1. Introduction
Members of the order Mucorales are the causative agents of the human infection mucormycosis [
1], a rare disease that is rising globally and has a high mortality rate especially in disseminated infections [
2]. These filamentous fungi affect mainly immunocompromised patients with impaired immune systems, such as neutropenia, hematological malignancies, and organ transplantation [
1]. Additionally, new risk factors, such as renal failure and post-tuberculosis, have emerged recently [
1]. Nevertheless, cases of apparently healthy people who suffered from burns, trauma, or surgeries have been reported [
2,
3]. In western countries, hematological malignancies and organ transplantation are the most common risk factors, whereas in Asian countries, diabetes mellitus is the most prevalent underlying condition [
2]. The infection process starts with the inhalation of spores into the lungs, consumption of contaminated food or through wounds, or disrupted skin [
4]. In patients with reduced phagocytic activity, including those with diabetic ketoacidosis, the Mucorales proliferate or persist, leading to serious consequences. Iron uptake also plays a crucial role in mucormycosis because patients with high levels of iron in the blood are more susceptible to suffering from mucormycosis [
4]. Over the last ten years, an increase in the incidence of mucormycosis has been registered, probably due to the rise in immunocompromised individuals [
5,
6]. Most recently, a huge increase in numbers of cases of mucormycosis in patients suffering from Coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been reported [
7]. Although COVID-19 itself may affect the immune system resulting in vulnerability of the patients to mucormycosis, poor diabetic control and the administration of systemic corticosteroids has greatly contributed to the occurrence of the disease in severe and critical COVID-19 patients [
6]. The current situation has led the World Health Organization (WHO) to include this group of fungi within the high group of importance in its list of first fungal priority pathogens [
8].
Mucor lusitanicus (formerly named
M. circinelloides f.
lusitanicus) [
9] belongs to the Mucorales group and has been extensively used as a model organism for the study of mucormycosis [
10]. Two main RNA interference (RNAi) mechanisms have been described in this fungus: the canonical pathway and the non-canonical RNAi pathway (NCRIP) [
11], also called the alternative [
12]. The first one operates in a classical way, where dsRNA is cleaved by the Dicer enzymes into small interfering RNAs (siRNAs) of 20-25-nt in length. These double-stranded RNA (dsRNAs) fragments are loaded in an RNA-induced silencing complex (RISC), whose main component is Argonaute, where a strand is degraded, while the other is used to direct the complex to the complementary sequences [
13]. In addition of protecting the genome from invading nucleic acids, this mechanism also controls gene expression by the production of endogenous short RNAs (esRNAs) derived from protein-coding genes, known as exonic-siRNAs (ex-siRNAs) [
14]. Several studies reveal this pathway is involved in the control of physiological and developmental processes, such as vegetative growth, sporulation and accelerated autolysis in response to nutrient starvation [
13,
15].
The NCRIP is characterized by the central role of the atypical RNase III R3B2 and the absence of Dicer and Argonaute components. This RNase is unique among the RNases III because although it can bind both single-stranded RNAs (ssRNAs) and dsRNAs, it cleaves only ssRNAs [
16]. In addition, it is conserved only among Mucorales, and its RNase III-like domain shows a limited similarity with the RNase III domain of several bacteria of the order Burkholderiales, suggesting that it could be originated from a horizontal transfer between
Burkholderia and an ancestor of the order Mucorales [
11]. In addition to R3B2, three RNA dependent RNA polymerases (RdRP) participate in the NCRIP. Their functions are not fully understood, although it is suggested that they bind to target transcripts and make a short complementary strand that mark them for degradation by R3B2, producing esRNAs that are sense to the mRNA, contrary to the canonical pathway that produces sense and antisense small RNAs (sRNAs) [
17]. These esRNAs are additionally characterized by their random size distribution in comparison with those generated by the canonical RNAi pathway and are enriched in uracil in the penultimate position. These esRNAs are known as
rdrp-dependent degraded RNAs (rdRNAs) and include most ex-siRNAs derived from the canonical pathway, specifically Class III [
11].
Interestingly, recent studies have shown the implication of the NCRIP in the virulence of
M. lusitanicus, since mutants lacking the main proteins involved in this RNAi pathway, RdRP1 and R3B2, exhibit a reduced virulence when assayed in mouse models. These mutants show a constitutive activation of the response to phagocytosis, indicating that NCRIP represses its target genes during saprophytic growth and activates its transcription during stressful conditions, such as during the phagocytosis of spores [
18].
An intriguing additional canonical RNAi pathway has been described in
M. lusitanicus, known as the epigenetic pathway, because it does not require any genomic change to promote resistance to antifungal drugs [
19,
20]. In response to exposure to the drug or prodrug, this mechanism produces siRNAs complementary to mRNAs of the target protein or proteins that metabolized the prodrug, resulting in drug-resistant epimutants that show low levels of the corresponding mRNAs. This resistance is transient because they restore the wild-type phenotype when grown in free-drug media. Interestingly, this epigenetic RNAi-dependent pathway is repressed by the NCRIP because mutants in the latter enhance the production of drug-resistant epimutants [
17,
19]. In addition, the NCRIP also modulates the activity of the canonical RNAi pathway in controlling the movement of the centromeric retrotransposon Grem-LINE1 to maintain genome stability [
12,
18].
