

Activation of Smooth Muscle Kir2.1 Channels and Na+/K+-ATPase Mediates Dilation of Porcine Coronary Arterioles at Physiological Levels of Potassium



Abstract
:1. Introduction
2. Results
2.1. Vasomotor Responses to Elevated Extraluminal K+
2.2. Role of Endothelium
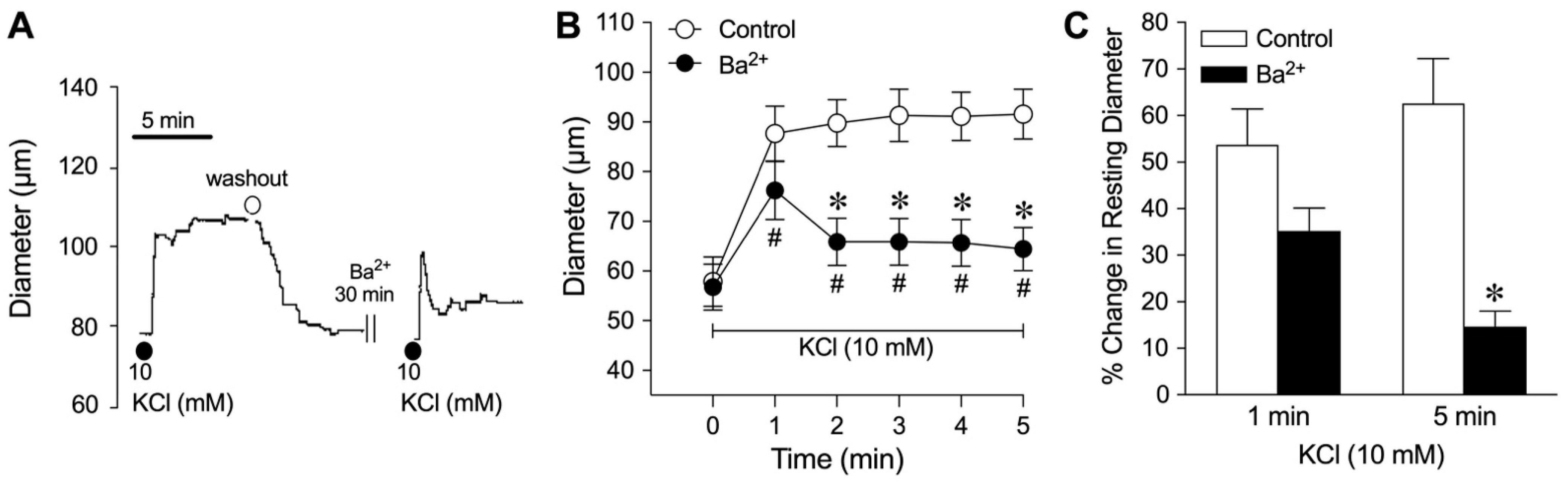
2.3. Role of K+ Channels
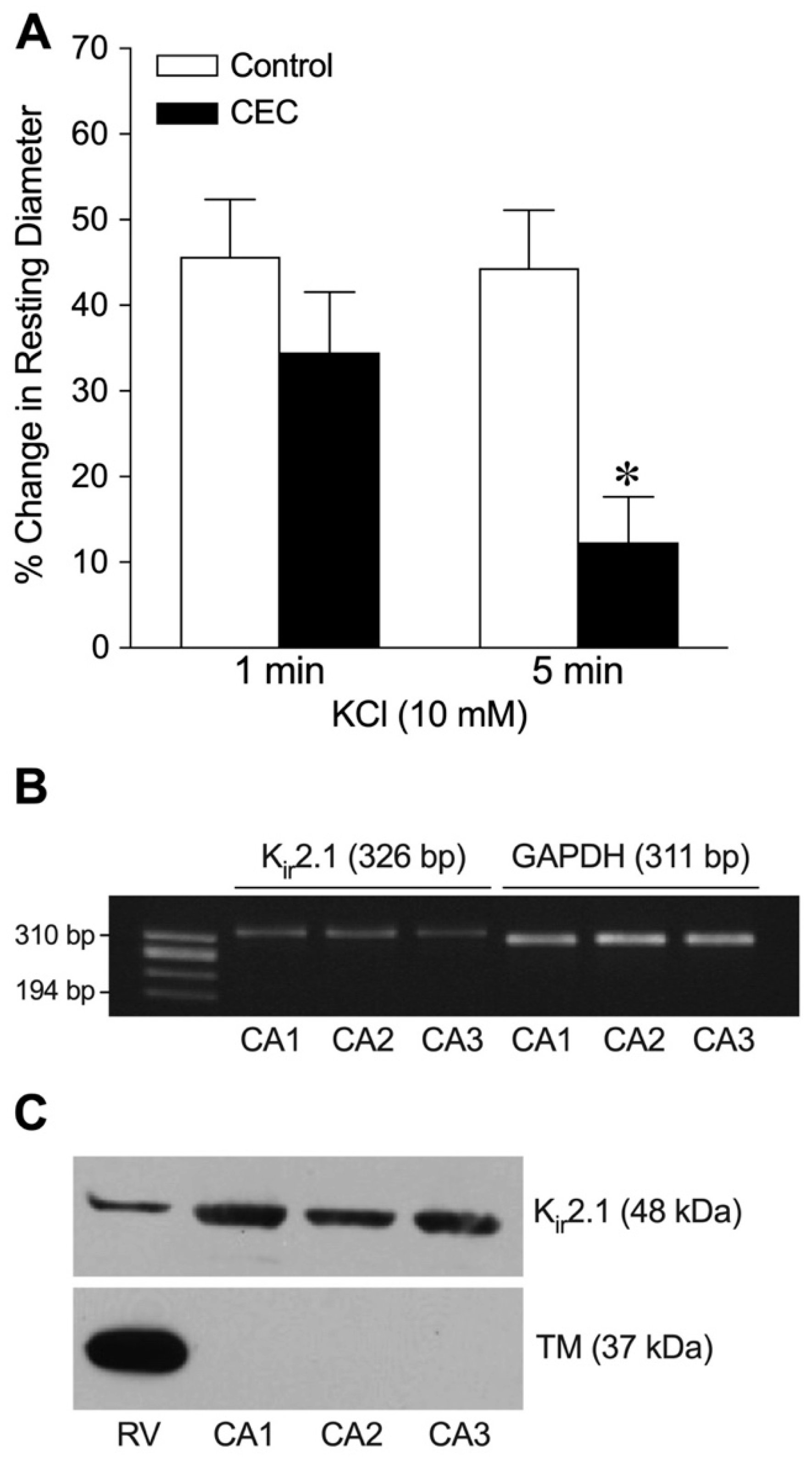
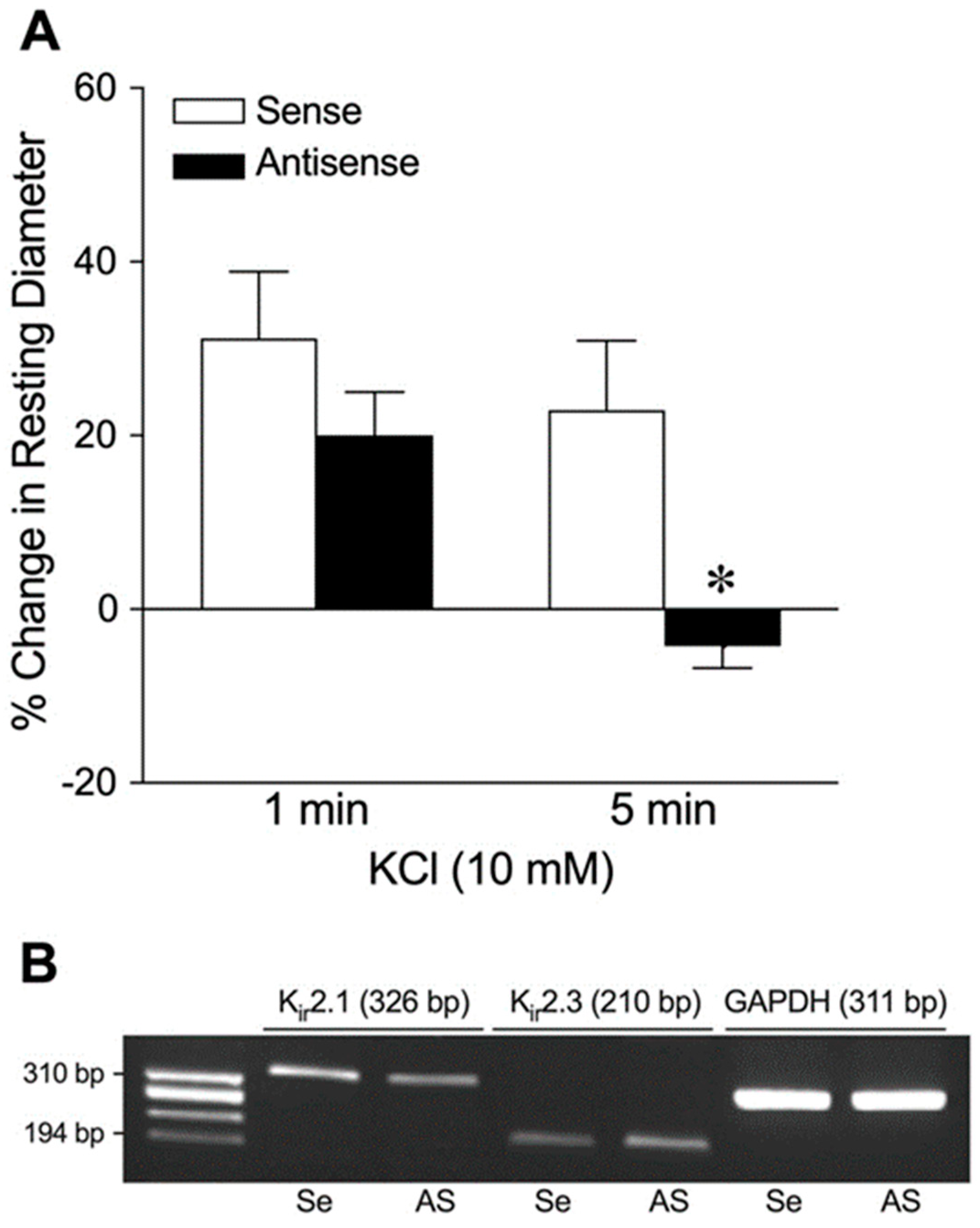
2.4. Role of Kir2.1 Channels
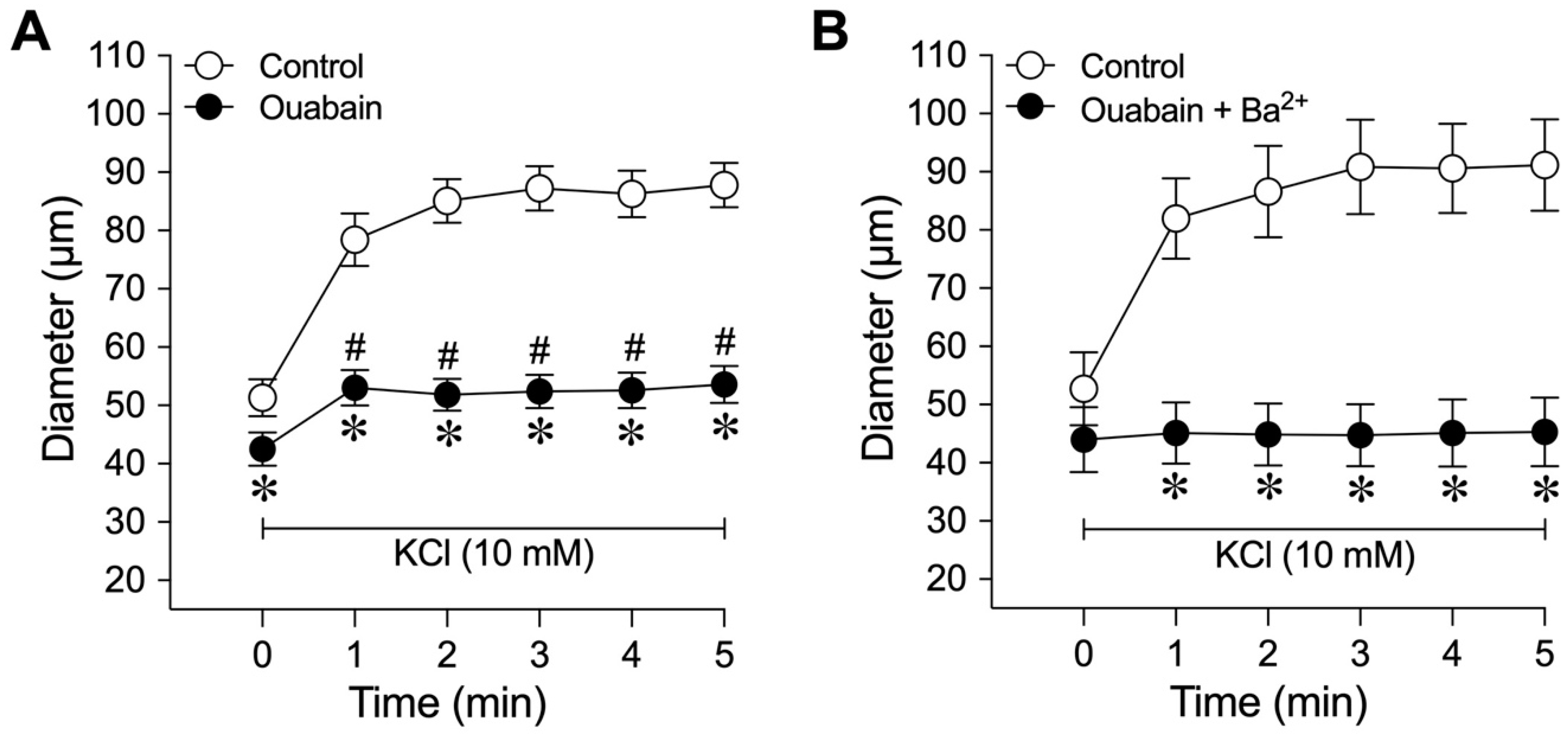
2.5. Role of Na+/K+-ATPase
3. Discussion
4. Materials and Methods
4.1. Animals and Chemicals
4.2. Isolation and Cannulation of Coronary Microvessels
4.3. Pharmacological Assessment of K+-Induced Vasodilation
4.4. Antisense Knockdown of Kir2.1 Channels
4.5. RNA Isolation and RT-PCR
4.6. Western Blot Analysis
4.7. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katz, L.N.; Lindner, E. The action of excess Na, Ca and K on the coronary vessels. Am. J. Physiol. 1938, 124, 155–160. [Google Scholar] [CrossRef]
- Murray, P.A.; Belloni, F.L.; Sparks, H.V. The role of potassium in the metabolic control of coronary vascular resistance of the dog. Circ. Res. 1979, 44, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Haddy, F.J. Potassium and blood vessels. Life Sci. 1975, 16, 1489–1497. [Google Scholar] [CrossRef]
- Hill, J.L.; Gettes, L.S. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation 1980, 61, 768–778. [Google Scholar] [CrossRef]
- Hirche, H.; Franz, C.; Bos, L.; Bissig, R.; Lang, R.; Schramm, M. Myocardial extracellular K+ and H+ increase and noradrenaline release as possible cause of early arrhythmias following acute coronary artery occlusion in pigs. J. Mol. Cell Cardiol. 1980, 12, 579–593. [Google Scholar] [CrossRef]
- Wiegand, V.; Guggi, M.; Meesmann, W.; Kessler, M.; Greitschus, F. Extracellular potassium activity changes in the canine myocardium after acute coronary occlusion and the influence of beta-blockade. Cardiovasc. Res. 1979, 13, 297–302. [Google Scholar] [CrossRef]
