
Proteasome Inhibitor MG-132 and PKC-ι-Specific Inhibitor ICA-1S Degrade Mutant p53 and Induce Apoptosis in Ovarian Cancer Cell Lines

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ICA-1S and MG-132 Have an Inhibitory Effect on Ovarian Cancer Cell Proliferation
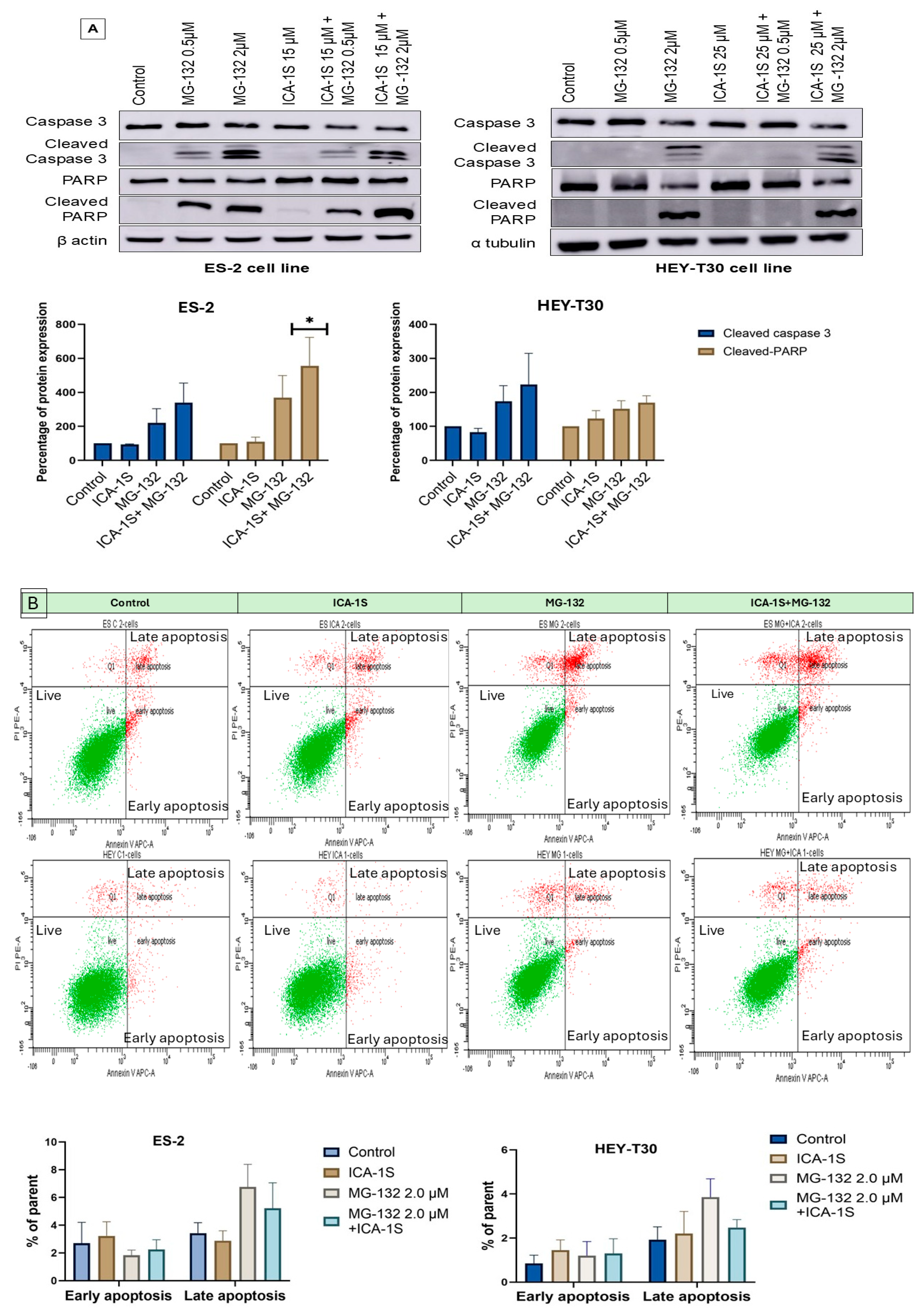
2.2. MG-132 Induces Ovarian Cancer Cell Death
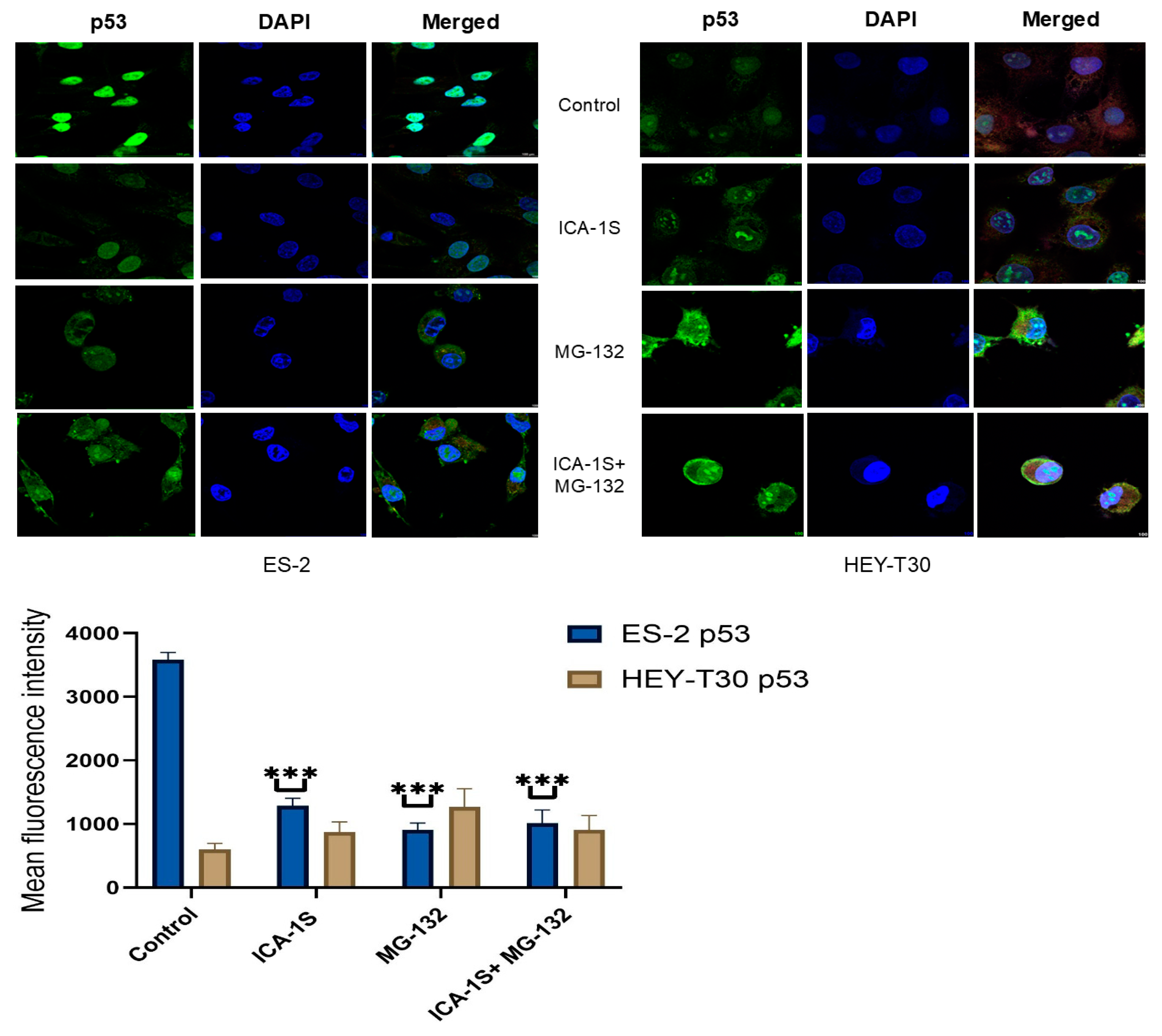
2.3. Level of WT p53 and Mutp53 Are Regulated Differently by MG-132 and ICA-1S
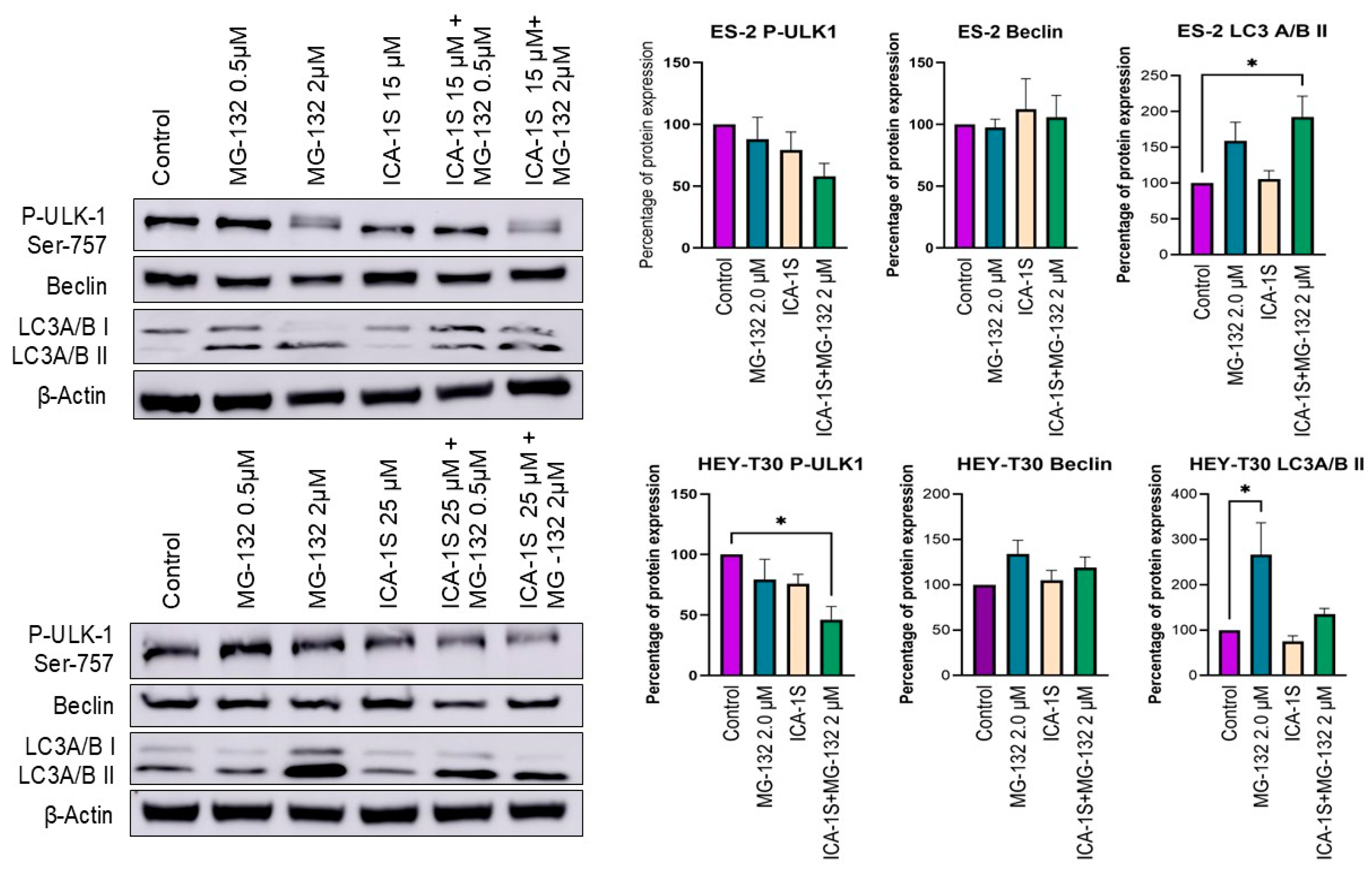
2.4. Autophagy May Play a Role in the Degradation of Mutp53 by MG-132
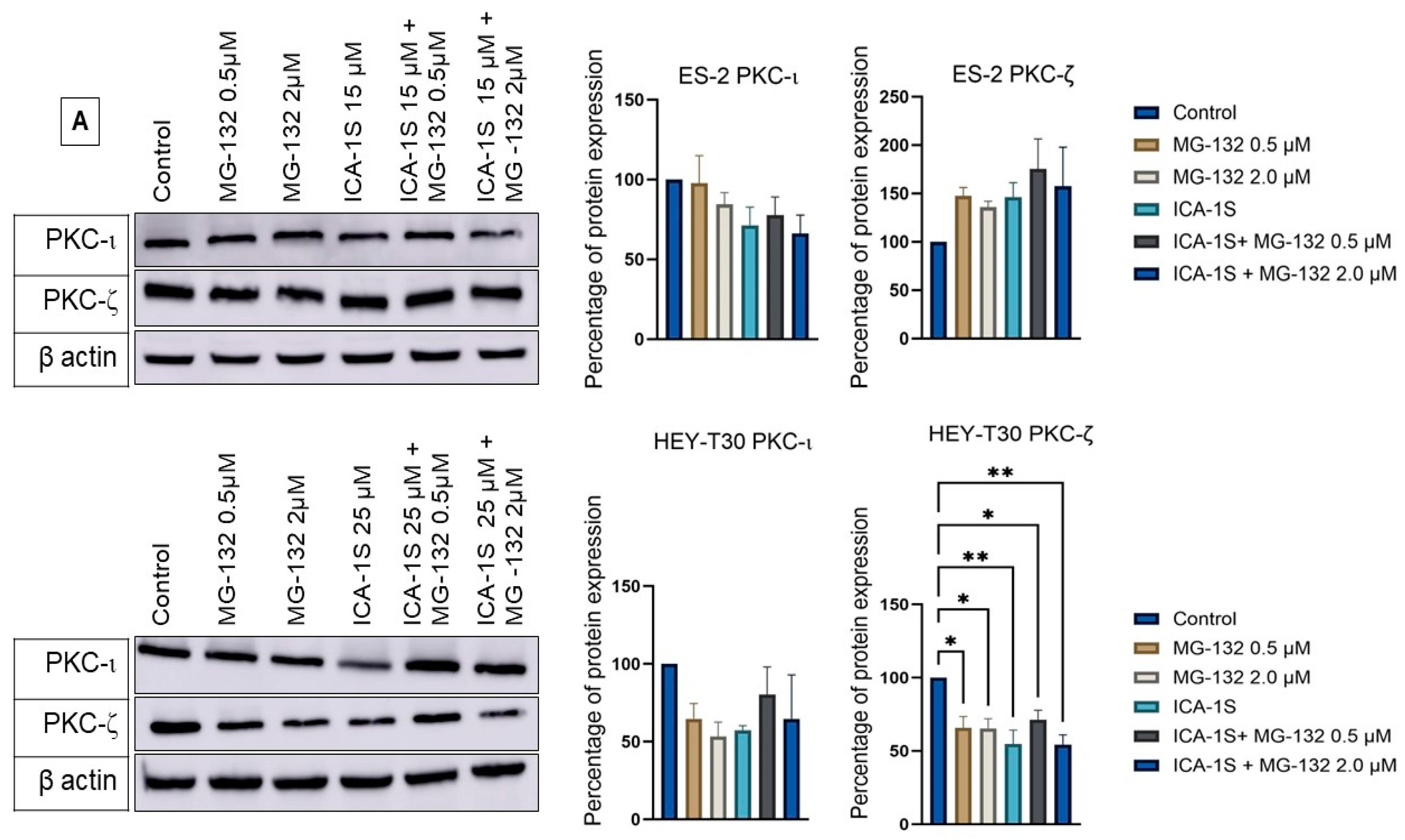
2.5. Level of Atypical Kinases and Association of PKC-ι with p53
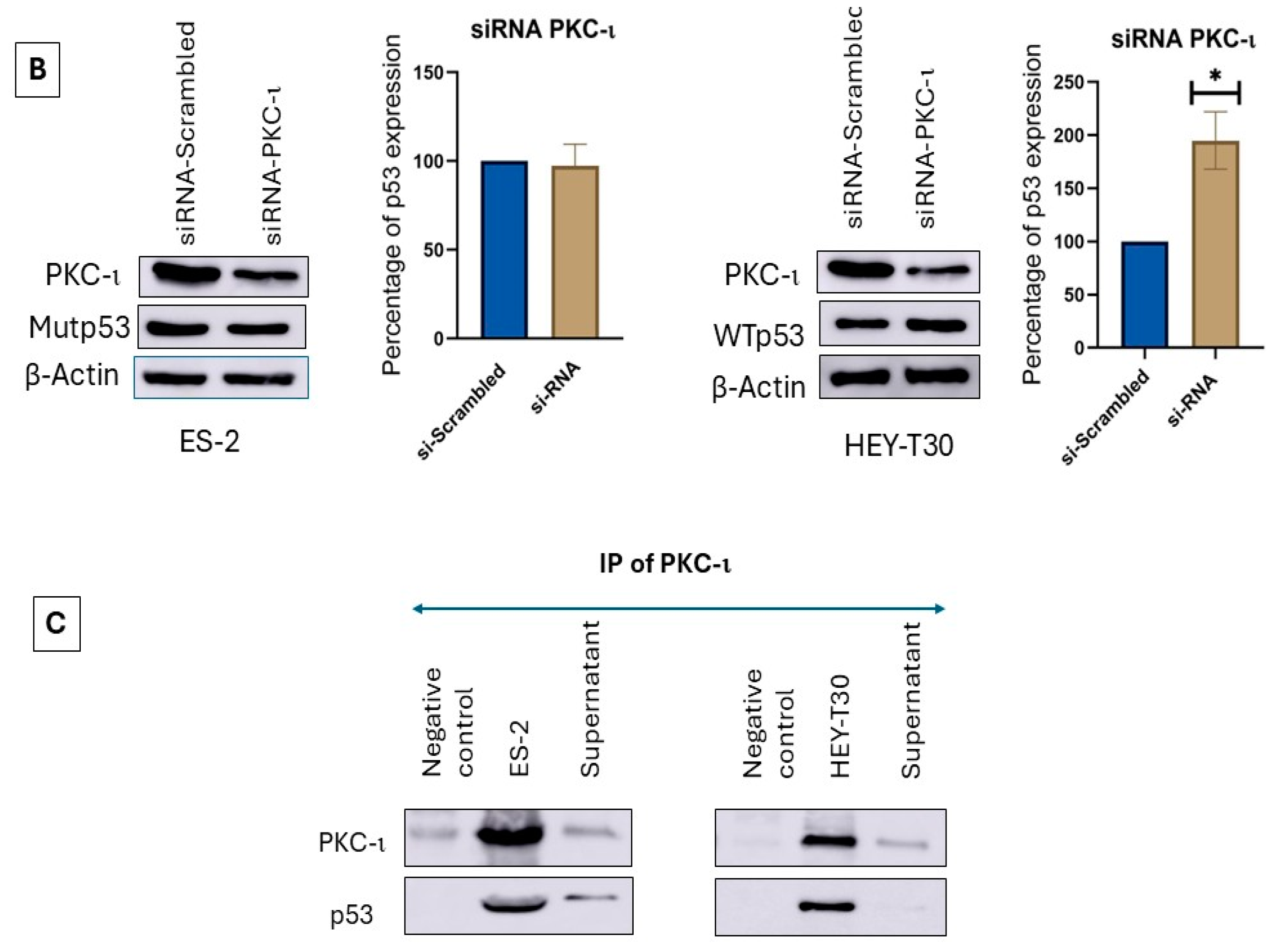
2.6. siRNA Inhibition of PKC-ι Does Not Affect the Mutp53 but Increases WTp53
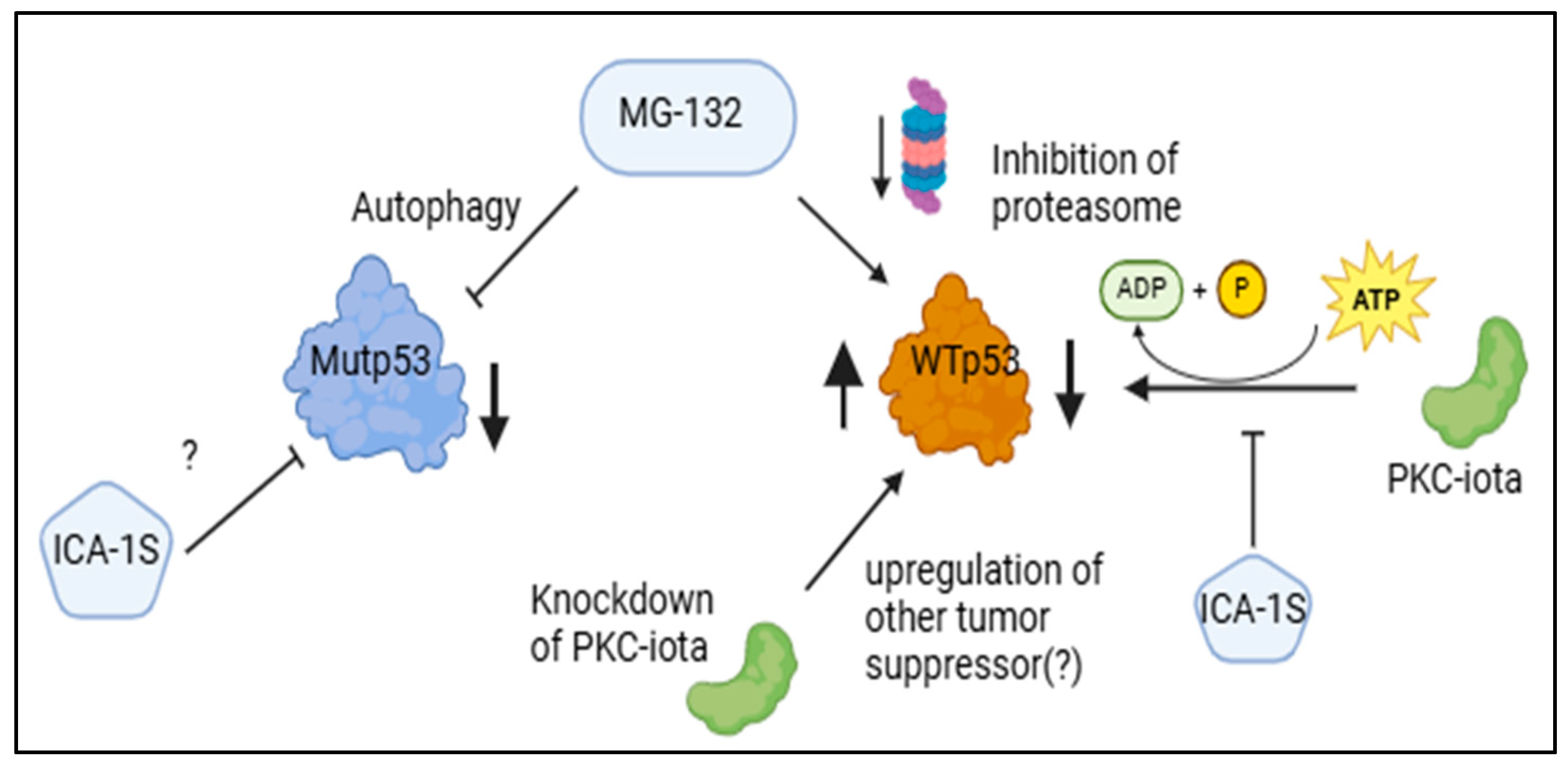
3. Discussion
4. Materials and Methods
4.1. Reagents, Antibodies, and Inhibitors
4.2. Ovarian Cancer Cell Lines
4.3. Determination of Cell Viability by WST-1 and Cell Proliferation Assay
4.4. Preparation of Cell Lysates and Western Blotting
4.5. Detection of p53 by Immunofluorescence
4.6. Co-Immunoprecipitation
4.7. Si-RNA-Based Knockdown of PKC-ι