Despite the importance of the NCRIP in gene regulation in
M. lusitanicus and probably other mucoralean fungi, no work has performed a comprehensive analysis to identify the direct targets of this RNAi pathway. Previous works have characterized the sRNAs produced and/or mRNA regulated by the NCRIP [
11,
12,
18], but they have not actively searched for direct targets of this RNAi pathway. In addition, the mechanism underlying the degradation of specific mRNA by the NCRIP remains largely unexplored. In this work, sRNAs and mRNAs from the
r3b2Δ mutant and a wild-type strain were sequenced in parallel and compared to identify the genes directly regulated by the NCRIP. In addition, analysis of the mRNA degradation using the luciferase gene as a reporter confirmed that the NCRIP regulates gene expression through targeted degradation of specific transcripts.
3. Discussion
The NCRIP is an exclusive RNAi pathway of Mucorales that is associated with virulence, making it attractive for new drug design [
18]. In this pathway, a unique RNase R3B2 degrades target mRNAs into sRNAs of undefined size [
11]. Identification of the direct targets of this RNAi pathway may help to characterize the regulation of NCRIP and to know the signals, or stimuli, involved in its activation or repression. In the present study, we sequenced both sRNAs and mRNAs from the same samples of an
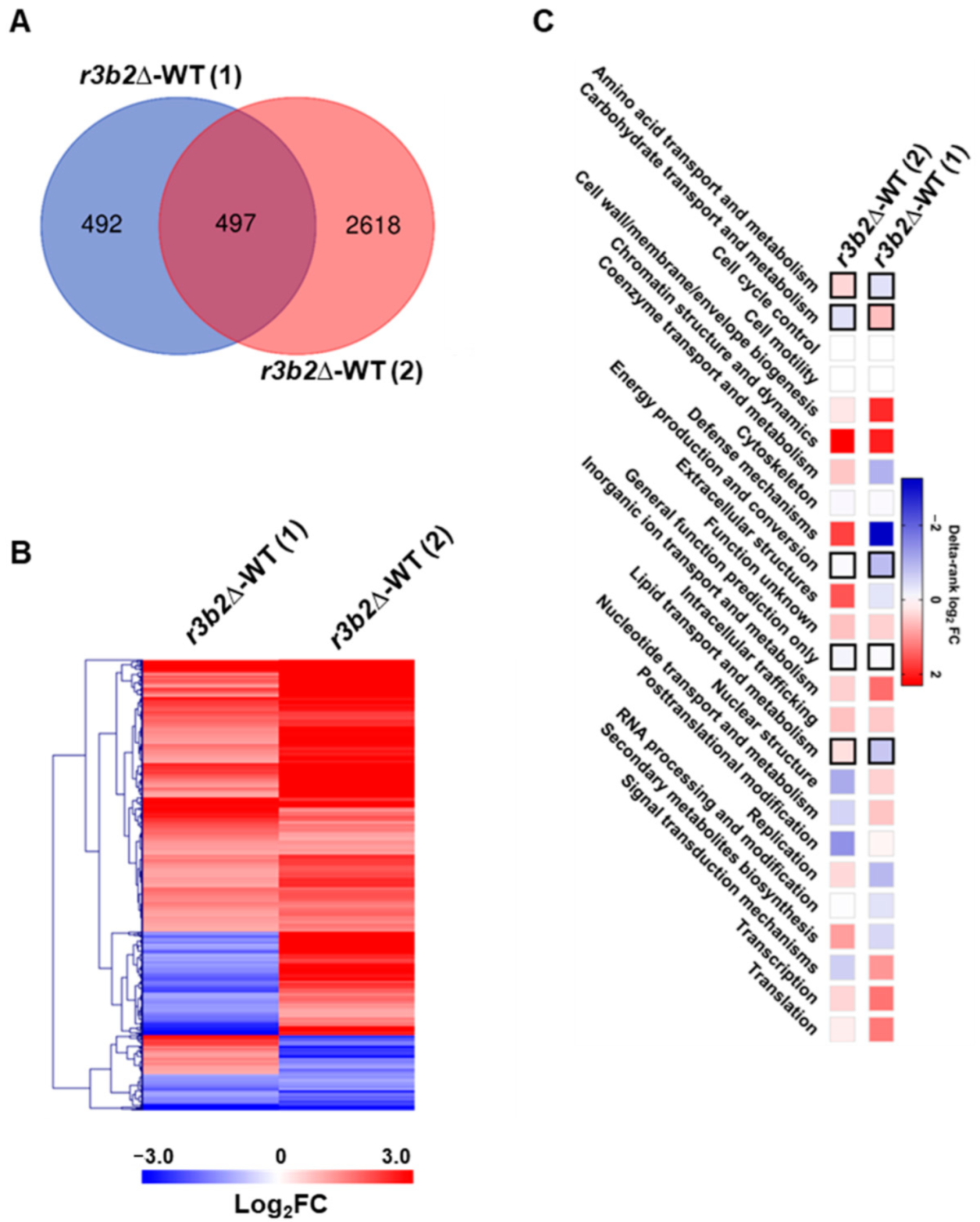
r3b2Δ mutant and a wild-type strain to identify genes directly regulated by NCRIP under non-stressful conditions, specifically solid rich media for 24 h. We identify nearly one thousand genes regulated by
r3b2 under these conditions, which are mainly involved in metabolic processes, as shown by previous studies [
18].