- Weiss, J.N.; Lamp, S.T.; Shine, K.I. Cellular K+ loss and anion efflux during myocardial ischemia and metabolic inhibition. Am. J. Physiol. 1989, 256, H1165–H1175. [Google Scholar] [CrossRef]
- Kleber, A.G. Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused guinea pig hearts. Circ. Res. 1983, 52, 442–450. [Google Scholar] [CrossRef]
- Gellai, M.; Detar, R. Evidence in support of hypoxia but against high potassium and hyperosmolarity as possible mediators of sustained vasodilation in rabbit cardiac and skeletal muscle. Circ. Res. 1974, 35, 681–691. [Google Scholar] [CrossRef]
- Konold, P.; Gebert, G.; Brecht, K. The mechanical response of isolated arteries to potassium. Experientia 1968, 24, 247–248. [Google Scholar] [CrossRef]
- Murray, P.A.; Sparks, H.V. The mechanism of K+-induced vasodilation of the coronary vascular bed of the dog. Circ. Res. 1978, 42, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bunger, R.; Haddy, R.J.; Querengasser, A.; Gerlach, E. Studies on potassium induced coronary dilation in the isolated guinea pig heart. Pflug. Arch. 1976, 363, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Knot, H.J.; Zimmermann, P.A.; Nelson, M.T. Extracellular K+-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K+ channels. J. Physiol. 1996, 492, 419–430. [Google Scholar] [CrossRef]
- Zaritsky, J.J.; Eckman, D.M.; Wellman, G.C.; Nelson, M.T.; Schwarz, T.L. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ. Res. 2000, 87, 160–166. [Google Scholar] [CrossRef]
- Haddy, F.J. Potassium effects on contraction in arterial smooth muscle mediated by Na+, K+-ATPase. Fed. Proc. 1983, 42, 239–245. [Google Scholar]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Myogenic activity in isolated subepicardial and subendocardial coronary arterioles. Am. J. Physiol. Heart Circ. Physiol. 1988, 255, H1558–H1562. [Google Scholar] [CrossRef]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am. J. Physiol. 1990, 259, H1063–H1070. [Google Scholar] [CrossRef]
