
Tissue-Specific Effects of the DNA Helicase FANCJ/BRIP1/BACH1 on Repeat Expansion in a Mouse Model of the Fragile X-Related Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
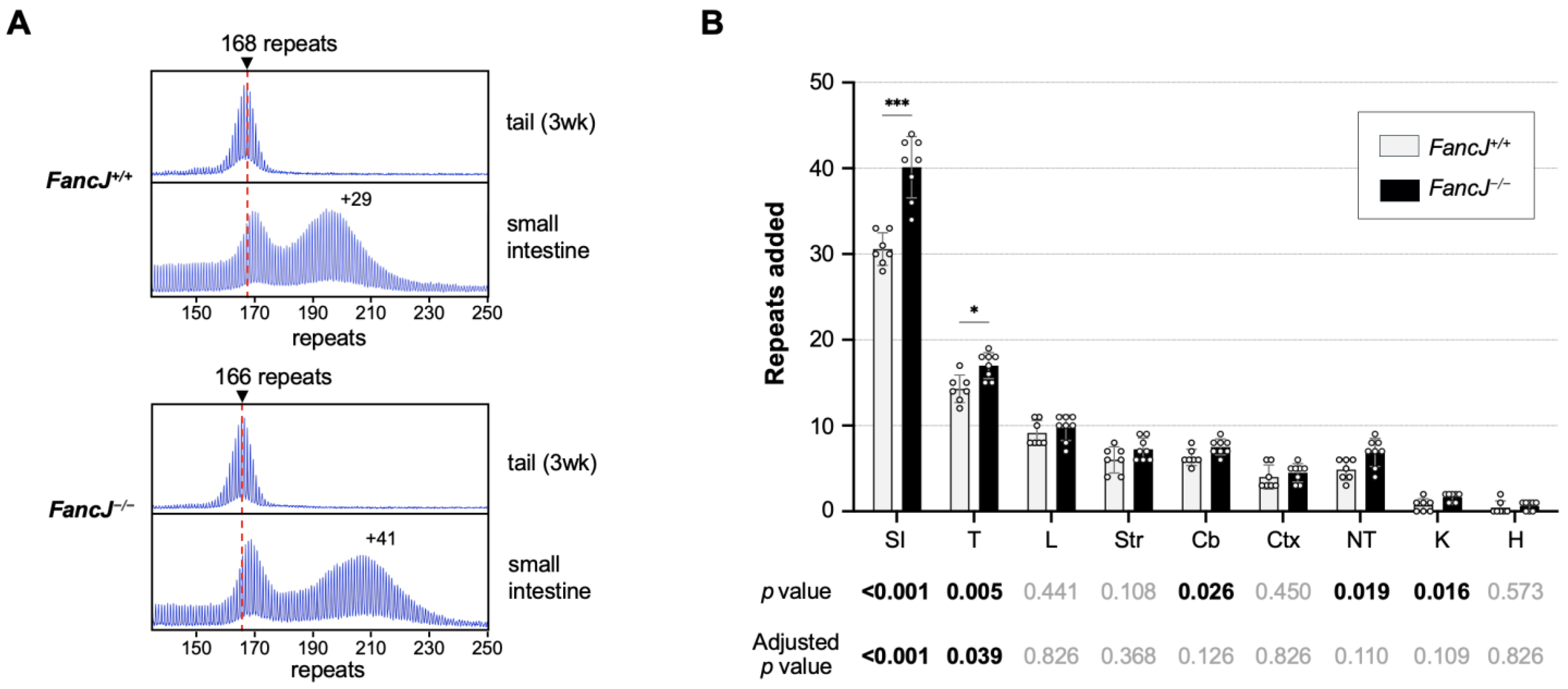
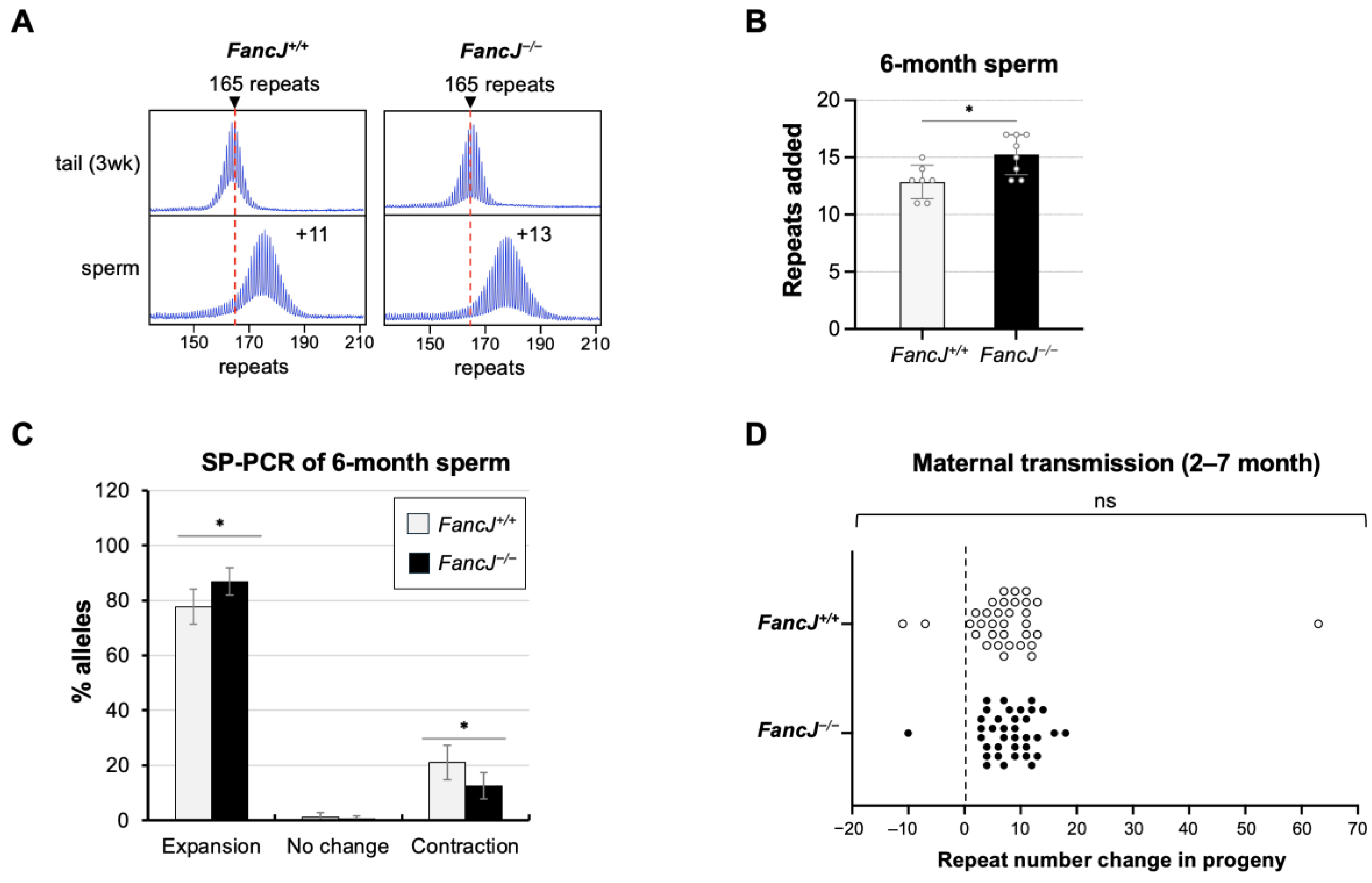
2.1. Loss of FANCJ Increases Somatic Expansion in the Small Intestine and Male Germline
2.2. Loss of FANCJ Has No Effect on Somatic Contractions in the Small Intestine
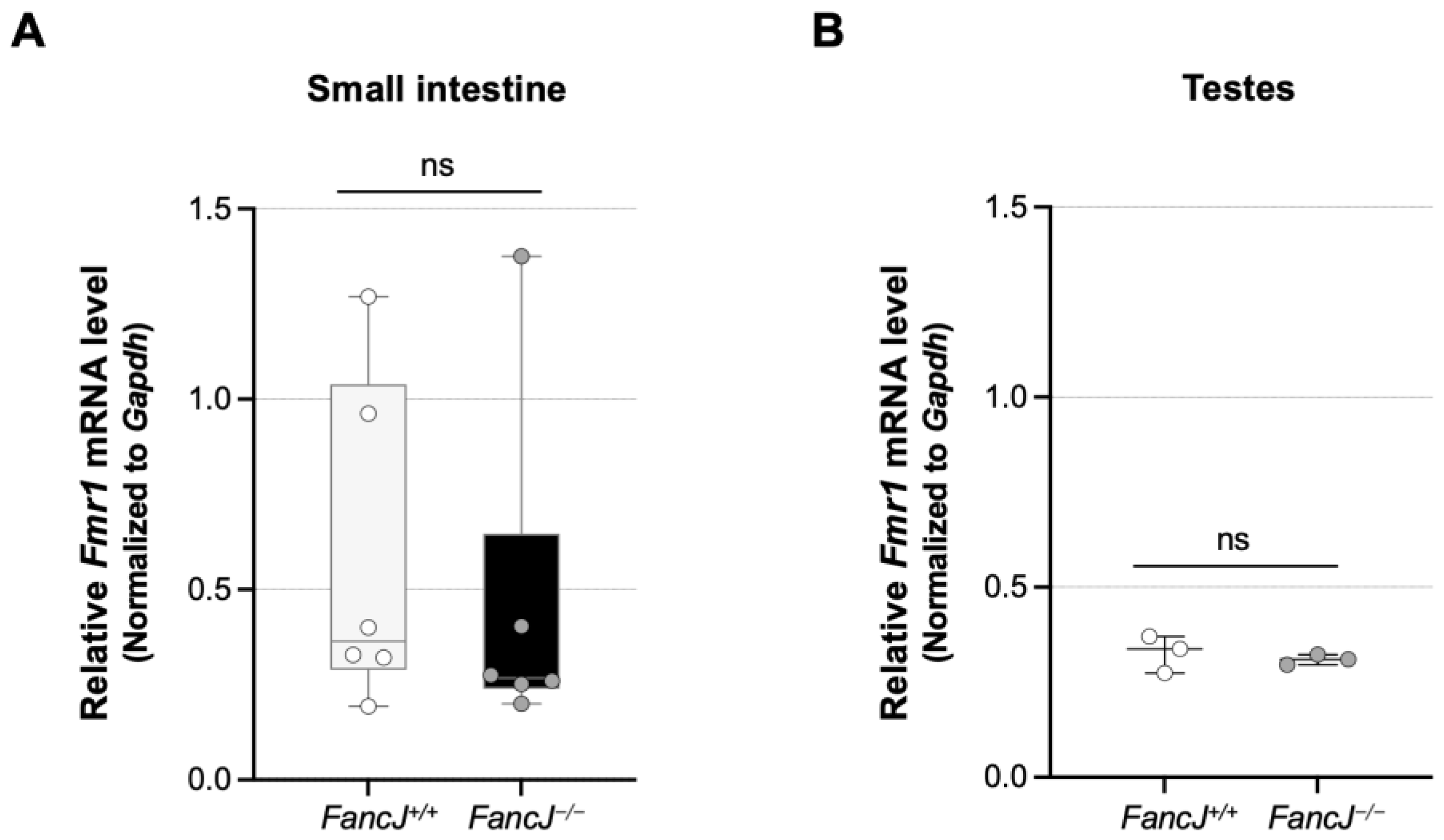
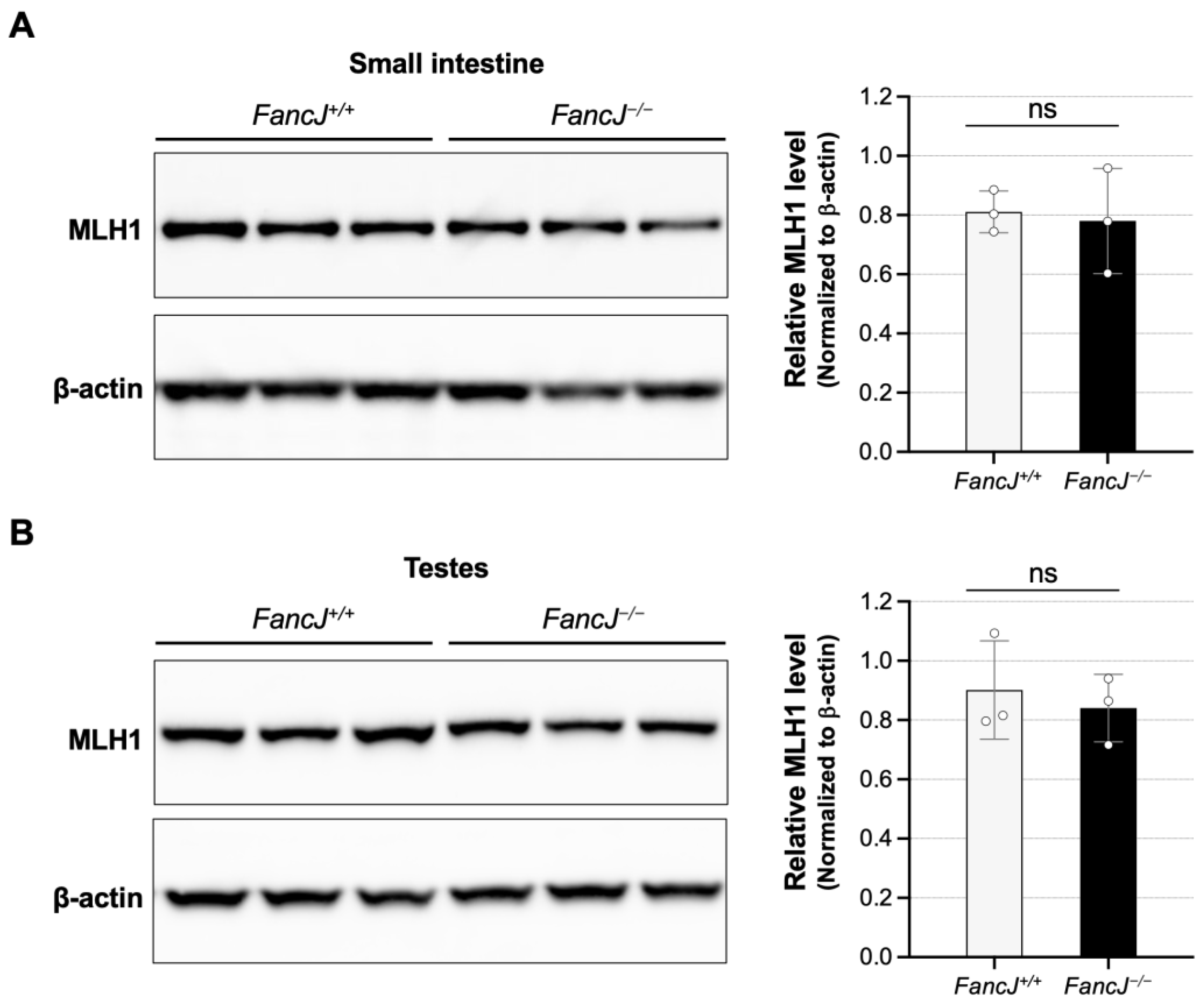
2.3. Loss of FANCJ Does Not Affect Fmr1 Transcription or the Levels of Key MMR Proteins Involved in Expansion
3. Discussion
4. Materials and Methods
4.1. Reagents and Services
4.2. Mouse Generation, Breeding, and Maintenance
4.3. DNA Isolation
4.4. Genotyping and Analysis of Repeat Number
4.5. Small-Pool PCR
4.6. Quantitation of mRNA
4.7. Western Blot
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hunter, J.E.; Berry-Kravis, E.; Hipp, H.; Todd, P.K. FMR1 Disorders. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lozano, R.; Rosero, C.A.; Hagerman, R.J. Fragile X spectrum disorders. Intractable Rare Dis. Res. 2014, 3, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Iong, K.P.; Tong, T.-H.; Lo, J.; Gane, L.W.; Berry-Kravis, E.; Nguyen, D.; Mu, L.Y.; Laffin, J.; Bailey, D.B.; et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012, 4, 100. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Kuhl, D.P.A.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkert, A.J.M.H.; Holden, J.J.A.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Usdin, K.; Hayward, B.E.; Kumari, D.; Lokanga, R.A.; Sciascia, N.; Zhao, X.-N. Repeat-mediated genetic and epigenetic changes at the FMR1 locus in the Fragile X-related disorders. Front. Genet. 2014, 5, 226. [Google Scholar] [CrossRef]
- Nolin, S.L.; Brown, W.T.; Glicksman, A.; Houck, J.G.E.; Gargano, A.D.; Sullivan, A.; Biancalana, V.; Bröndum-Nielsen, K.; Hjalgrim, H.; Holinski-Feder, E.; et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 2003, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Protic, D.; Allen, E.G.; Archibald, A.D.; Baud, A.; Brown, T.W.; Budimirovic, D.B.; Cohen, J.; Dufour, B.; Eiges, R.; et al. Insight and Recommendations for Fragile X-Premutation-Associated Conditions from the Fifth International Conference on FMR1 Premutation. Cells 2023, 12, 2330. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Tortora, N.; Allen, E.; Macpherson, J.; Mila, M.; Vianna-Morgante, A.M.; Sherman, S.L.; Dobkin, C.; Latham, G.J.; et al. Expansions and contractions of the FMR1 CGG repeat in 5508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. Part A 2019, 179, 1148–1156. [Google Scholar] [CrossRef]
