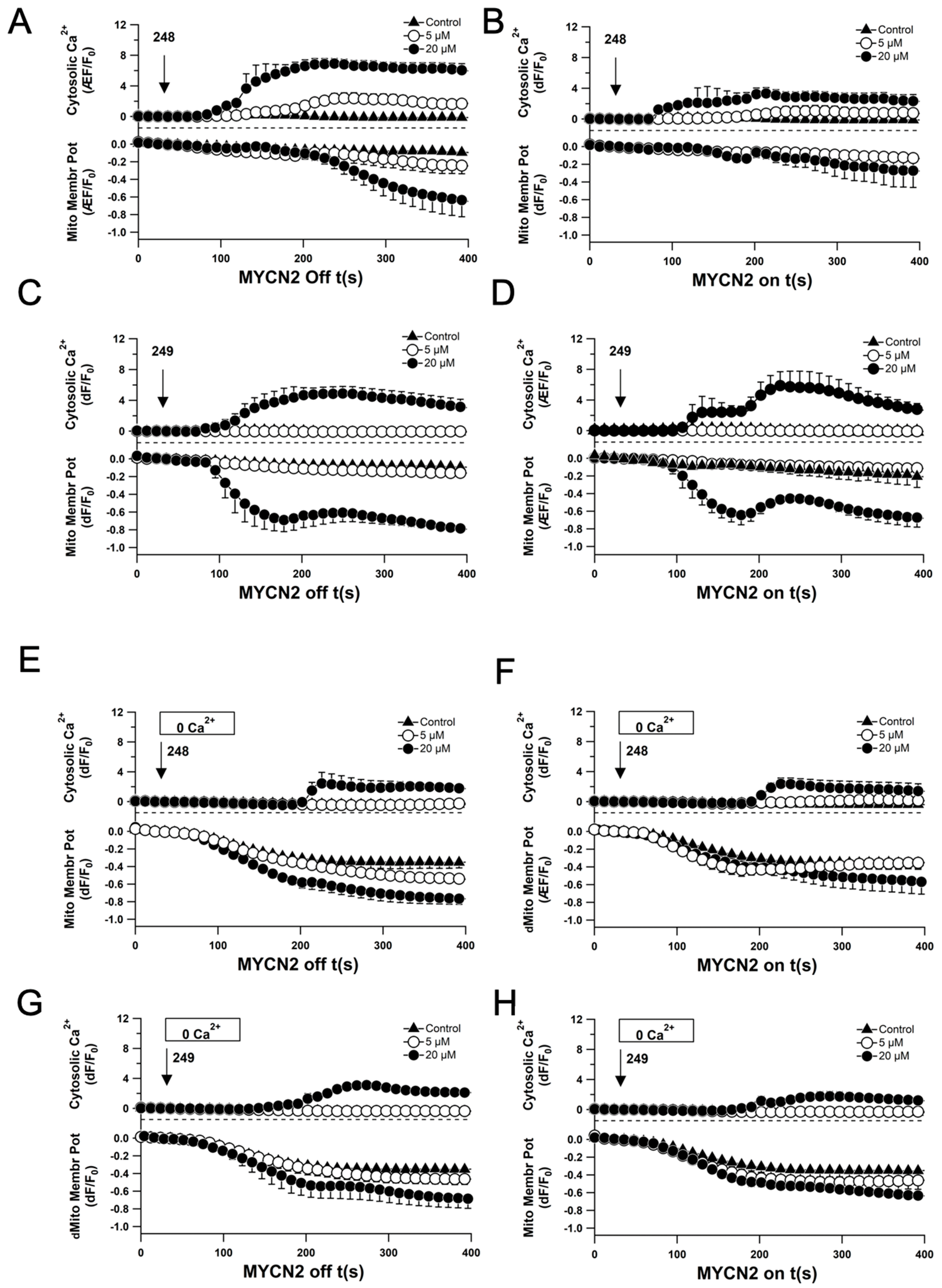
2.5. Compounds 248 and 249 Induce Ca2+ Signaling and Loss of Mitochondrial Membrane Potential
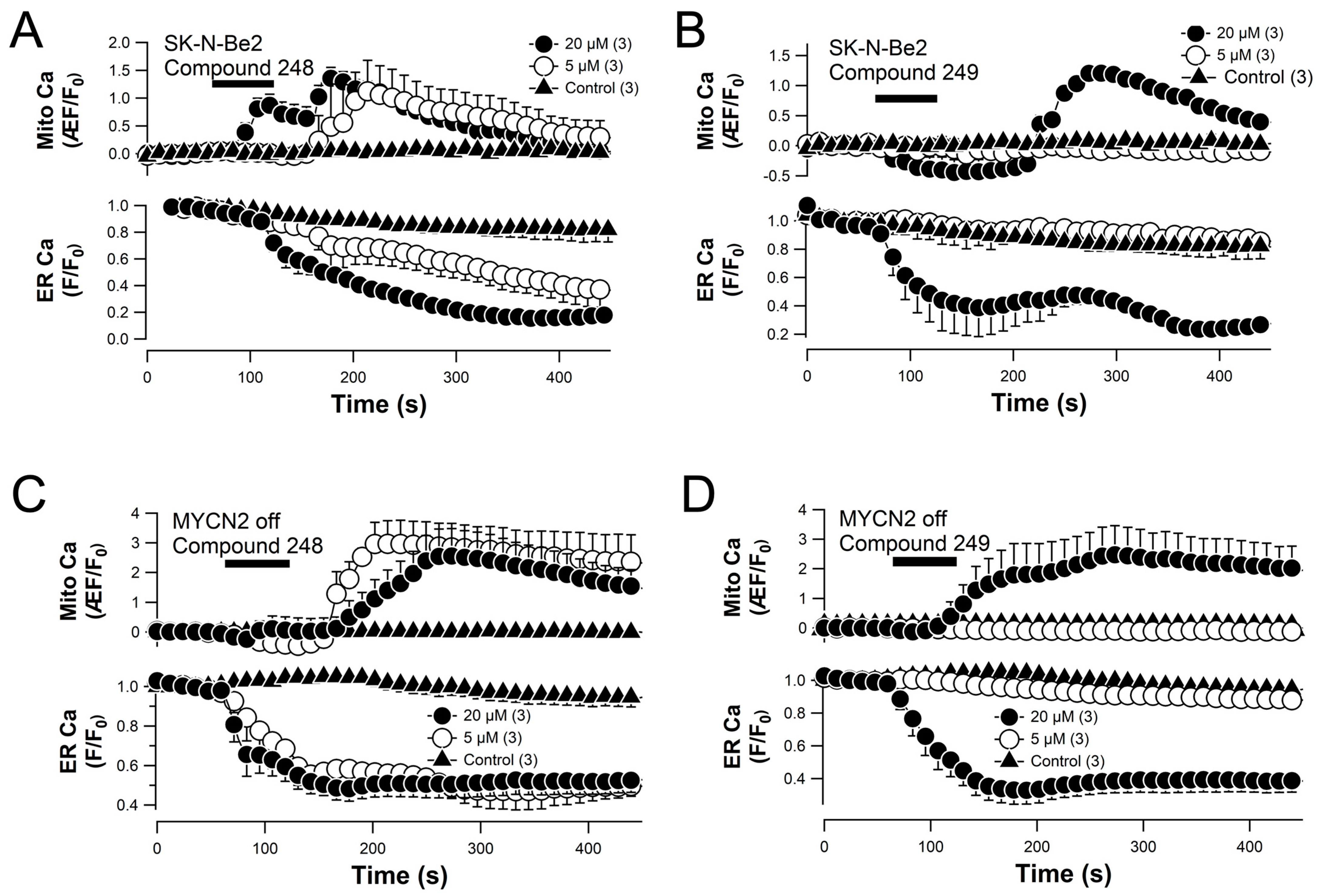
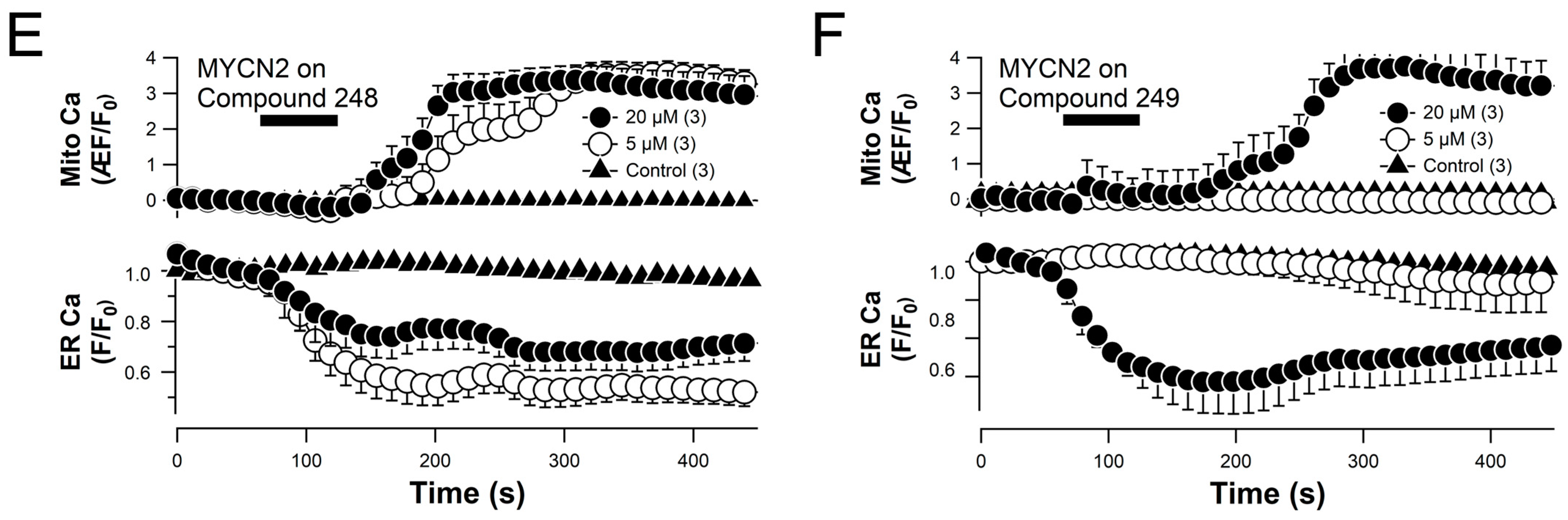
2.6. Compounds 248 and 249 Promote ER Calcium Release and Mitochondrial Calcium Uptake
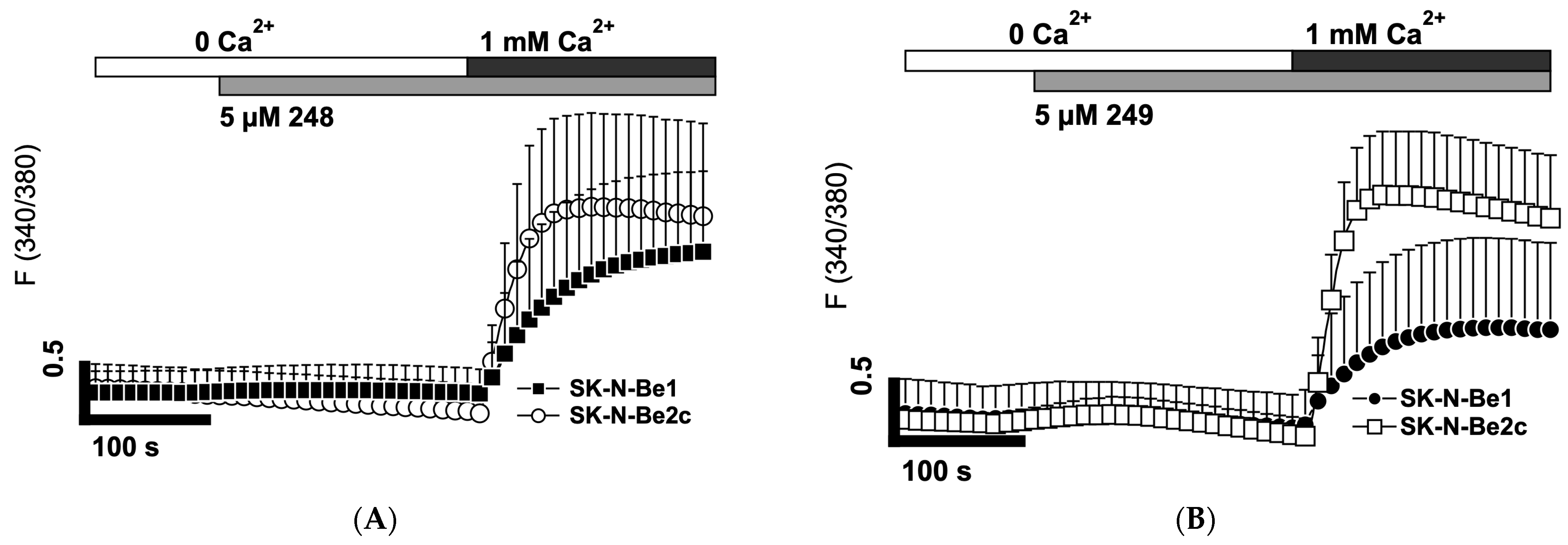
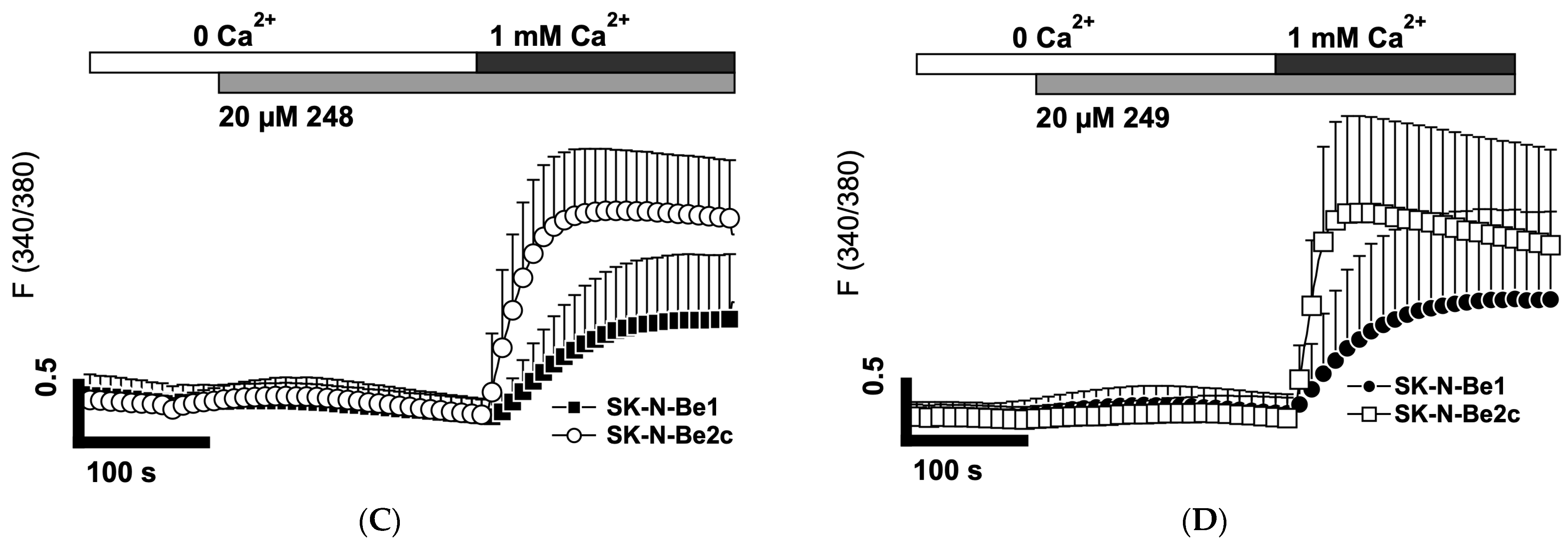
2.7. Compounds 248 and 249 Activate Store-Operated Calcium Entry
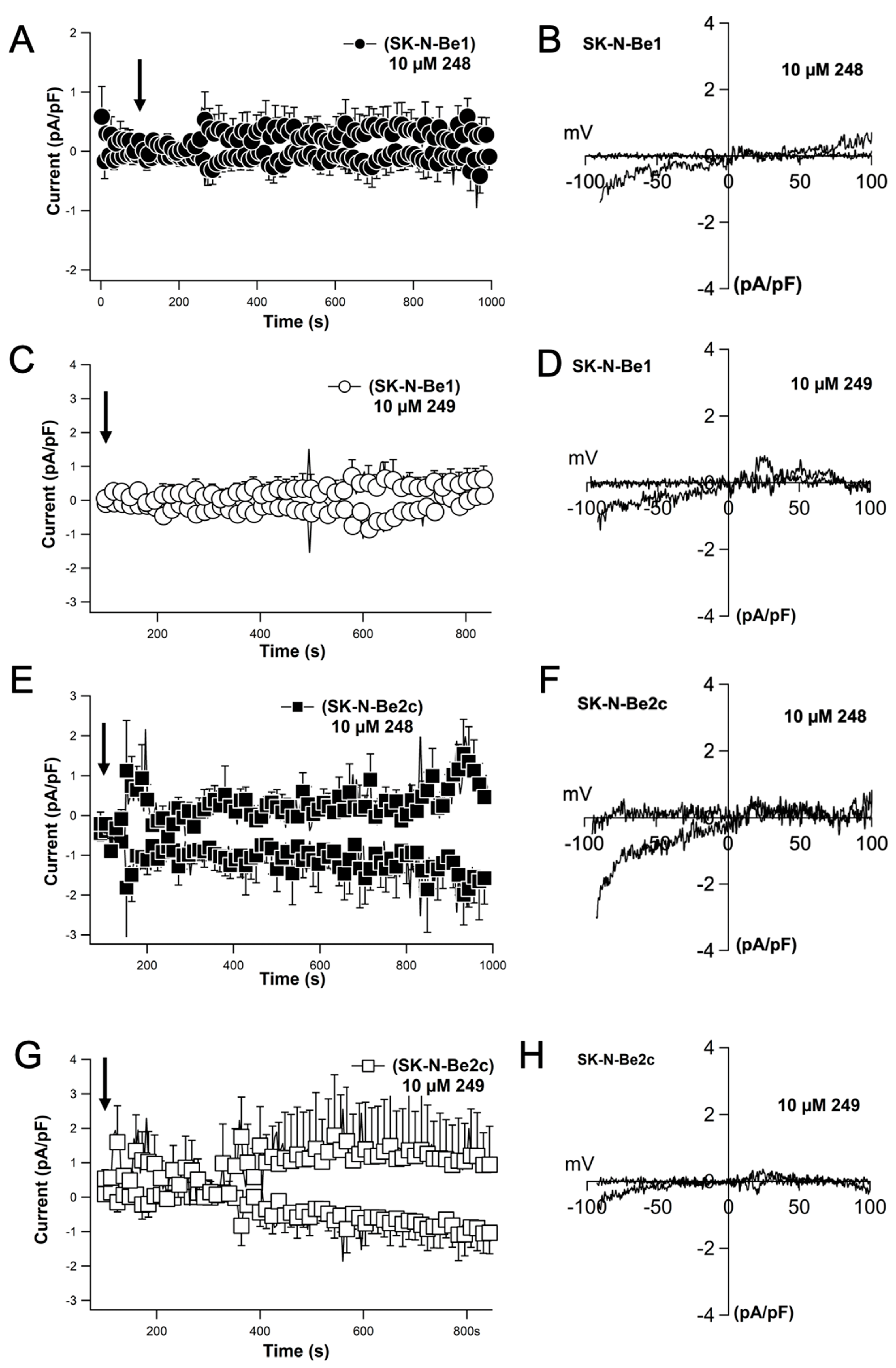
2.8. Compounds 248 and 249 Activate Store-Operated Calcium Entry Resembling a Current That Is Characteristic of ORAI Channels
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals
4.3. Synthesis of Compounds
4.4. Changes in Intracellular Free Calcium and Mitochondrial Membrane Potential
4.5. Time Lapsed Live-Cell Imaging
4.6. Electrophysiology
4.7. Sulforhodamine B Assay
4.8. Western Blot Analysis
4.9. Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brodeur, G.M.; Fong, C.T. Molecular biology and genetics of human neuroblastoma. Cancer Genet. Cytogenet. 1989, 41, 153–174. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Varela, C.R.; Light, J.E.; Kolla, V.; Evans, A.E. Trk Receptor Expression and Inhibition in Neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [Google Scholar] [CrossRef]
- Carlson, L.-M.; De Geer, A.; Sveinbjørnsson, B.; Orrego, A.; Martinsson, T.; Kogner, P.; Levitskaya, J. The microenvironment of human neuroblastoma supports the activation of tumor-associated T lymphocytes. OncoImmunology 2013, 2, e23618. [Google Scholar] [CrossRef]
- Pistoia, V.; Morandi, F.; Bianchi, G.; Pezzolo, A.; Prigione, I.; Raffaghello, L. Immunosuppressive Microenvironment in Neuroblastoma. Front. Oncol. 2013, 3, 167. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; Baruchel, S.; Irwin, M.S. Current and future strategies for relapsed neuroblastoma: Challenges on the road to precision therapy. J. Pediatr. Hematol. Oncol. 2013, 35, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Lange, I.; Espinoza-Fuenzalida, I.; Ali, M.W.; Serrano, L.E.; Koomoa, D.T. FTY-720 induces apoptosis in neuroblastoma via multiple signaling pathways. Oncotarget 2017, 8, 109985–109999. [Google Scholar] [CrossRef] [PubMed]
- Lange, I.; Koomoa, D.L. MycN promotes TRPM7 expression and cell migration in neuroblastoma through a process that involves polyamines. FEBS Open Bio. 2014, 4, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Lange, I.; Koster, J.; Koomoa, D.T. Calcium signaling regulates fundamental processes involved in Neuroblastoma progression. Cell Calcium 2019, 82, 102052. [Google Scholar] [CrossRef]
- Hirschler-Laszkiewicz, I.; Chen, S.-J.; Bao, L.; Wang, J.; Zhang, X.-Q.; Shanmughapriya, S.; Keefer, K.; Madesh, M.; Cheung, J.Y.; Miller, B.A. The human ion channel TRPM2 modulates neuroblastoma cell survival and mitochondrial function through Pyk2, CREB, and MCU activation. Am. J. Physiol. Cell Physiol. 2018, 315, C571–C586. [Google Scholar] [CrossRef]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef]
- Park, Y.J.; Yoo, S.A.; Kim, M.; Kim, W.U. The Role of Calcium-Calcineurin-NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef]
- Go, C.K.; Hooper, R.; Aronson, M.R.; Schultz, B.; Cangoz, T.; Nemani, N.; Zhang, Y.; Madesh, M.; Soboloff, J. The Ca(2+) export pump PMCA clears near-membrane Ca(2+) to facilitate store-operated Ca(2+) entry and NFAT activation. Sci. Signal. 