- Hein, T.W.; Kuo, L. cAMP-independent dilation of coronary arterioles to adenosine: Role of nitric oxide, G proteins, and KATP channels. Circ. Res. 1999, 85, 634–642. [Google Scholar] [CrossRef]
- Haddy, F.J.; Scott, J.B. Metabolically linked vasoactive chemicals in local regulation of blood flow. Physiol. Rev. 1968, 48, 688–707. [Google Scholar] [CrossRef]
- Scott, J.B.; Daugherty, R.M., Jr.; Overbeck, H.W.; Haddy, F.J. Vascular effects of ions. Fed. Proc. 1968, 27, 1403–1407. [Google Scholar]
- Ishizaka, H.; Kuo, L. Acidosis-induced coronary arteriolar dilation is mediated by ATP-sensitive potassium channels in vascular smooth muscle. Circ. Res. 1996, 78, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Ishizaka, H.; Kuo, L. Endothelial ATP-sensitive potassium channels mediate coronary microvascular dilation to hyperosmolarity. Am. J. Physiol. 1997, 273, H104–H112. [Google Scholar] [CrossRef]
- Ishizaka, H.; Gudi, S.R.; Frangos, J.A.; Kuo, L. Coronary arteriolar dilation to acidosis: Role of ATP-sensitive potassium channels and pertussis toxin-sensitive G proteins. Circulation 1999, 99, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.W.; Belardinelli, L.; Kuo, L. Adenosine A2A receptors mediate coronary microvascular dilation to adenosine: Role of nitric oxide and ATP-sensitive potassium channels. J. Pharmacol. Exp. Ther. 1999, 291, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Hill, M.; Kuo, L. Local Regulation of Blood Flow. In Handbook of Physiology Section 2: The Cardiovascular System: Microcirculation, 2nd ed.; Tuma, R.F., Duran, W.N., Ley, K., Eds.; The American Physiological Society and Elsevier: Bethesda, MD, USA, 2008; pp. 159–284. [Google Scholar]
- Hein, T.W.; Wang, W.; Zoghi, B.; Muthuchamy, M.; Kuo, L. Functional and molecular characterization of receptor subtypes mediating coronary microvascular dilation to adenosine. J. Mol. Cell Cardiol. 2001, 33, 271–282. [Google Scholar] [CrossRef]