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallis, B.; Bowman, K.R.; Lu, P.; Lim, C.S. The Challenges and Prospects of p53-Based Therapies in Ovarian Cancer. Biomolecules 2023, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Trimble, E.L.; Wright, J.; Christian, M.C. Treatment of platinum-resistant ovarian cancer. Expert Opin. Pharmacother. 2001, 2, 1299–1306. [Google Scholar] [PubMed]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef]
- Maioru, O.-V.; Radoi, V.-E.; Coman, M.-C.; Hotinceanu, I.-A.; Dan, A.; Eftenoiu, A.-E.; Burtavel, L.-M.; Bohiltea, L.-C.; Severin, E.-M. Developments in Genetics: Better Management of Ovarian Cancer Patients. Int. J. Mol. Sci. 2023, 24, 15987. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar]
- Visintin, I.; Feng, Z.; Longton, G.; Ward, D.C.; Alvero, A.B.; Lai, Y.; Tenthorey, J.; Leiser, A.; Flores-Saaib, R.; Yu, H.; et al. Diagnostic markers for early detection of ovarian cancer. Clin. Cancer Res. 2008, 14, 1065–1072. [Google Scholar] [PubMed]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.-A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer. 2014, 14, 359–370. [Google Scholar]
- Parrales, A.; Ranjan, A.; Iyer, S.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar]
- Sato, H.; Hiraki, M.; Namba, T.; Egawa, N.; Baba, K.; Tanaka, T.; Noshiro, H. Andrographolide induces degradation of mutant p53 via activation of Hsp70. Int. J. Oncol. 2018, 53, 761–770. [Google Scholar] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar]