2019, 12, p.eaaw2627. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, C.; He, L. ORAI Ca(2+) Channels in Cancers and Therapeutic Interventions. Biomolecules 2024, 14, 417. [Google Scholar] [CrossRef] [PubMed]
- Rubaiy, H.N. ORAI Calcium Channels: Regulation, Function, Pharmacology, and Therapeutic Targets. Pharmaceuticals 2023, 16, 162. [Google Scholar] [CrossRef]
- Garcia-Sancho, J. The coupling of plasma membrane calcium entry to calcium uptake by endoplasmic reticulum and mitochondria. J. Physiol. 2014, 592, 261–268. [Google Scholar] [CrossRef]
- Noble, M.; Lin, Q.-T.; Sirko, C.; Houpt, J.A.; Novello, M.J.; Stathopulos, P.B. Structural Mechanisms of Store-Operated and Mitochondrial Calcium Regulation: Initiation Points for Drug Discovery. Int. J. Mol. Sci. 2020, 21, 3642. [Google Scholar] [CrossRef]
- Deak, A.T.; Blass, S.; Khan, M.J.; Groschner, L.N.; Waldeck-Weiermair, M.; Hallström, S.; Graier, W.F.; Malli, R. Inositol-1,4,5-trisphosphate (IP3)-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J. Cell Sci. 2014, 127, 2944–2955. [Google Scholar] [CrossRef]