- Liu, Q.; Flavahan, N.A. Hypoxic dilatation of porcine small coronary arteries: Role of the endothelium and KATP-channels. Br. J. Pharmacol. 1997, 120, 728–734. [Google Scholar] [CrossRef]
- Jones, C.J.; Kuo, L.; Davis, M.J.; DeFily, D.V.; Chilian, W.M. Role of nitric oxide in the coronary microvascular responses to adenosine and increased metabolic demand. Circulation 1995, 91, 1807–1813. [Google Scholar] [CrossRef]
- Kuo, L.; Davis, M.J.; Chilian, W.M. Longitudinal gradients for endothelium-dependent and -independent vascular responses in the coronary microcirculation. Circulation 1995, 92, 518–525. [Google Scholar] [CrossRef]
- Kuo, L.; Chilian, W.M.; Davis, M.J. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. Am. J. Physiol. Heart Circ. Physiol. 1991, 261, H1706–H1715. [Google Scholar] [CrossRef]
- Jones, C.J.H.; Kuo, L.; Davis, M.J.; Chilian, W.M. Distribution and control of coronary microvascular resistance. Adv. Exp. Med. Biol. 1993, 346, 181–188. [Google Scholar]
- Rivers, R.J.; Hein, T.W.; Zhang, C.; Kuo, L. Activation of barium-sensitive inward rectifier potassium channels mediates remote dilation of coronary arterioles. Circulation 2001, 104, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Chancellor, J.D. Adenosine potentiates flow-induced dilation of coronary arterioles by activating KATP channels in endothelium. Am. J. Physiol. Heart Circ. Physiol. 1995, 269, H541–H549. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.C.; Kuo, L. Interaction between adenosine and flow-induced dilation in coronary microvascular network. Am. J. Physiol. Heart Circ. Physiol. 1997, 272, H1571–H1581. [Google Scholar] [CrossRef] [PubMed]
- Dora, K.A.; Garland, C.J. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2424–H2429. [Google Scholar] [CrossRef]
- McCarron, J.G.; Halpern, W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am. J. Physiol. 1990, 259, H902–H908. [Google Scholar] [CrossRef]