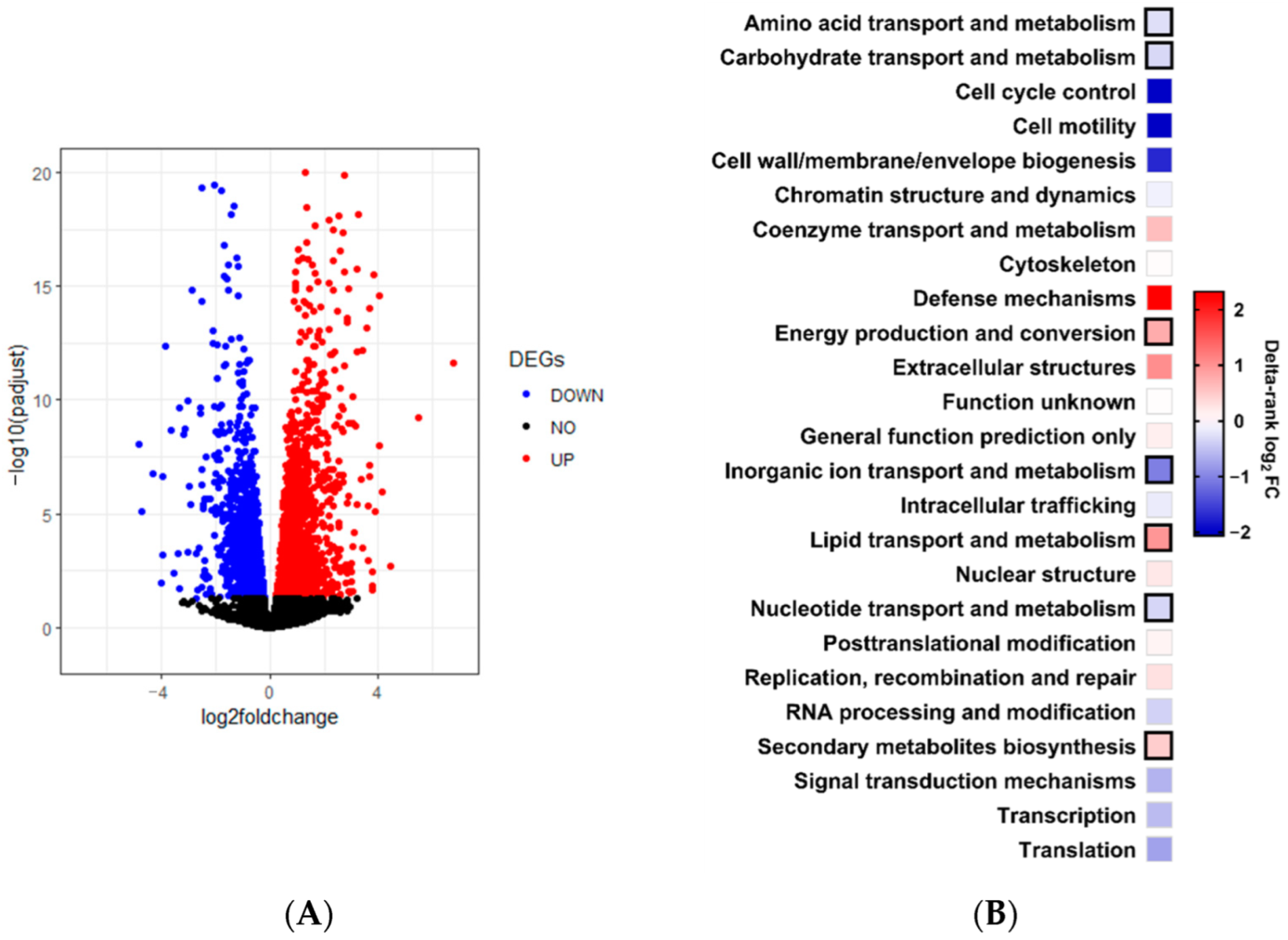
To identify direct targets of the NCRIP, we compared mRNA and sRNA analysis. We define direct targets as genes showing increased mRNA levels and decreased sRNA levels in the
r3b2Δ mutant compared to the wild-type strain, as the absence of
r3b2 should result in lower targeted mRNA degradation. Under non-stressful conditions, we detected 35 genes that met these criteria, suggesting that they are NCRIP direct targets. The lack of a direct linear correlation between mRNA and sRNAs accumulation suggests that after 24 h, we may be observing the consequences of early NCRIP action. The small number of identified direct targets indicates that most genes regulated by
r3b2 are secondary NCRIP targets or targets of the canonical RNAi pathway as
r3b2 is also required in this RNAi pathway [
11]. Half of these DEGs were found differentially expressed in another transcriptomic analysis conducted under entirely different conditions (liquid culture and 48 h of growth), suggesting that they are regulated by this RNAi pathway independently of growth conditions. Again, most of these genes are involved in metabolism, indicating that NCRIP plays an important role in the regulation of metabolism, including the response to oxidative stress [
11,
18].
