
The Organogermanium Compound 3-(trihydroxygermyl)propanoic Acid Exerts Anti-Inflammatory Effects via Adenosine-NR4A2 Signaling
 ,
,
Abstract
:1. Introduction
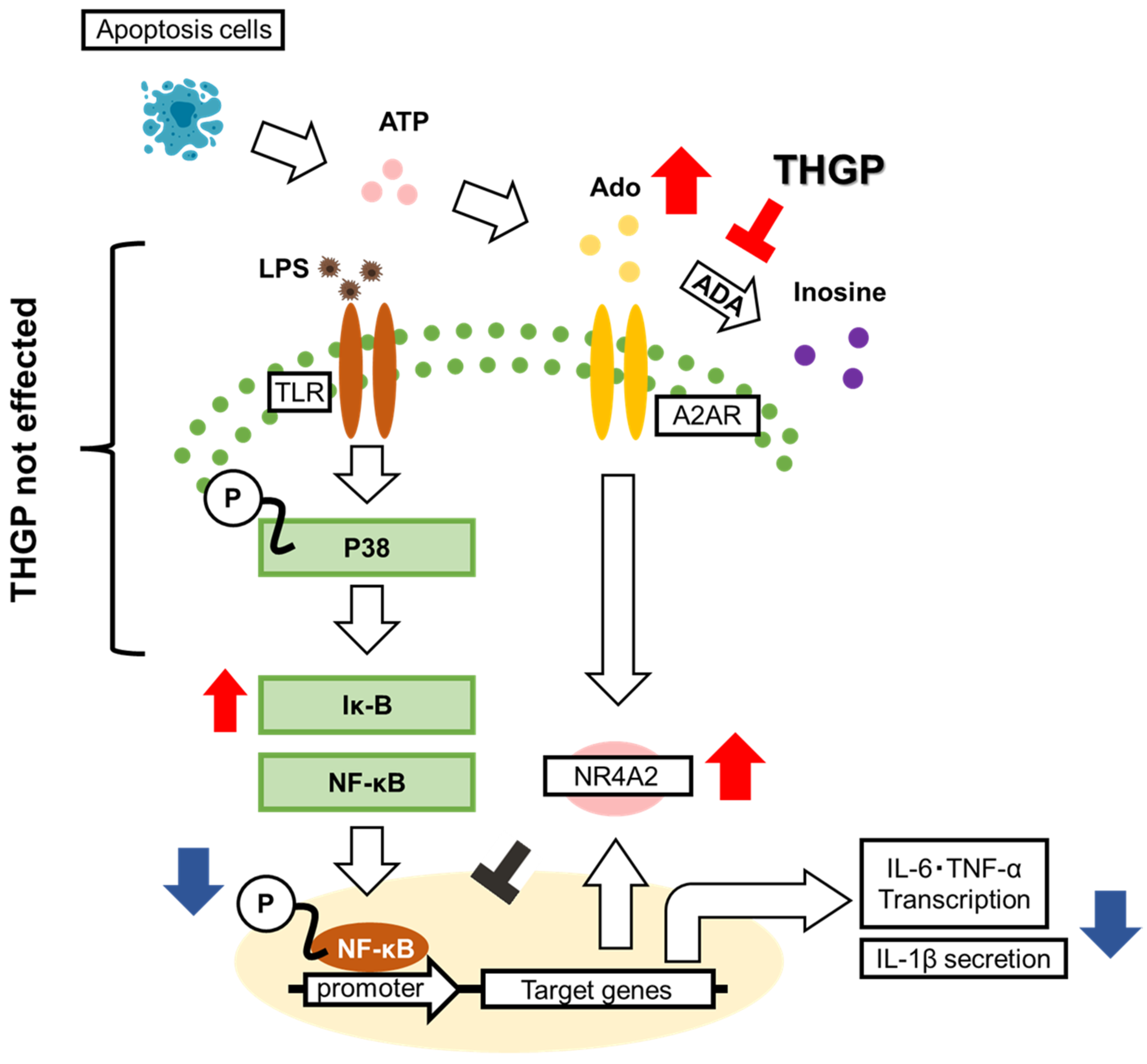
2. Results
2.1. THGP Suppresses the LPS-Induced Nuclear Translocation of NF-κB
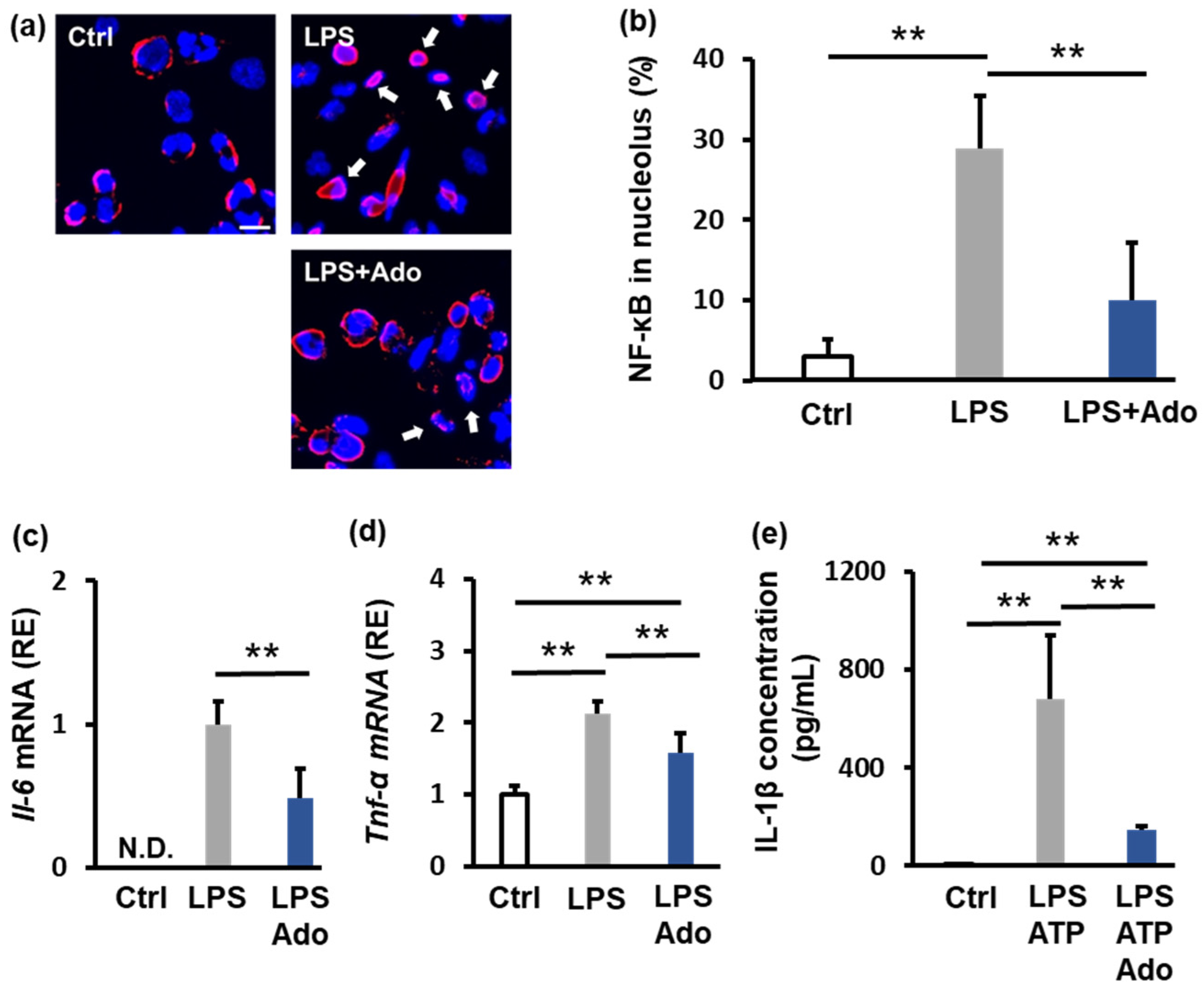
2.2. Adenosine Suppresses Inflammation
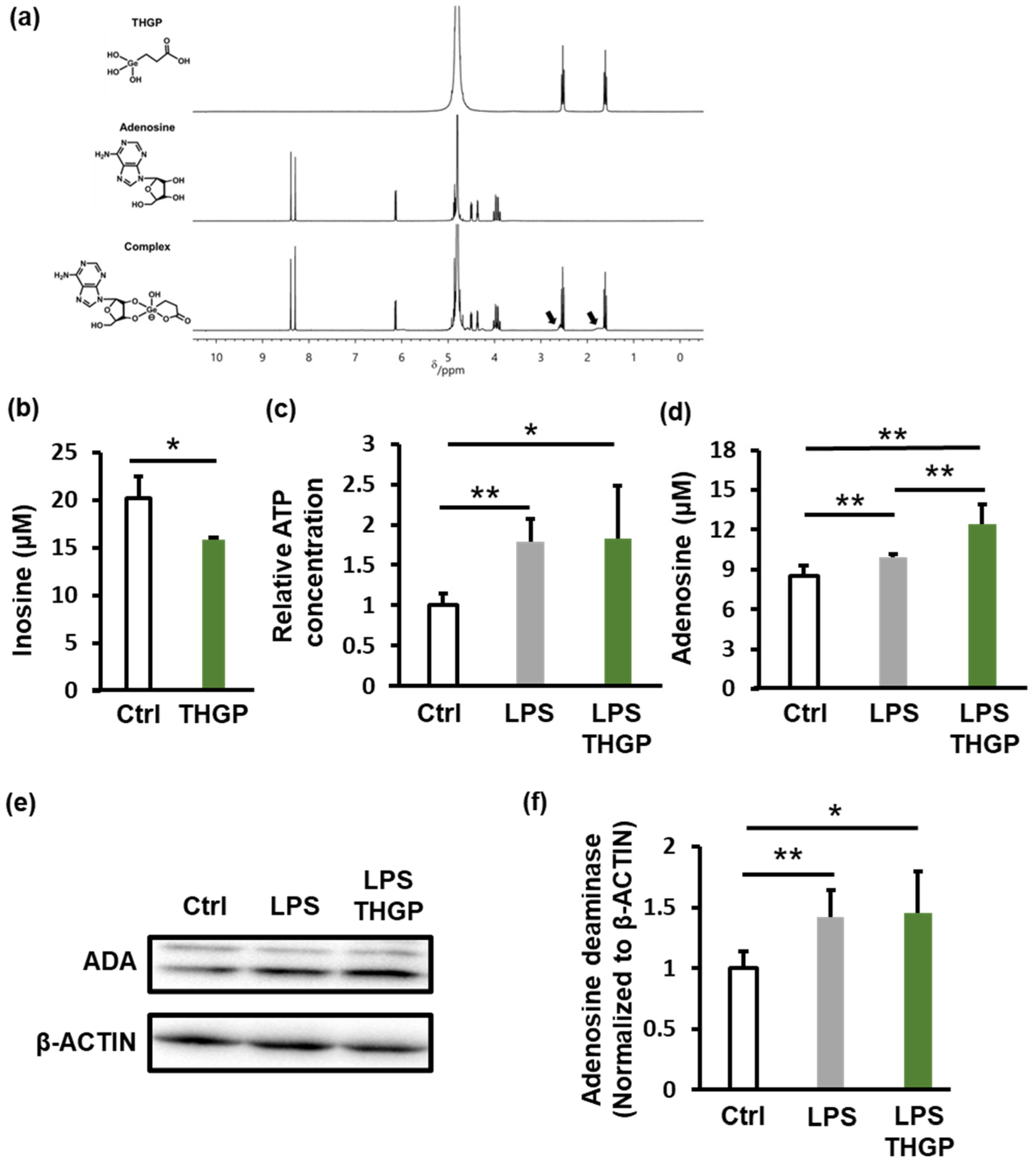
2.3. THGP Forms a Complex with Adenosine and Inhibits Its Degradation
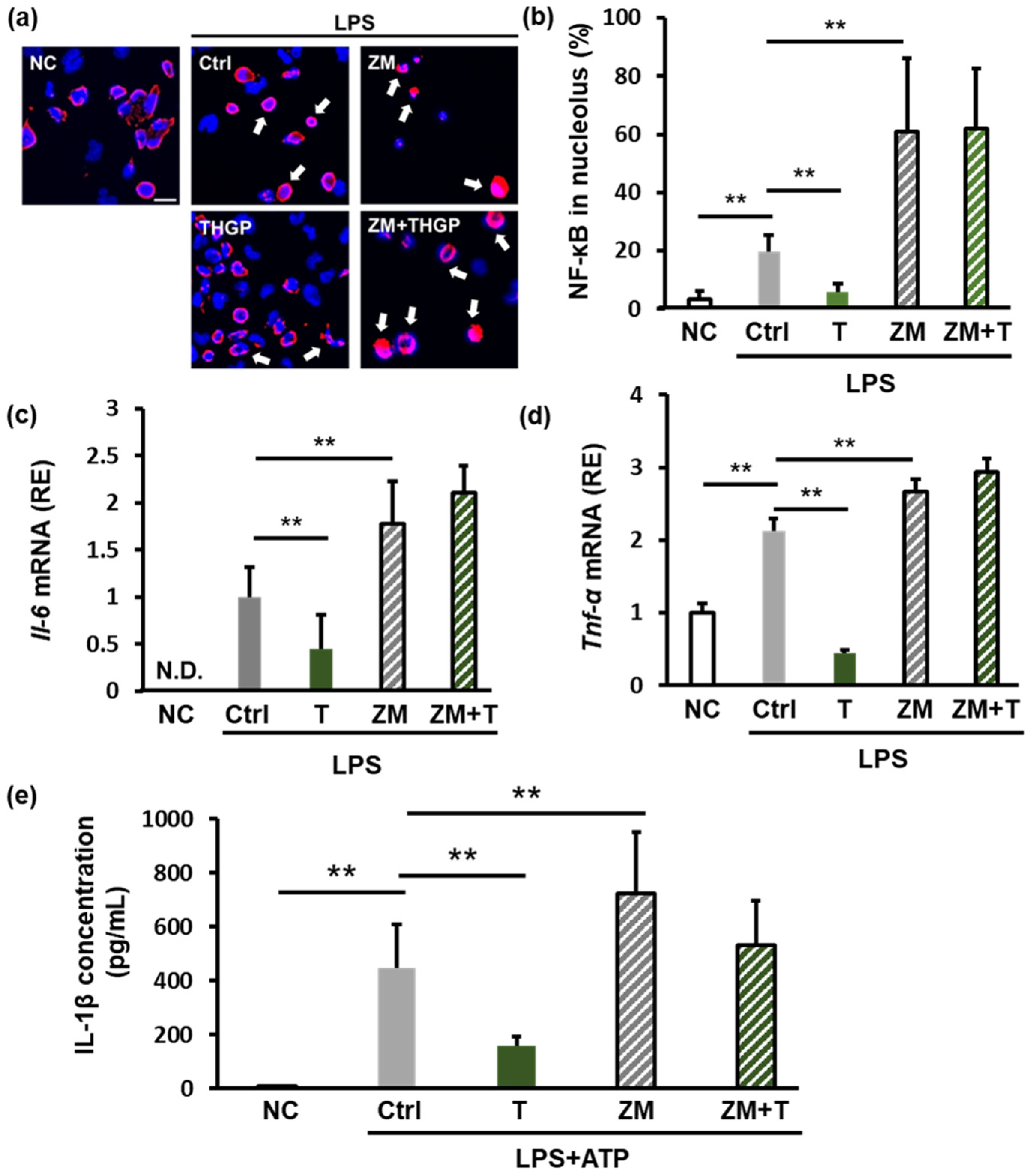
2.4. THGP Suppresses Inflammation by Forming a Complex with Adenosine
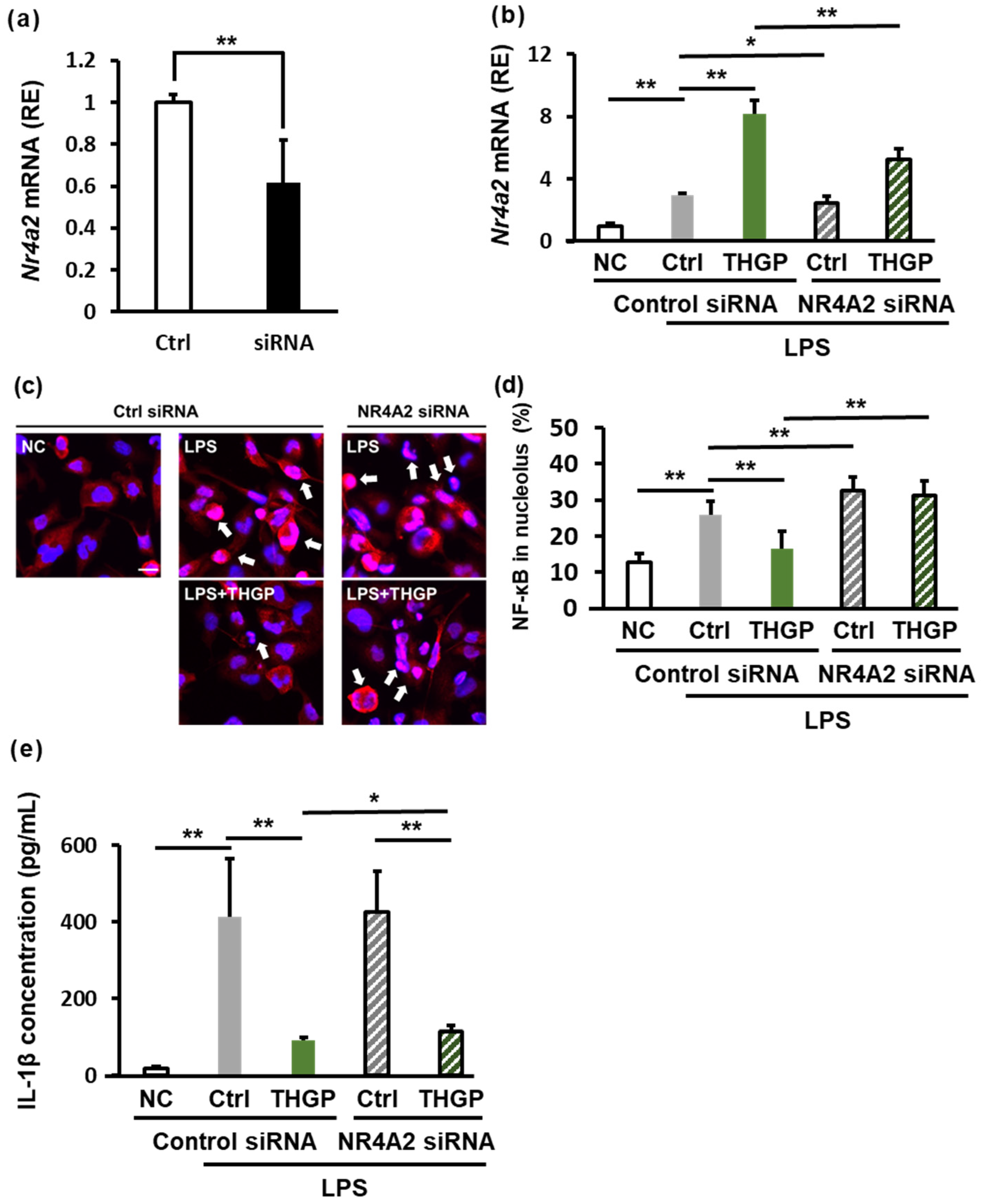
2.5. THGP Increases NR4A2 Expression
2.6. THGP Suppresses Inflammation by Regulating NR4A2 Expression
3. Discussion
4. Materials and Methods
4.1. Preparation of Reagents
4.2. Cell Culture
4.3. Western Blotting
4.4. Immunofluorescence Staining
4.5. RT‒PCR
4.6. Measurement of ATP Levels
4.7. Measurement of Adenosine Levels
4.8. Measurement of Inosine Levels
4.9. 1H-NMR Analysis
4.10. ELISA for IL-1β Quantification
4.11. Measurement of Inflammatory Markers After A2R Inhibitor Treatment