- De Clerck, I.; Boussery, K.; Pannier, J.L.; Van De Voorde, J. Potassium potently relaxes small rat skeletal muscle arteries. Med. Sci. Sports Exerc. 2003, 35, 2005–2012. [Google Scholar] [CrossRef]
- Burns, W.R.; Cohen, K.D.; Jackson, W.F. K+-induced dilation of hamster cremasteric arterioles involves both the Na+/K+-ATPase and inward-rectifier K+ channels. Microcirculation 2004, 11, 279–293. [Google Scholar] [CrossRef]
- Chilton, L.; Loutzenhiser, R. Functional evidence for an inward rectifier potassium current in rat renal afferent arterioles. Circ. Res. 2001, 88, 152–158. [Google Scholar] [CrossRef]
- Quayle, J.M.; Dart, C.; Standen, N.B. The properties and distribution of inward rectifier potassium currents in pig coronary arterial smooth muscle. J. Physiol. 1996, 494, 715–726. [Google Scholar] [CrossRef]
- Quayle, J.M.; McCarron, J.G.; Brayden, J.E.; Nelson, M.T. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am. J. Physiol. 1993, 265, C1363–C1370. [Google Scholar] [CrossRef]
- Hirst, G.D.; Edwards, F.R. Sympathetic neuroeffector transmission in arteries and arterioles. Physiol. Rev. 1989, 69, 546–604. [Google Scholar] [CrossRef] [PubMed]
- Gradel, A.K.J.; Salomonsson, M.; Sorensen, C.M.; Holstein-Rathlou, N.H.; Jensen, L.J. Long-term diet-induced hypertension in rats is associated with reduced expression and function of small artery SKCa IKCa, and Kir2.1 channels. Clin. Sci. 2018, 132, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Yin, M.Z.; Kim, H.J.; Vorn, R.; Yoo, H.Y.; Kim, S.J. Decreased inward rectifier and voltage-gated K+ currents of the right septal coronary artery smooth muscle cells in pulmonary arterial hypertensive rats. Korean J. Physiol. Pharmacol. 2020, 24, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Sancho, M.; Gao, Y.; Hald, B.O.; Yin, H.; Boulton, M.; Steven, D.A.; MacDougall, K.W.; Parrent, A.G.; Pickering, J.G.; Welsh, D.G. An assessment of KIR channel function in human cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H794–H800. [Google Scholar] [CrossRef]