- Lokanga, R.A.; Entezam, A.; Kumari, D.; Yudkin, D.; Qin, M.; Smith, C.B.; Usdin, K. Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum. Mutat. 2013, 34, 157–166. [Google Scholar] [CrossRef]
- Depienne, C.; Mandel, J.L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 2021, 108, 764–785. [Google Scholar] [CrossRef]
- Swami, M.; Hendricks, A.E.; Gillis, T.; Massood, T.; Mysore, J.; Myers, R.H.; Wheeler, V.C. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet. 2009, 18, 3039–3047. [Google Scholar] [CrossRef]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900.e14. [Google Scholar] [CrossRef]
- Morales, F.; Vásquez, M.; Corrales, E.; Vindas-Smith, R.; Santamaría-Ulloa, C.; Zhang, B.; Sirito, M.; Estecio, M.R.; Krahe, R.; Monckton, D.G. Longitudinal increases in somatic mosaicism of the expanded CTG repeat in myotonic dystrophy type 1 are associated with variation in age-at-onset. Hum. Mol. Genet. 2020, 29, 2496–2507. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.P.; MacDonald, M.E.; Wheeler, V.C.; Jones, L.; Holmans, P.; Orth, M.; Monckton, D.G.; Long, J.D.; Kwak, S.; Gusella, J.F.; et al. Huntington’s Disease Pathogenesis: Two Sequential Components. J. Huntington’s Dis. 2021, 10, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Aishworiya, R.; Hwang, Y.H.; Santos, E.; Hayward, B.; Usdin, K.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. Clinical implications of somatic allele expansion in female FMR1 premutation carriers. Sci. Rep. 2023, 13, 7050. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Benn, C.L.; Gibson, K.R.; Reynolds, D.S. Drugging DNA Damage Repair Pathways for Trinucleotide Repeat Expansion Diseases. J. Huntington’s Dis. 2021, 10, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Donaldson, J.; Flower, M.; Moss, D.J.H.; Tabrizi, S.J. Genetic modifiers of repeat expansion disorders. Emerg. Top. Life Sci. 2023, 7, 325–337. [Google Scholar]
- Zhao, X.; Kumari, D.; Miller, C.J.; Kim, G.-Y.; Hayward, B.; Vitalo, A.G.; Pinto, R.M.; Usdin, K. Modifiers of Somatic Repeat Instability in Mouse Models of Friedreich Ataxia and the Fragile X-Related Disorders: Implications for the Mechanism of Somatic Expansion in Huntington’s Disease. J. Huntington’s Dis. 2021, 10, 149–163. [Google Scholar] [CrossRef]
- Lokanga, R.A.; Zhao, X.N.; Usdin, K. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2014, 35, 129–136. [Google Scholar] [CrossRef]