- Gualdani, R.; de Clippele, M.; Ratbi, I.; Gailly, P.; Tajeddine, N. Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma. Cancers 2019, 11, 430. [Google Scholar] [CrossRef]
- Poletto, V.; Dragoni, S.; Lim, D.; Biggiogera, M.; Aronica, A.; Cinelli, M.; De Luca, A.; Rosti, V.; Porta, C.; Guerra, G.; et al. Endoplasmic Reticulum Ca(2+) Handling and Apoptotic Resistance in Tumor-Derived Endothelial Colony Forming Cells. J. Cell. Biochem. 2016, 117, 2260–2271. [Google Scholar] [CrossRef]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, L.; Matsuda, T.; Zhao, Y.; Rebane, A.; Drobizhev, M.; Chang, Y.F.; Araki, S.; Arai, Y.; March, K.; et al. Improved orange and red Ca2+ indicators and photophysical considerations for optogenetic applications. ACS Chem. Neurosci. 2013, 4, 963–972. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Slack, A.; Chen, Z.; Tonelli, R.; Pule, M.; Hunt, L.; Pession, A.; Shohet, J.M. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc. Natl. Acad. Sci. USA 2005, 102, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Tweddle, D.A.; Malcolm, A.J.; Bown, N.; Pearson, A.D.; Lunec, J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. 2001, 61, 8–13. [Google Scholar]