- Qamirani, E.; Razavi, H.M.; Wu, X.; Davis, M.J.; Kuo, L.; Hein, T.W. Sodium azide dilates coronary arterioles via activation of inward rectifier K+ channels and Na+-K+-ATPase. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1617–H1623. [Google Scholar] [CrossRef]
- Cheng, C.H. In vitro and in vivo inhibitory actions of morin on rat brain phosphatidylinositolphosphate kinase activity. Life Sci. 1997, 61, 2035–2047. [Google Scholar] [CrossRef]
- Hansen, S.B. Lipid agonism: The PIP2 paradigm of ligand-gated ion channels. Biochim. Biophys. Acta 2015, 1851, 620–628. [Google Scholar] [CrossRef]
- Huang, C.L.; Feng, S.; Hilgemann, D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature 1998, 391, 803–806. [Google Scholar] [CrossRef]
- Rohacs, T.; Chen, J.; Prestwich, G.D.; Logothetis, D.E. Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J. Biol. Chem. 1999, 274, 36065–36072. [Google Scholar] [CrossRef]
- Nakao, M.; Gadsby, D.C. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. J. Gen. Physiol. 1989, 94, 539–565. [Google Scholar] [CrossRef]
- Hexum, T.D. Characterization of NaK-ATPase from vascular smooth muscle. Gen. Pharmacol. 1981, 12, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Weston, A.H.; Richards, G.R.; Burnham, M.P.; Feletou, M.; Vanhoutte, P.M.; Edwards, G. K+-induced hyperpolarization in rat mesenteric artery: Identification, localization and role of Na+/K+-ATPases. Br. J. Pharmacol. 2002, 136, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.W.; Liao, J.C.; Kuo, L. oxLDL specifically impairs endothelium-dependent, NO-mediated dilation of coronary arterioles. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H175–H183. [Google Scholar] [CrossRef]
- Chilian, W.M.; Eastham, C.L.; Marcus, M.L. Microvascular distribution of coronary vascular resistance in beating left ventricle. Am. J. Physiol. 1986, 251, H779–H788. [Google Scholar] [CrossRef]