- Zhang, B.; Guo, N. The role of proteasome inhibitor MG132 in cisplatin resistant ovarian cancer. Eur. J. Gynaecol. Oncol. 2023, 44, 28–36. [Google Scholar]
- Guo, N.; Peng, Z.; Zhang, J. Proteasome Inhibitor MG132 Enhances Sensitivity to Cisplatin on Ovarian Carcinoma Cells in Vitro and In Vivo. Int. J. Gynecol. Cancer 2016, 26, 839–844. [Google Scholar]
- Halasi, M.; Pandit, B.; Gartel, A.L. Proteasome inhibitors suppress the protein expression of mutant p53. Cell Cycle 2014, 13, 3202–3206. [Google Scholar] [PubMed]
- Wang, Y.; Hill, K.S.; Fields, A.P. PKCiota maintains a tumor-initiating cell phenotype that is required for ovarian tumor-igenesis. Mol. Cancer Res. 2013, 11, 1624–1635. [Google Scholar]
- Tyagi, K.; Roy, A.; Mandal, S. Protein kinase C iota promotes glycolysis via PI3K/AKT/mTOR signalling in high grade serous ovarian cancer. Mol. Biol. Rep. 2024, 51, 983. [Google Scholar]
- Zhang, L.; Huang, J.; Yang, N.; Liang, S.; Barchetti, A.; Giannakakis, A.; Cadungog, M.G.; O’Brien-Jenkins, A.; Massobrio, M.; Roby, K.F.; et al. Integrative genomic analysis of protein kinase C (PKC) family identifies PKCι as a biomarker and potential Oncogene in ovarian carcinoma. Cancer Res. 2006, 66, 4627–4635. [Google Scholar]