- Feng, L.; Maddox, M.M.; Alam, Z.; Tsutsumi, L.S.; Narula, G.; Bruhn, D.F.; Wu, X.; Sandhaus, S.; Lee, R.B.; Simmons, C.J.; et al. Synthesis, Structure–Activity Relationship Studies, and Antibacterial Evaluation of 4-Chromanones and Chalcones, as Well as Olympicin A and Derivatives. J. Med. Chem. 2014, 57, 8398–8420. [Google Scholar] [CrossRef]
- Koomoa, D.-L.T.; Geerts, D.; Lange, I.; Koster, J.; Pegg, A.E.; Feith, D.J.; Bachmann, A.S. DFMO/eflornithine inhibits migration and invasion downstream of MYCN and involves p27Kip1 activity in neuroblastoma. Int. J. Oncol. 2013, 42, 1219–1228. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 248 (µM) | 249 (µM) | |
|---|---|---|
| MYCN2 (− doxy) | 10.2 | 11.6 |
| MYCN2 (+ doxy) | 11.1 | 11.2 |
| SK-N-Be1 | 10.6 | 24.2 |
| SK-N-Be2(c) | 7.9 | 12.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koomoa, D.-L.T.; Sunada, N.; Espinoza-Fuenzalida, I.; Tacdol, D.; Shackleford, M.; Feng, L.; Sun, D.; Lange, I. Novel Compounds Target Aberrant Calcium Signaling in the Treatment of Relapsed High-Risk Neuroblastoma. Int. J. Mol. Sci. 2025, 26, 3180. https://doi.org/10.3390/ijms26073180