- Zhao, X.N.; Kumari, D.; Gupta, S.; Wu, D.; Evanitsky, M.; Yang, W.; Usdin, K. Mutsβ generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum. Mol. Genet. 2015, 24, 7087–7096. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Lokanga, R.; Allette, K.; Gazy, I.; Wu, D.; Usdin, K. A MutSβ-Dependent Contribution of MutSα to Repeat Expansions in Fragile X Premutation Mice? PLoS Genet. 2016, 12, e1006190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Wilkins, K.; Edelmann, W.; Usdin, K. MutLγ promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. 2018, 14, e1007719. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.J.; Kim, G.-Y.; Zhao, X.; Usdin, K. All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the Fragile X-related disorders. PLoS Genet. 2020, 16, e1008902. [Google Scholar] [CrossRef]
- Walker, A.; Jimenez, D.A.; Usdin, K.; Zhao, X. PMS2 has both pro-mutagenic and anti-mutagenic effects on repeat instability in the Repeat Expansion Diseases. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zhao, X.N.; Usdin, K. Gender and cell-type-specific effects of the transcription-coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the fragile X-related disorders. Hum. Mutat. 2014, 35, 341–349. [Google Scholar] [CrossRef]
- Wheeler, V.C.; Dion, V. Modifiers of CAG/CTG Repeat Instability: Insights from Mammalian Models. J. Huntington’s Dis. 2021, 10, 123–148. [Google Scholar] [CrossRef]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef]
- Flower, M.; Lomeikaite, V.; Ciosi, M.; Cumming, S.; Morales, F.; Lo, K.; Moss, D.H.; Jones, L.; Holmans, P.; Monckton, D.G.; et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain 2019, 142, 1876–1886. [Google Scholar] [CrossRef]
- Bettencourt, C.; Hensman-Moss, D.; Flower, M.; Wiethoff, S.; Brice, A.; Goizet, C.; Stevanin, G.; Koutsis, G.; Karadima, G.; Panas, M.; et al. DNA repair pathways underlie a common genetic mechanism modulating onset in polyglutamine diseases. Ann. Neurol. 2016, 79, 983–990. [Google Scholar] [CrossRef]
- Hwang, Y.H.; Hayward, B.E.; Zafarullah, M.; Kumar, J.; Johnson, B.D.; Holmans, P.; Usdin, K.; Tassone, F. Both cis and trans-acting genetic factors drive somatic instability in female carriers of the FMR1 premutation. Sci. Rep. 2022, 12, 10419. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.J.; Usdin, K. Mismatch repair is a double-edged sword in the battle against microsatellite instability. Expert Rev. Mol. Med. 2022, 24, e32. [Google Scholar] [CrossRef] [PubMed]
- Palombo, F.; Iaccarino, I.; Nakajima, E.; Ikejima, M.; Shimada, T.; Jiricny, J. hMutSβ, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr. Biol. 1996, 6, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Habraken, Y.; Sung, P.; Prakash, L.; Prakash, S. Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Curr. Biol. 1996, 6, 1185–1187. [Google Scholar] [CrossRef]