- Nanos-Webb, A.; Bui, T.; Karakas, C.; Zhang, D.; Carey, J.P.W.; Mills, G.B.; Hunt, K.K.; Keyomarsi, K. PKCiota promotes ovarian tumor progression through deregulation of cyclin E. Oncogene 2016, 35, 2428–2440. [Google Scholar]
- Dinkins, K.; Barton, W.; Wheeler, L.; Smith, H.J.; Mythreye, K.; Arend, R.C. Targeted therapy in high grade serous ovarian Cancer: A literature review. Gynecol. Oncol. Rep. 2024, 54, 101450. [Google Scholar] [CrossRef] [PubMed]
- Khalid, K.M.; Ratnayake, W.S.; Apostolatos, C.A.; Acevedo-Duncan, M. Dual inhibition of atypical PKC signaling and PI3K/Akt signaling dysregulates c-Myc to induce apoptosis in clear cell Renal Cell Carcinoma. Front. Oncol. 2024, 13, 1213715. [Google Scholar] [CrossRef] [PubMed]
- Breedy, S.; Ratnayake, W.S.; Lajmi, L.; Hill, R.; Acevedo-Duncan, M. 14-3-3 and Smad2/3 are crucial mediators of atypical-PKCs: Implications for neuroblastoma progression. Front. Oncol. 2023, 13, 1051516. [Google Scholar] [CrossRef]
- Apostolatos, A.H.; Apostolatos, C.A.; Ratnayake, W.S.; Neuger, A.; Sansil, S.; Bourgeois, M.; Acevedo-Duncan, M. Preclinical testing of 5-amino-1-((1R,2S,3S,4R)-2,3-dihydroxy-4-methylcyclopentyl)-1H-imidazole-4-carboxamide: A potent protein kinase C-iota in-hibitor as a potential prostate carcinoma therapeutic. Anticancer Drugs 2019, 30, 65–71. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Borzdzilowska, P.; Bednarek, I. The Effect of alpha-Mangostin and Cisplatin on Ovarian Cancer Cells and the Microenvi-ronment. Biomedicines 2022, 10, 1116. [Google Scholar] [CrossRef]
- Pillai, P.; Desai, S.; Patel, R.; Sajan, M.; Farese, R.; Ostrov, D.; Acevedo-Duncan, M. A novel PKC-iota inhibitor abrogates cell proliferation and induces apoptosis in neuroblastoma. Int. J. Biochem. Cell Biol. 2011, 43, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Wang, X.; Chen, D. Proteasome inhibitor MG132 enhances the sensitivity of human OSCC cells to cisplatin via a ROS/DNA damage/p53 axis. Exp. Ther. Med. 2023, 25, 224. [Google Scholar] [CrossRef]
- Dang, L.; Wen, F.; Yang, Y.; Liu, D.; Wu, K.; Qi, Y.; Li, X.; Zhao, J.; Zhu, D.; Zhang, C.; et al. Proteasome inhibitor MG132 inhibits the proliferation and promotes the cisplatin-induced apoptosis of human esophageal squamous cell carcinoma cells. Int. J. Mol. Med. 2014, 33, 1083–1088. [Google Scholar] [CrossRef]
- Sun, F.; Zhang, Y.; Xu, L.; Li, S.; Chen, X.; Zhang, L.; Wu, Y.; Li, J. Proteasome Inhibitor MG132 Enhances Cisplatin-Induced Apoptosis in Osteosarcoma Cells and Inhibits Tumor Growth. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 655–664. [Google Scholar] [CrossRef]