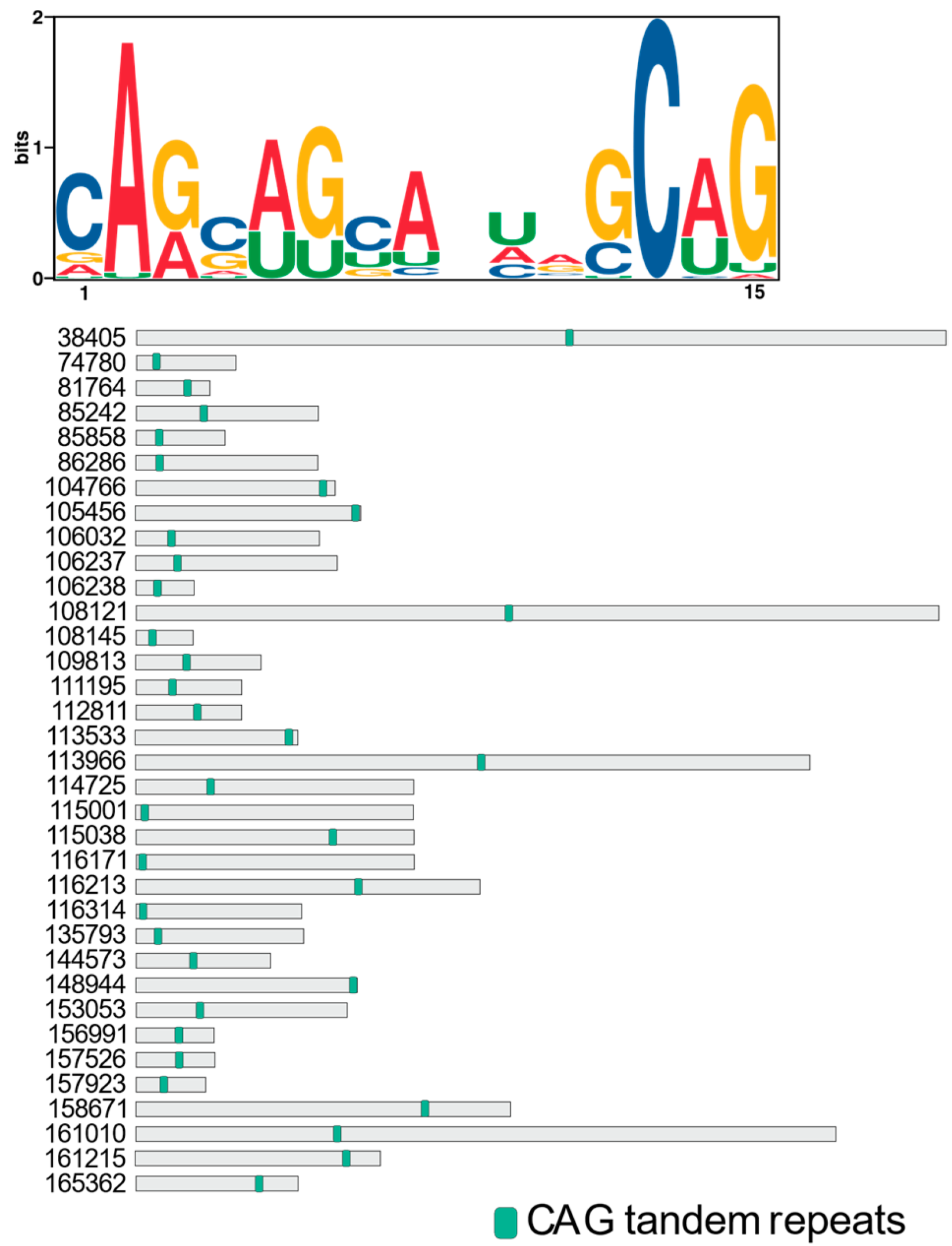
The discovery of CAG tandem repeats in the direct targets of R3B2 suggests a potential mechanism for the specific cleavage and regulation of these RNAs within the NCRIP. The CAG repeats may facilitate recognition or binding by R3B2, possibly through the formation of secondary structures such as hairpins, which are known to influence RNA stability and interactions with RNA-binding proteins. Similar to what has been observed in the mutant transcripts of the
huntingtin (HTT) gene, where expanded CAG repeats fold into hairpin structures. These structures are not stable enough to evade siRNA targeting, leading to efficient silencing of the mutant transcripts compared to normal counterparts [
28]. This suggests that the hairpin formations of CAG repeats make them accessible to RNA interference mechanisms, which may be mirrored in the way R3B2 targets its substrates.
For R3B2, which is unique among RNase III enzymes due to its specificity for ssRNAs despite binding to both ssRNAs and dsRNAs [
16], the presence of CAG repeats could enhance substrate specificity by promoting the formation of partially double-stranded regions within otherwise single-stranded transcripts. This structural adaptability might not