4.12. Measurement of Inflammatory Markers After NR4A2 Knockdown Using an siRNA
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nathan, C. Points of Control in Inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C. The Dual Roles of Neutrophils and Macrophages in Inflammation: A Critical Balance Between Tissue Damage and Repair. Yearb. Sports Med. 2008, 2008, 119–120. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Zou, X.-B.; Chai, Y.-F.; Yao, Y.-M. Macrophage Polarization in Inflammatory Diseases. Int. J. Biol. Sci. 2014, 10, 520–529. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Libby, P. Inflammatory Mechanisms: The Molecular Basis of Inflammation and Disease. Nutr. Rev. 2007, 65, S140–S146. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A. Mechanisms and Functions of P38 MAPK Signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-κB and IκB Proteins: Implications in Cancer and Inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Rathinam, V.A.K.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of Inflammasome Signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef]
- Gombault, A.; Baron, L.; Couillin, I. ATP Release and Purinergic Signaling in NLRP3 Inflammasome Activation. Front. Immunol. 2013, 3, 414. [Google Scholar] [CrossRef] [PubMed]
- Kumano, N.; Ishikawa, T.; Koinumaru, S.; Kikumoto, T.; Suzuki, S.; Nakai, Y.; Konno, K. Antitumor Effect of the Organogermanium Compound Ge-132 on the Lewis Lung Carcinoma (3LL) in C57Bl/6 (B6) Mice. Tohoku J. Exp. Med. 1985, 146, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Azumi, J.; Takeda, T.; Shimada, Y.; Zhuang, T.; Tokuji, Y.; Sakamoto, N.; Aso, H.; Nakamura, T. Organogermanium THGP Induces Differentiation into M1 Macrophages and Suppresses the Proliferation of Melanoma Cells via Phagocytosis. Int. J. Mol. Sci. 2023, 24, 1885. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, M.; Ohba, S.; Yukawa, M. Effect of Germanium, Poly-Trans-[2-Carboxyethyl]Germasesquioxane on Natural Killer(NK)Activity in Dog. J. Vet. Med. Sci. 2002, 64, 719–721. [Google Scholar] [CrossRef]
- Shimada, Y.; Sato, K.; Tokuji, Y.; Nakamura, T. Nuclear Magnetic Resonance Studies of the Interactions between the Organic Germanium Compound Ge-132 and Saccharides. Carbohydr. Res. 2015, 407, 10–15. [Google Scholar] [CrossRef]