- Gupta, S.; Gellert, M.; Yang, W. Mechanism of mismatch recognition revealed by human MutSbeta bound to unpaired DNA loops. Nat. Struct. Mol. Biol. 2011, 19, 72–78. [Google Scholar] [CrossRef]
- Adihe Lokanga, R.; Zhao, X.-N.; Entezam, A.; Usdin, K. X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: Implications for the mechanism of repeat expansion. Hum. Mol. Genet. 2014, 23, 4985–4994. [Google Scholar] [CrossRef]
- Goula, A.V.; Stys, A.; Chan, J.P.K.; Trottier, Y.; Festenstein, R.; Merienne, K. Transcription elongation and tissue-specific somatic CAG instability. PLoS Genet. 2012, 8, e1003051. [Google Scholar] [CrossRef]
- Khristich, A.N.; Mirkin, S.M. On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J. Biol. Chem. 2020, 295, 4134–4170. [Google Scholar] [CrossRef]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Borel, V.; Adelman, C.A.; Schindler, D.; Boulton, S.J. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015, 29, 2532–2546. [Google Scholar] [CrossRef]
- Gupta, R.; Sharma, S.; Sommers, J.A.; Jin, Z.; Cantor, S.B.; Brosh, R.M. Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J. Biol. Chem. 2005, 280, 25450–25460. [Google Scholar] [CrossRef]
- Sommers, J.A.; Rawtani, N.; Gupta, R.; Bugreev, D.V.; Mazin, A.V.; Cantor, S.B.; Brosh, R.M., Jr. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J. Biol. Chem. 2009, 284, 7505–7517. [Google Scholar] [CrossRef]
- London, T.B.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J. Biol. Chem. 2008, 283, 36132–36139. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shin-Ya, K.; Brosh, R.M., Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol. Cell. Biol. 2008, 28, 4116–4128. [Google Scholar] [CrossRef] [PubMed]
- Levitus, M.; Waisfisz, Q.; Godthelp, B.C.; de Vries, Y.; Hussain, S.; Wiegant, W.W.; Elghalbzouri-Maghrani, E.; Steltenpool, J.; A Rooimans, M.; Pals, G.; et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat. Genet. 2005, 37, 934–935. [Google Scholar] [CrossRef] [PubMed]
- Bridge, W.L.; Vandenberg, C.J.; Franklin, R.J.; Hiom, K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat. Genet. 2005, 37, 953–957. [Google Scholar] [CrossRef]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar] [CrossRef]
- Horan, T.S.; Ascenção, C.F.R.; Mellor, C.A.; Wang, M.; Smolka, M.B.; Cohen, P.E. The DNA helicase FANCJ (BRIP1) functions in double strand break repair processing, but not crossover formation during prophase I of meiosis in male mice. PLoS Genet. 2024, 20, e1011175. [Google Scholar] [CrossRef]