only make the RNAs susceptible to cleavage by R3B2 but also facilitate the generation of esRNAs that are sense to the mRNA, distinct from the canonical RNAi pathways which produce both sense and antisense sRNAs.
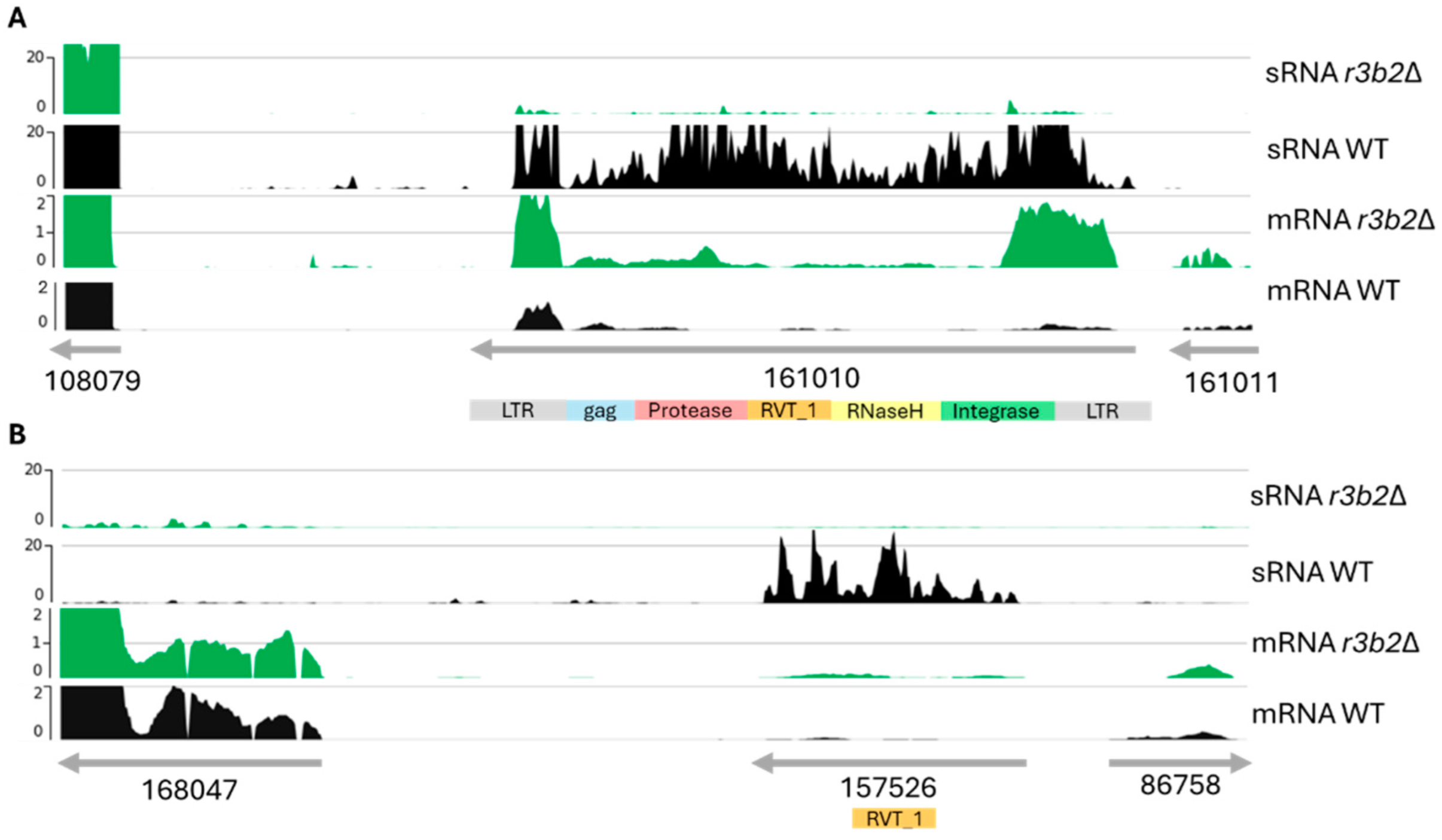
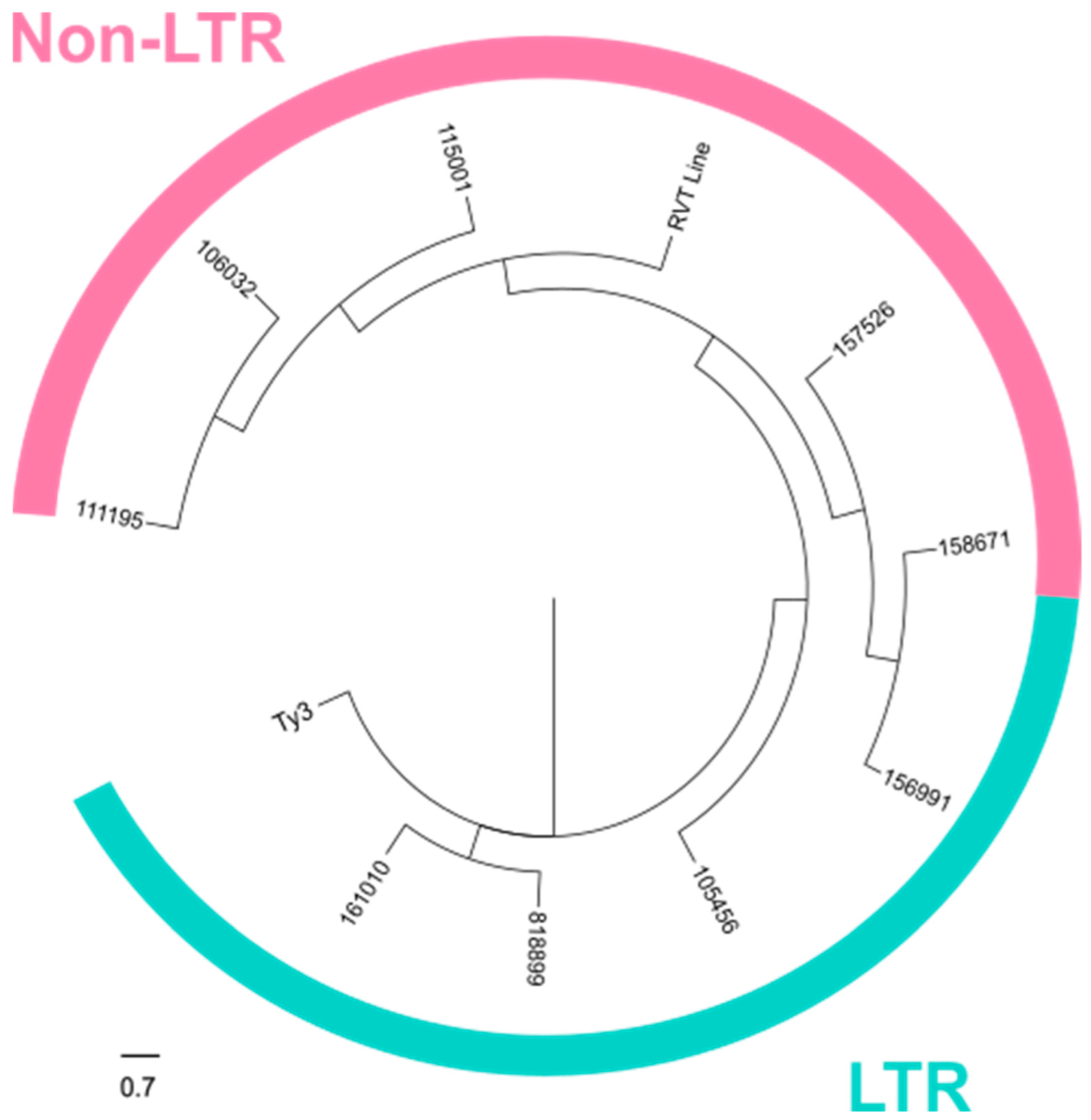
In our analysis of the direct NCRIP targets, we identified nine loci containing transposon-related domains. The structural study together with the phylogenetic analysis strongly suggests that these direct targets correspond to transposable elements (TEs) from the LINE-like and LTR transposon families. This observation aligns with the known role of the interference pathways in regulating TE activity, particularly through mechanisms that silence or limit their transposition. The presence of both LINE-like and LTR transposon domains indicates that the NCRIP may target a diverse range of transposon families, which could play a significant role in genome stability and gene regulation under specific conditions. These findings highlight the potential of NCRIP to act as a key regulator not only in controlling transposon proliferation from the LINE family, as was previously described [
18], but also in regulating LTR transposons, thus maintaining genomic integrity.