- Azumi, J.; Shimada, Y.; Takeda, T.; Aso, H.; Nakamura, T. The Organogermanium Compound 3-(Trihydroxygermyl) Propanoic Acid (THGP) Suppresses Inflammasome Activation Via Complexation with ATP. Int. J. Mol. Sci. 2022, 23, 13364. [Google Scholar] [CrossRef]
- Nakamura, T.; Shimada, Y.; Takeda, T.; Sato, K.; Akiba, M.; Fukaya, H. Organogermanium Compound, Ge-132, Forms Complexes with Adrenaline, ATP and Other Physiologocal Cis-Diol Compounds. Future Med. Chem. 2015, 7, 1233–1246. [Google Scholar] [CrossRef]
- Azumi, J.; Takeda, T.; Shimada, Y.; Aso, H.; Nakamura, T. The Organogermanium Compound THGP Suppresses Melanin Synthesis via Complex Formation with L-DOPA on Mushroom Tyrosinase and in B16 4A5 Melanoma Cells. Int. J. Mol. Sci. 2019, 20, 4785. [Google Scholar] [CrossRef]
- Crean, D.; Cummins, E.P.; Bahar, B.; Mohan, H.; McMorrow, J.P.; Murphy, E.P. Adenosine Modulates NR4A Orphan Nuclear Receptors To Attenuate Hyperinflammatory Responses in Monocytic Cells. J. Immunol. 2015, 195, 1436–1448. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular Function and Molecular Structure of Ecto-Nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Murphree, L.J.; Sullivan, G.W.; Marshall, M.A.; Linden, J. Lipopolysaccharide Rapidly Modifies Adenosine Receptor Transcripts in Murine and Human Macrophages: Role of NF-κB in A2A Adenosine Receptor Induction. Biochem. J. 2005, 391, 575–580. [Google Scholar] [CrossRef]
- Haskó, G. Adenosine: An Endogenous Regulator of Innate Immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.-G. Adenosine Receptors as Therapeutic Targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Sato, K.; Takeda, T.; Tokuji, Y. The Organogermanium Compound Ge-132 Interacts with Nucleic Acid Components and Inhibits the Catalysis of Adenosine Substrate by Adenosine Deaminase. Biol. Trace Element Res. 2018, 181, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 Cell Line: An in Vitro Cell Model for Immune Modulation Approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef]
- Zito, G.; Buscetta, M.; Cimino, M.; Dino, P.; Bucchieri, F.; Cipollina, C. Cellular Models and Assays to Study NLRP3 Inflammasome Biology. Int. J. Mol. Sci. 2020, 21, 4294. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Doiyama, S.; Azumi, J.; Shimada, Y.; Tokuji, Y.; Yamaguchi, H.; Nagata, K.; Sakamoto, N.; Aso, H.; Nakamura, T. Organogermanium Suppresses Cell Death Due to Oxidative Stress in Normal Human Dermal Fibroblasts. Sci. Rep. 2019, 9, 13637. [Google Scholar] [CrossRef]
- Rodríguez-Calvo, R.; Tajes, M.; Vázquez-Carrera, M. The NR4A Subfamily of Nuclear Receptors: Potential New Therapeutic Targets for the Treatment of Inflammatory Diseases. Expert Opin. Ther. Targets 2017, 21, 291–304. [Google Scholar] [CrossRef]
- Wang, Y.; Teng, G.; Zhou, H.; Dong, C. Germanium Reduces Inflammatory Damage in Mammary Glands During Lipopolysaccharide-Induced Mastitis in Mice. Biol. Trace Element Res. 2020, 198, 617–626. [Google Scholar] [CrossRef]
- Aso, H.; Suzuki, F.; Yamaguchi, T.; Hayashi, Y.; Ebina, T.; Ishida, N. Induction of Interferon and Activation of NK Cells and Macrophages in Mice by Oral Administration of Ge-132, an Organic Germanium Compound. Microbiol. Immunol. 1985, 29, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, F.; Brutkiewicz, R.; Pollard, R. Cooperation of Lymphokine(s) in the Macrophages in Expression of Antitumor Activity of Carboxyethylgermanium Sesquioxide (Ge-132). Anticancer Res. 1986, 6, 177–182. [Google Scholar]