- Barthelemy, J.; Hanenberg, H.; Leffak, M. FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res. 2016, 44, 6803–6816. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Desetty, R.; Saito, T.T.; Schlabach, M.; Lach, F.P.; Sowa, M.E.; Clark, A.B.; Kunkel, T.A.; Harper, J.W.; Colaiácovo, M.P.; et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 2010, 39, 36–47. [Google Scholar] [CrossRef]
- Zhao, X.N.; Usdin, K. FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair 2018, 69, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lu, H.; Usdin, K. FAN1’s protection against CGG repeat expansion requires its nuclease activity and is FANCD2-independent. Nucleic Acids Res. 2021, 49, 11643–11652. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.M.; Dragileva, E.; Kirby, A.; Lloret, A.; Lopez, E.; Claire, J.S.; Panigrahi, G.B.; Hou, C.; Holloway, K.; Gillis, T.; et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome-wide and candidate approaches. PLoS Genet. 2013, 9, e1003930. [Google Scholar] [CrossRef]
- Hayward, B.; Kumari, D.; Santra, S.; van Karnebeek, C.D.M.; van Kuilenburg, A.B.P.; Usdin, K. All three MutL complexes are required for repeat expansion in a human stem cell model of CAG-repeat expansion mediated glutaminase deficiency. Sci. Rep. 2024, 14, 13772. [Google Scholar] [CrossRef]
- Cantor, S.B.; Xie, J. Assessing the link between BACH1/FANCJ and MLH1 in DNA crosslink repair. Environ. Mol. Mutagen. 2010, 51, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Guillemette, S.; Peng, M.; Gilbert, C.; Buermeyer, A.; Cantor, S.B. An MLH1 mutation links BACH1/FANCJ to colon cancer, signaling, and insight toward directed therapy. Cancer Prev. Res. 2010, 3, 1409–1416. [Google Scholar] [CrossRef]
- Peng, M.; Xie, J.; Ucher, A.; Stavnezer, J.; Cantor, S.B. Crosstalk between BRCA-Fanconi anemia and mismatch repair pathways prevents MSH2-dependent aberrant DNA damage responses. EMBO J. 2014, 33, 1698–1712. [Google Scholar] [CrossRef]
- Guillemette, S.; Branagan, A.; Peng, M.; Dhruva, A.; Schärer, O.D.; Cantor, S.B. FANCJ localization by mismatch repair is vital to maintain genomic integrity after UV irradiation. Cancer Res. 2014, 74, 932–944. [Google Scholar] [CrossRef]
- Isik, E.; Shukla, K.; Pospisilova, M.; König, C.; Andrs, M.; Rao, S.; Rosano, V.; Dobrovolna, J.; Krejci, L.; Janscak, P. MutSβ-MutLβ-FANCJ axis mediates the restart of DNA replication after fork stalling at cotranscriptional G4/R-loops. Sci. Adv. 2024, 10, eadk2685. [Google Scholar] [CrossRef]