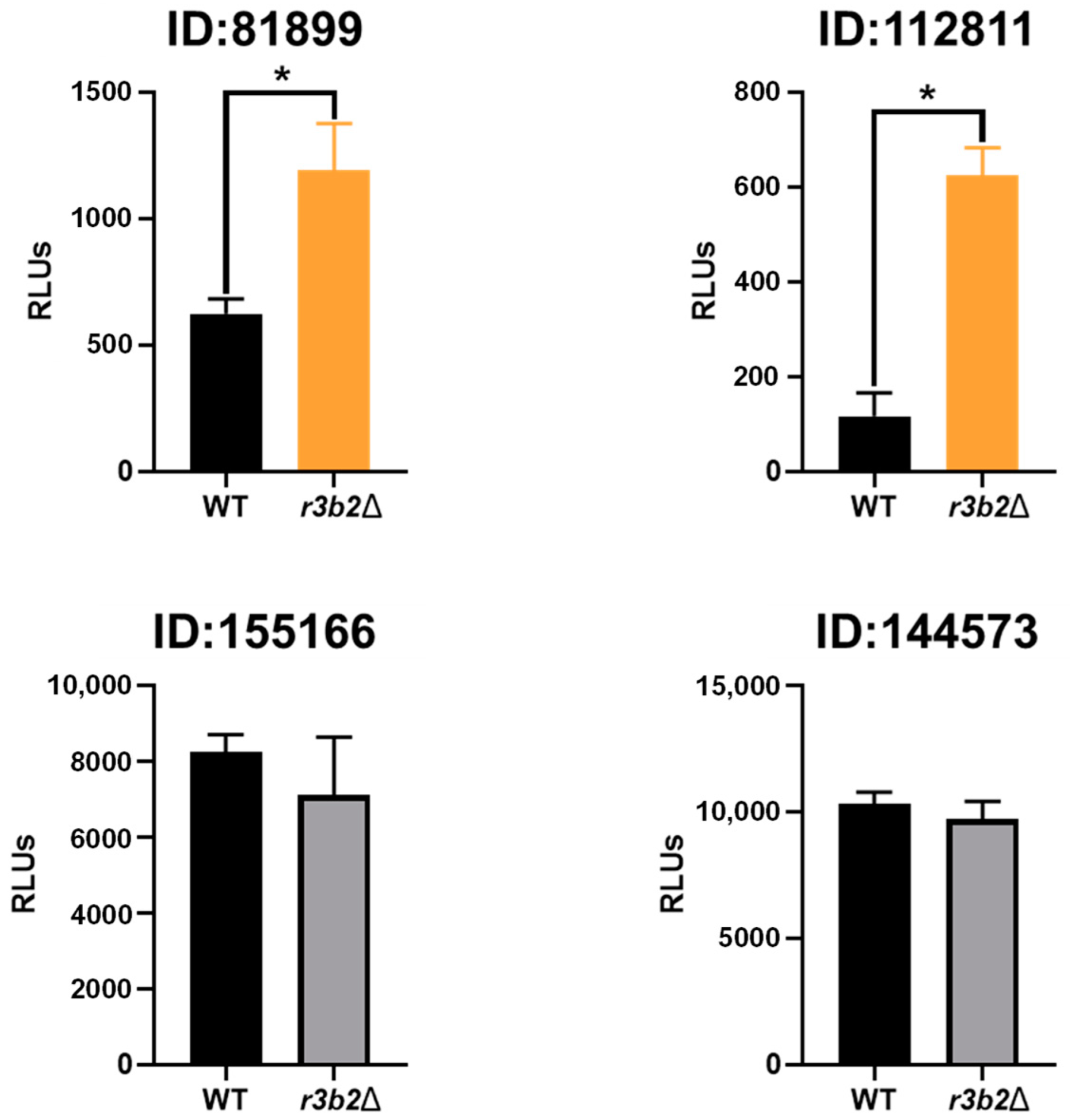
To demonstrate the ability of NCRIP to degrade target mRNAs, we expressed polycistronic mRNAs containing the luciferase gene as a reporter along with different direct NCRIP targets in the r3b2Δ mutant and the wild-type strain. Light emission analysis confirmed that mRNAs containing two of three analyzed direct NCRIP targets, including a target with transposable element domains, were degraded in the wild-type strain but not in the r3b2Δ mutant. Interestingly, the polycistronic mRNA containing the third direct NCRIP target (ID:144573) did not show significant degradation in the wild-type strain. One possible explanation for this result is that the hybrid mRNA may have acquired a secondary structure that hindered its recognition by NCRIP, preventing its degradation. Alternatively, this target (ID:144573) might require additional sequence elements, RNA-binding factors or specific cellular conditions to be efficiently processed by the pathway. Further analyses, such as structural modeling of the hybrid mRNA or identification of additional cis-regulatory elements, could help clarify the underlying mechanism governing NCRIP specificity. Overall, these experiments suggest that NCRIP is a true degradation pathway that uses RNAi components to regulate the levels of specific mRNAs, mainly those derived from TEs belonging to the LINE and LTR transposon families. Interestingly, direct targets of the NCRIP are characterized by the presence of a motif consisting of CAG tandem repeats that could contribute to the secondary structure formation and contribute to the direct specific mRNAs to be degraded by NCRIP.