- Heaps, C.L.; Bowles, D.K. Gender-specific K+-channel contribution to adenosine-induced relaxation in coronary arterioles. J. Appl. Physiol. 2002, 92, 550–558. [Google Scholar] [CrossRef]
- Barrett-Jolley, R.; Dart, C.; Standen, N.B. Direct block of native and cloned (Kir2.1) inward rectifier K+ channels by chloroethylclonidine. Br. J. Pharmacol. 1999, 128, 760–766. [Google Scholar] [CrossRef]
- Wang, S.Y.; Friedman, M.; Johnson, R.G.; Weintraub, R.M.; Sellke, F.W. Adrenergic regulation of coronary microcirculation after extracorporeal circulation and crystalloid cardioplegia. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H2462–H2470. [Google Scholar] [CrossRef]
- Rosa Jr, R.H.; Hein, T.W.; Yuan, Z.; Xu, W.; Pechal, M.I.; Geraets, R.L.; Newman, J.M.; Kuo, L. Brimonidine evokes heterogeneous vasomotor response of retinal arterioles: Diminished nitric oxide-mediated vasodilation when size goes small. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H231–H238. [Google Scholar] [CrossRef]
- Bolz, S.S.; Fisslthaler, B.; Pieperhoff, S.; De Wit, C.; Fleming, I.; Busse, R.; Pohl, U. Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca2+ changes and dilation in isolated resistance arteries. FASEB J. 2000, 14, 255–260. [Google Scholar] [CrossRef]
- Fisslthaler, B.; Popp, R.; Kiss, L.; Potente, M.; Harder, D.R.; Fleming, I.; Busse, R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999, 401, 493–497. [Google Scholar] [CrossRef]
- Nakamura, T.Y.; Artman, M.; Rudy, B.; Coetzee, W.A. Inhibition of rat ventricular IK1 with antisense oligonucleotides targeted to Kir2.1 mRNA. Am. J. Physiol. 1998, 274, H892–H900. [Google Scholar] [PubMed]
- Zhang, C.; Hein, T.W.; Wang, W.; Chang, C.I.; Kuo, L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J. 2001, 15, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.B.; Santos-Bezerra, D.P.; Thieme, K.; Passarelli, M.; Machado, U.F.; Lin, C.J.; Correa-Giannella, M.L. Optimization of total RNA isolation from human urinary sediment. Clin. Chim. Acta 2016, 462, 158–161. [Google Scholar] [CrossRef]