- Xu, J.; Wu, H.; Sun, J.; Gong, Z.; Lu, X.; Yang, E.; Chen, Z.; Huang, S.; Nong, X.; Zhang, D. The anti-tumor effect of proteasome inhibitor MG132 for human adenoid cystic carcinoma: Correlate with the emerging role of Nrf2/Keap1 signaling pathway. Head Face Med. 2022, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Langland, G.T.; Yannone, S.M.; Langland, R.A.; Nakao, A.; Guan, Y.; Long, S.B.; Vonguyen, L.; Chen, D.J.; Gray, J.W.; Chen, F. Radiosensitivity profiles from a panel of ovarian cancer cell lines exhibiting genetic alterations in p53 and disparate DNA-dependent protein kinase activities. Oncol. Rep. 2010, 23, 1021–1026. [Google Scholar]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures, and advances in therapy. Signal Transduct. Target Ther. 2023, 8, 92. [Google Scholar]
- Mullany, L.K.; Wong, K.-K.; Marciano, D.C.; Katsonis, P.; King-Crane, E.R.; Ren, Y.A.; Lichtarge, O.; Richards, J.S. Specific TP53 Mutants Overrepresented in Ovarian Cancer Impact CNV, TP53 Activity, Responses to Nutlin-3a, and Cell Survival. Neoplasia 2015, 17, 789–803. [Google Scholar] [PubMed]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [PubMed]