Koomoa D-LT, Sunada N, Espinoza-Fuenzalida I, Tacdol D, Shackleford M, Feng L, Sun D, Lange I. Novel Compounds Target Aberrant Calcium Signaling in the Treatment of Relapsed High-Risk Neuroblastoma. International Journal of Molecular Sciences. 2025; 26(7):3180. https://doi.org/10.3390/ijms26073180
Chicago/Turabian StyleKoomoa, Dana-Lynn T., Nathan Sunada, Italo Espinoza-Fuenzalida, Dustin Tacdol, Madeleine Shackleford, Li Feng, Dianqing Sun, and Ingo Lange. 2025. "Novel Compounds Target Aberrant Calcium Signaling in the Treatment of Relapsed High-Risk Neuroblastoma" International Journal of Molecular Sciences 26, no. 7: 3180. https://doi.org/10.3390/ijms26073180
APA StyleKoomoa, D.-L. T., Sunada, N., Espinoza-Fuenzalida, I., Tacdol, D., Shackleford, M., Feng, L., Sun, D., & Lange, I. (2025). Novel Compounds Target Aberrant Calcium Signaling in the Treatment of Relapsed High-Risk Neuroblastoma. International Journal of Molecular Sciences, 26(7), 3180. https://doi.org/10.3390/ijms26073180