- van den Broek, W.J.A.A.; Nelen, M.R.; Wansink, D.G.; Coerwinkel, M.M.; te Riele, H.; Groenen, P.J.T.A.; Wieringa, B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum. Mol. Genet. 2002, 11, 191–198. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Thornhill, A.R.; McMurray, C.T. Somatic deletion events occur during early embryonic development and modify the extent of CAG expansion in subsequent generations. Hum. Mol. Genet. 2004, 13, 3057–3068. [Google Scholar] [CrossRef]
- Higham, C.F.; Morales, F.; Cobbold, C.A.; Haydon, D.T.; Monckton, D.G. High levels of somatic DNA diversity at the myotonic dystrophy type 1 locus are driven by ultra-frequent expansion and contraction mutations. Hum. Mol. Genet. 2012, 21, 2450–2463. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kumari, D.; Sciascia, N.; Usdin, K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism 2016, 7, 42. [Google Scholar] [CrossRef]
- Zhao, X.; Gazy, I.; Hayward, B.; Pintado, E.; Hwang, Y.H.; Tassone, F.; Usdin, K. Repeat Instability in the Fragile X-Related Disorders: Lessons from a Mouse Model. Brain Sci. 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, M.; Pearson, C.E.; Thornton, C.A. Bidirectional transcription stimulates expansion and contraction of expanded (CTG)*(CAG) repeats. Hum. Mol. Genet. 2011, 20, 580–588. [Google Scholar] [CrossRef] [PubMed]
- McLean, Z.L.; Gao, D.; Correia, K.; Roy, J.C.L.; Shibata, S.; Farnum, I.N.; Valdepenas-Mellor, Z.; Kovalenko, M.; Rapuru, M.; Morini, E.; et al. Splice modulators target PMS1 to reduce somatic expansion of the Huntington’s disease-associated CAG repeat. Nat. Commun. 2024, 15, 3182. [Google Scholar] [CrossRef]
- Mathews, E.W.; Coffey, S.R.; Gärtner, A.; Belgrad, J.; Bragg, R.M.; O’Reilly, D.; Cantle, J.P.; McHugh, C.; Summers, A.; Fentz, J.; et al. Suppression of Huntington’s Disease Somatic Instability by Transcriptional Repression and Direct CAG Repeat Binding. bioRxiv 2024. [Google Scholar] [CrossRef]
- Brosh, R.M., Jr.; Cantor, S.B. Molecular and cellular functions of the FANCJ DNA helicase defective in cancer and in Fanconi anemia. Front. Genet. 2014, 5, 372. [Google Scholar] [CrossRef]
- Sun, X.; Brieño-Enríquez, M.A.; Cornelius, A.; Modzelewski, A.J.; Maley, T.T.; Campbell-Peterson, K.M.; Holloway, J.K.; Cohen, P.E. FancJ (Brip1) loss-of-function allele results in spermatogonial cell depletion during embryogenesis and altered processing of crossover sites during meiotic prophase I in mice. Chromosoma 2016, 125, 237–252. [Google Scholar] [CrossRef]
- Gomes-Pereira, M.; Fortune, M.T.; Ingram, L.; McAbney, J.P.; Monckton, D.G. Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: Implications for the mechanism of triplet repeat expansion. Hum. Mol. Genet. 2004, 13, 1815–1825. [Google Scholar] [CrossRef]