- Anwar, A.; Norris, D.A.; Fujita, M. Ubiquitin proteasomal pathway mediated degradation of p53 in melanoma. Arch. Biochem. Biophys. 2011, 508, 198–203. [Google Scholar]
- Choundhury, S.; Kolukula, V.; Preet, A.; Albanese, C.; Avantaggiati, M. Dissecting the pathways that destabilize mutant p53: The proteasome or autophagy? Cell Cycle 2013, 12, 1022–1029. [Google Scholar] [CrossRef]
- Regala, R.P.; Weems, C.; Jamieson, L.; Khoor, A.; Edell, E.S.; Lohse, C.M.; Fields, A.P. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005, 65, 8905–8911. [Google Scholar] [CrossRef]
- Paul, A.; Gunewardena, S.; Stecklein, S.R.; Saha, B.; Parelkar, N.; Danley, M.; Rajendran, G.; Home, P.; Ray, S.; Jokar, I.; et al. PKClambda/iota signaling promotes triple-negative breast cancer growth and metastasis. Cell Death Differ. 2014, 21, 1469–1481. [Google Scholar] [CrossRef]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [PubMed]
- Shihan, M.H.; Novo, S.G.; Le Marchand, S.J.; Wang, Y.; Duncan, M.K. A simple method for quantitating confocal fluorescent images. Biochem. Biophys. Rep. 2021, 25, 100916. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzan, M.; Nowshin Oishee, N.; Olatunji, A.O.; Hasib Shourav, A.; Noor, R.E.; Astalos, A.J.; Leahy, J.W.; Acevedo-Duncan, M. Proteasome Inhibitor MG-132 and PKC-ι-Specific Inhibitor ICA-1S Degrade Mutant p53 and Induce Apoptosis in Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2025, 26, 3035. https://doi.org/10.3390/ijms26073035