- Hein, T.W.; Kuo, L. LDLs impair vasomotor function of the coronary microcirculation: Role of superoxide anions. Circ. Res. 1998, 83, 404–414. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intervention | n | Resting Diameter (µm) 5 mM KCl | % Change in Resting Diameter 10 mM KCl |
|---|---|---|---|
| Group 1 | 5 | ||
| Control Denudation | 57 ± 6 57 ± 7 | 48 ± 8 43 ± 6 | |
| Group 2 | 5 | ||
| Control Glibenclamide | 63 ± 9 60 ± 10 | 38 ± 6 47 ± 13 | |
| Group 3 | 3 | ||
| Control Iberiotoxin | 72 ± 12 70 ± 13 | 38 ± 5 41 ± 8 | |
| Group 4 | 4 | ||
| Control 4-AP | 60 ± 9 45 ± 5 * | 33 ± 7 55 ± 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hein, T.W.; Razavi, H.M.; Xu, X.; Somvanshi, S.; Muthuchamy, M.; Kuo, L. Activation of Smooth Muscle Kir2.1 Channels and Na+/K+-ATPase Mediates Dilation of Porcine Coronary Arterioles at Physiological Levels of Potassium. Int. J. Mol. Sci. 2025, 26, 2654. https://doi.org/10.3390/ijms26062654
Hein TW, Razavi HM, Xu X, Somvanshi S, Muthuchamy M, Kuo L. Activation of Smooth Muscle Kir2.1 Channels and Na+/K+-ATPase Mediates Dilation of Porcine Coronary Arterioles at Physiological Levels of Potassium. International Journal of Molecular Sciences. 2025; 26(6):2654. https://doi.org/10.3390/ijms26062654
Chicago/Turabian StyleHein, Travis W., Habib M. Razavi, Xin Xu, Sonal Somvanshi, Mariappan Muthuchamy, and Lih Kuo. 2025. "Activation of Smooth Muscle Kir2.1 Channels and Na+/K+-ATPase Mediates Dilation of Porcine Coronary Arterioles at Physiological Levels of Potassium" International Journal of Molecular Sciences 26, no. 6: 2654. https://doi.org/10.3390/ijms26062654
APA StyleHein, T. W., Razavi, H. M., Xu, X., Somvanshi, S., Muthuchamy, M., & Kuo, L. (2025). Activation of Smooth Muscle Kir2.1 Channels and Na+/K+-ATPase Mediates Dilation of Porcine Coronary Arterioles at Physiological Levels of Potassium. International Journal of Molecular Sciences, 26(6), 2654. https://doi.org/10.3390/ijms26062654