- Bourn, R.L.; De Biase, I.; Pinto, R.M.; Sandi, C.; Al-Mahdawi, S.; Pook, M.A.; Bidichandani, S.I. Pms2 suppresses large expansions of the (GAA.TTC)n sequence in neuronal tissues. PLoS ONE 2012, 7, e47085. [Google Scholar] [CrossRef] [PubMed]
- Halabi, A.; Fuselier, K.T.B.; Grabczyk, E. GAA*TTC repeat expansion in human cells is mediated by mismatch repair complex MutLgamma and depends upon the endonuclease domain in MLH3 isoform one. Nucleic Acids Res. 2018, 46, 4022–4032. [Google Scholar] [CrossRef]
- Mouro Pinto, R.; Murtha, R.; Azevedo, A.; Douglas, C.; Kovalenko, M.; Ulloa, J.; Crescenti, S.; Burch, Z.; Oliver, E.; Vitalo, A.; et al. Identification of genetic modifiers of Huntington’s disease somatic CAG repeat instability by in vivo CRISPR-Cas9 genome editing. bioRxiv 2024. [Google Scholar] [CrossRef]
- Deshmukh, A.L.; Porro, A.; Mohiuddin, M.; Lanni, S.; Panigrahi, G.B.; Caron, M.-C.; Masson, J.-Y.; Sartori, A.A.; Pearson, C.E. FAN1, a DNA Repair Nuclease, as a Modifier of Repeat Expansion Disorders. J. Huntington’s Dis. 2021, 10, 95–122. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Hong, E.P.; Shin, J.W.; Chao, M.J.; Loupe, J.; Gillis, T.; Mysore, J.S.; Holmans, P.; Jones, L.; Orth, M.; et al. Genetic and Functional Analyses Point to FAN1 as the Source of Multiple Huntington Disease Modifier Effects. Am. J. Hum. Genet. 2020, 107, 96–110. [Google Scholar] [CrossRef]
- Cannavo, E.; Gerrits, B.; Marra, G.; Schlapbach, R.; Jiricny, J. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 2007, 282, 2976–2986. [Google Scholar] [CrossRef]
- Segui, N.; Mina, L.B.; Lázaro, C.; Sanz-Pamplona, R.; Pons, T.; Navarro, M.; Bellido, F.; López-Doriga, A.; Valdés-Mas, R.; Pineda, M.; et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 2015, 149, 563–566. [Google Scholar] [CrossRef]
- Dhar, S.; Datta, A.; Brosh, R.M., Jr. DNA helicases and their roles in cancer. DNA Repair 2020, 96, 102994. [Google Scholar] [CrossRef]
- Frizzell, A.; Nguyen, J.H.; Petalcorin, M.I.; Turner, K.D.; Boulton, S.J.; Freudenreich, C.H.; Lahue, R.S. RTEL1 inhibits trinucleotide repeat expansions and fragility. Cell Rep. 2014, 6, 827–835. [Google Scholar] [CrossRef]
- Rastokina, A.; Cebrián, J.; Mozafari, N.; Mandel, N.H.; Smith, C.I.E.; Lopes, M.; Zain, R.; Mirkin, S.M. Large-scale expansions of Friedreich’s ataxia GAA*TTC repeats in an experimental human system: Role of DNA replication and prevention by LNA-DNA oligonucleotides and PNA oligomers. Nucleic Acids Res. 2023, 51, 8532–8549. [Google Scholar] [CrossRef]