4. Materials and Methods
4.1. Fungal Strains and Cell Cultures
The fungal strains used in this work were derived from
M. lusitanicus CBS77.49. The wild-type control strain was MU636, and the
r3b2Δ mutant strain was MU412. These strains are auxotrophic for leucine due to the same mutation in the
leuA gene and share the same genetic background [
11,
29].
M. circinelloides cultures were grown in rich YPG media at pH 4.5 and 26 °C for optimal growth and sporulation. YNBS 3.2 was used to obtain the transformants [
27], and YNB 3.2 was used for luciferase activity assay. The transformants expressing the mRNAs containing the luciferase gene and each of the three direct NCRIP targets and control are indicated in
Supplementary Table S1.
The E. coli strains were grown in lysogeny broth (LB) medium, supplemented with ampicillin (0.1 mg/mL) if necessary. Solid bacterial cultures were incubated at 37 °C overnight, and the liquid cultures were under the same conditions and at 250 rpm.
4.2. RNA-Seq
For RNA extraction, 2.5 × 105 spores were grown in solid YPG at pH 4.5 and 26 °C for 24 h under light conditions. After incubation, three replicates of each sample were pooled to ensure reproducibility. Total RNA, including both sRNA and mRNA, was extracted using the mirVana RNA Isolation Kit (AM1561, Thermo Fisher Scientific, Waltham, MA, USA), following the manufacturer’s procedure. RNA integrity and concentration were assessed through Agilent 2100 Bioanalyzer, (Agilent Technologies, Santa Clara, CA, USA) before proceeding to library preparation.
Library preparation was performed at Novogene using the TruSeq Stranded mRNA HT Sample Prep Kit (Illumina, San Diego, CA, USA), which enriches mRNA via poly(A) selection. The total RNA input was 1 µg per sample, and 8 cycles of PCR were used for library amplification. Libraries were quantified using KAPA Biosystems qPCR Kit (Roche, Basel, Switzerland) and analyzed on a Roche LightCycler 480 real-time PCR instrument (Roche). The libraries were then multiplexed and sequenced using the Illumina NovaSeq 6000 platform (Illumina) with NovaSeq XP v1.5 reagent Kits on an S4 flow cell, following a 2 × 150 bp paired-end sequencing strategy.
For sRNA sequencing, library preparation was conducted at Novogene using Small RNA Sample Prep Kit (Illumina). Unlike mRNA-seq, sRNA molecules do not require fragmentation before library construction. Instead, adapters were ligated directly to the 5′ and 3′ ends of the small RNA sequences. Reverse transcription was then performed to generate a complementary DNA (cDNA) library, followed by PCR amplification to enrich the sRNA sequences. The prepared libraries were multiplexed and sequenced using the Illumina NovaSeq 6000 platform (Illumina), employing a single-end 50 bp (SE50) sequencing strategy to capture short RNA fragments efficiently. The sequencing depth was optimized to achieve a comprehensive representation of the sRNA transcriptome.
4.3. Bioinformatic Analyses
4.3.1. sRNA Analysis
Following preprocessing, clean reads were aligned to the
M. circinelloides f. lucitanicus v2.0 genome using Bowtie2 v2.5.3 [
30] The reference genome was indexed using Bowtie2 prior to alignment to facilitate efficient and accurate read mapping.
For the quantification of reads mapped to
Mucor genes, FeatureCounts v2.0.1 [
31] tool was used. The software produced a count table file (with several reads from each library that defined each locus). This count table served as input for the differential expression (DE) analysis between the mutant and wild-type strains using DESeq2 v2.11.40.8 tool [
32]. We considered as differentially expressed the loci with a false discovery rate (FDR) adjusted
p-value ≤ 0.05 and log2 fold change [log
2FC] >= |1.0|.
4.3.2. mRNA Analysis
Raw mRNA datasets were checked for quality with FASTQC before and after removing adapter and contaminant sequences with TrimGalore! v.0.4.3.1 excluding reads with a Phred quality score Q ≤ 32 and/or a total length ≤ 20 nucleotides as well as adapter sequences with an overlap ≥ 4 bases. The mRNA clean reads were aligned to the
M. circinelloides f. lucitanicus v2.0 genome using HISAT2 aligner [
33], which uses a graph-based alignment approach to achieve high sensitivity and accuracy. HISAT2 employs both a global GFM index and a large set of smaller, local indexes that cover the whole genome, allowing for efficient alignment, especially for reads spanning multiple exons. Gene expression levels were quantified by counting the number of mapped reads to specific genes. However, read count alone is not sufficient to represent gene expression levels as it is influenced by factors such as gene length and sequencing depth. Therefore, the FPKM (fragments per kilobase of transcript per million mapped reads) metric was used to normalize for these factors, making gene expression estimates comparable across different genes and experiments.