Marzan M, Nowshin Oishee N, Olatunji AO, Hasib Shourav A, Noor RE, Astalos AJ, Leahy JW, Acevedo-Duncan M. Proteasome Inhibitor MG-132 and PKC-ι-Specific Inhibitor ICA-1S Degrade Mutant p53 and Induce Apoptosis in Ovarian Cancer Cell Lines. International Journal of Molecular Sciences. 2025; 26(7):3035. https://doi.org/10.3390/ijms26073035
Chicago/Turabian StyleMarzan, Mahfuza, Nuzhat Nowshin Oishee, Abigail Oluwafisayo Olatunji, Abiral Hasib Shourav, Radwan Ebna Noor, Aaron Joshua Astalos, James W. Leahy, and Mildred Acevedo-Duncan. 2025. "Proteasome Inhibitor MG-132 and PKC-ι-Specific Inhibitor ICA-1S Degrade Mutant p53 and Induce Apoptosis in Ovarian Cancer Cell Lines" International Journal of Molecular Sciences 26, no. 7: 3035. https://doi.org/10.3390/ijms26073035
APA StyleMarzan, M., Nowshin Oishee, N., Olatunji, A. O., Hasib Shourav, A., Noor, R. E., Astalos, A. J., Leahy, J. W., & Acevedo-Duncan, M. (2025). Proteasome Inhibitor MG-132 and PKC-ι-Specific Inhibitor ICA-1S Degrade Mutant p53 and Induce Apoptosis in Ovarian Cancer Cell Lines. International Journal of Molecular Sciences, 26(7), 3035. https://doi.org/10.3390/ijms26073035