- Yue, F.; Mouse Encode Consortium; Cheng, Y.; Breschi, A.; Vierstra, J.; Wu, W.; Ryba, T.; Sandstrom, R.; Samantha, K.; Davis, C.; et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014, 515, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Entezam, A.; Biacsi, R.; Orrison, B.; Saha, T.; Hoffman, G.E.; Grabczyk, E.; Nussbaum, R.L.; Usdin, K. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene 2007, 395, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; McHugh, C.; Coffey, S.R.; Jimenez, D.A.; Adams, E.; Carroll, J.B.; Usdin, K. Stool is a sensitive and noninvasive source of DNA for monitoring expansion in repeat expansion disease mouse models. Dis. Model. Mech. 2022, 15, dmm049453. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lu, H.; Dagur, P.K.; Usdin, K. Isolation and Analysis of the CGG-Repeat Size in Male and Female Gametes from a Fragile X Mouse Model. Methods Mol. Biol. 2020, 2056, 173–186. [Google Scholar]
- Hayward, B.E.; Zhou, Y.; Kumari, D.; Usdin, K. A Set of Assays for the Comprehensive Analysis of FMR1 Alleles in the Fragile X-Related Disorders. J. Mol. Diagn. 2016, 18, 762–774. [Google Scholar] [CrossRef]
- Ciosi, M.; Cumming, S.A.; Chatzi, A.; Larson, E.; Tottey, W.; Lomeikaite, V.; Hamilton, G.; Wheeler, V.C.; Pinto, R.M.; Kwak, S.; et al. Approaches to Sequence the HTT CAG Repeat Expansion and Quantify Repeat Length Variation. J. Huntington’s Dis. 2021, 10, 53–74. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimenez, D.A.; Walker, A.; Usdin, K.; Zhao, X. Tissue-Specific Effects of the DNA Helicase FANCJ/BRIP1/BACH1 on Repeat Expansion in a Mouse Model of the Fragile X-Related Disorders. Int. J. Mol. Sci. 2025, 26, 2655. https://doi.org/10.3390/ijms26062655
Jimenez DA, Walker A, Usdin K, Zhao X. Tissue-Specific Effects of the DNA Helicase FANCJ/BRIP1/BACH1 on Repeat Expansion in a Mouse Model of the Fragile X-Related Disorders. International Journal of Molecular Sciences. 2025; 26(6):2655. https://doi.org/10.3390/ijms26062655
Chicago/Turabian StyleJimenez, Diego Antonio, Alexandra Walker, Karen Usdin, and Xiaonan Zhao. 2025. "Tissue-Specific Effects of the DNA Helicase FANCJ/BRIP1/BACH1 on Repeat Expansion in a Mouse Model of the Fragile X-Related Disorders" International Journal of Molecular Sciences 26, no. 6: 2655. https://doi.org/10.3390/ijms26062655
APA StyleJimenez, D. A., Walker, A., Usdin, K., & Zhao, X. (2025). Tissue-Specific Effects of the DNA Helicase FANCJ/BRIP1/BACH1 on Repeat Expansion in a Mouse Model of the Fragile X-Related Disorders. International Journal of Molecular Sciences, 26(6), 2655. https://doi.org/10.3390/ijms26062655