To further analyze gene expression, the HTSeq software v2.0.5 [
34] was used in union mode to calculate read counts, and FPKM values were derived for each gene. For differential gene expression analysis, several steps were carried out. Read count normalization: normalization was applied to adjust for differences in sequencing depth and other technical factors that could skew expression measurements. Model-dependent
p-value estimation: differential expression between conditions was tested using a statistical model, and
p-values were calculated to determine the significance of gene expression changes. False discovery rate (FDR) estimation: to account for multiple hypothesis testing, the FDR was calculated, and genes with an FDR ≤ 0.05 were considered significantly differentially expressed. Additionally, genes with a log2 fold change (log
2FC) ≥ |1.0| were considered to exhibit significant expression changes.
Transposons and their adjacent gene expression analysis were performed using bamCoverage tool v3.3.2.0.0 to normalize coverage to bins per million reads (BPM) in 25 bp bins, thus, dividing the genome into 25 bp bins. The bigWig files produced by bamCoverage were visualized using pyGenomeTracks (v3.6).
4.3.3. Functional Analysis
KOG class enrichment analyses were performed in Prism; delta-ranks were computed as the difference between the mean of all genes within the KOG class and the mean rank of all other genes in a Mann–Whitney U-test. A KOG class was considered over-represented if p ≤ 0.05 in one-sided Fisher’s exact evaluation of the DEGs compared to the total number of genes in each KOG class.
GO terms related to the molecular biology (MB) and biological processes (BP) of the direct targets of the NCRIP were obtained from the GO annotation. To visualize the relationships and GO terms of these genes, a network was constructed using Cytoscape v3.10.3 [
35], incorporating expression data from the transcriptomic analysis of sRNA and mRNAs.
4.3.4. Prediction of RNA Motifs in the Direct Targets of the NCRIP
DNA sequences of the direct targets were retrieved using bedtools getfasta tool. Subsequently, DNA was converted to RNA. The RNA sequences were then analyzed for conserved motifs using the MEME suite [
36].
4.3.5. Phylogenetic Analysis
The reverse transcriptase (RVT) domains found in the transposable elements were aligned with MUSCLE [
37]. A neighbor-joining tree was inferred from this alignment using the JTT substitution model and a bootstrap procedure of 1000 iterations (MEGA v 10.0.5) [
38].
4.4. RT-qPCR Quantification
For RT-qPCR, about 5 µg of total RNA of wild-type and mutant strain samples were treated with Turbo DNase (Thermo Fisher). The RNA samples were routinely checked for DNA contamination by a PCR analysis using primers for the housekeeping elongation factor gene. For cDNA synthesis, 1 µg of total RNA was reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) at 25 °C for 10 min, 42 °C for 50 min, and 70 °C for 15 min. The RT-qPCR was performed in triplicate using 5X SYBER green PCR master mix (Applied Biosystems, Thermo Fisher Scientific Inc., Waltham, MA, USA) with a QuantStudio TM 5 flex system (Applied Biosystems) following the supplier’s recommendations. To ensure non-specific amplification, non-template control and melting curve were tested. The primer sequences used for the quantification of genes are listed (in
Supplementary Table S2). The efficiencies of every pair of primers were approximately identical; the relative expression of target genes was obtained by the delta–delta cycle threshold (ΔΔCt) method, normalizing for the endogenous control elongation factor (
ef).
4.5. Plasmid Constructions
The plasmid pMAT1904 carries the luciferase gene under the control of the strong promotor
zrt1 gene (P
zrt1) from
M. lusitanicus and the selection marker
leuA. Selected genes were cloned using primers with restriction sites for the enzyme SacII for subsequent gene digestion and cloning in pMAT1904. On both sides of the luciferase gene and the each NCRIP target, there are
carRP gene sequences to facilitate the integration of the whole construct in this
M. lusitanicus locus. The plasmids obtained are in
Supplementary Table S3.
4.6. Mucor Transformation
In this work, two recipient strains were used: MU636 (
leuA−) and MU412 (leuA
−,
r3b2Δ). Transformants were obtained following the previous transformation procedure [
27]. In short, harvested fresh spores were incubated for 2–4 h, ensuring their correct germination. After, protoplasts were achieved by cell wall digestion of the germinated spores with lysing enzymes (Merck, Darmstadt, Germany) and chitosanase (Merck). Protoplast transformation was performed by electroporation with SalI linearized plasmids and later incubated in poor agar medium YNBS 3.2 and checked after 3–4 days for colonies. The right construct integration disrupts the
carRP gene, involved in carotenoid biosynthesis, producing albino colonies. Those white colonies were severally transferred to fresh YNB agar plates (5–10 cycles) to obtain homokaryons.
4.7. Transformants Screening by PCR
DNA from transformants was extracted as previously described. Integration of the gene constructions was checked by PCR using primers that aligned to the luciferase gene and the carRP.
4.8. Luciferase Assay
Luciferase activity was tested in vitro following the procedure developed by Urlike et al. [
26]. Light emission was measured by FLUOstar Omega luminometer (Thermo Fisher). Bioluminescent strains were cultured in YNB 3.2 in a total of 100 µL at 2 × 10
5 spores/mL for 24 h in 96-well microtiter plates (Nunc, Thermo Fisher), at 26 °C and under light, 50 µL of luciferin (Synchem, D-luciferin, synthetic luciferin) were added to the cultures and light emission was immediately measured, before the measuring plate was shaken orbitally at 100 rpm.
To normalize light emission against fungal biomass, we used a 3.5% (vol/vol) calcofluor-white stain (18909-100ML-F, Sigma Aldrich, Burlington, MA, USA) and measured fluorescence using the Omega luminometer with excitation and emission lengths of 360 nm and 485 nm, respectively.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}