- Prónai, L.; Arimori, S. Decreased Plasma Superoxide Scavenging Activity in Immunological Disorders—Carboxyethylgermanium Sesquioxide (Ge-132) as a Promoter of Prednisolone. Biotherapy 1992, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Alunni, S.; Orrù, M.; Ottavi, L. A Study on the Inhibition of Adenosine Deaminase. J. Enzym. Inhib. Med. Chem. 2008, 23, 182–189. [Google Scholar] [CrossRef]
- Cory, J.G.; Suhadolnik, R.J. Structural Requirements of Nucleosides for Binding by Adenosine Deaminase. Biochemistry 1965, 4, 1729–1732. [Google Scholar] [CrossRef]
- García-Yagüe, Á.J.; Rada, P.; Rojo, A.I.; Lastres-Becker, I.; Cuadrado, A. Nuclear Import and Export Signals Control the Subcellular Localization of Nurr1 Protein in Response to Oxidative Stress. J. Biol. Chem. 2013, 288, 5506–5517. [Google Scholar] [CrossRef]
- Watanabe, T.; Sekine, S.; Naguro, I.; Sekine, Y.; Ichijo, H. Apoptosis Signal-Regulating Kinase 1 (ASK1)-P38 Pathway-Dependent Cytoplasmic Translocation of the Orphan Nuclear Receptor NR4A2 Is Required for Oxidative Stress-Induced Necrosis. J. Biol. Chem. 2015, 290, 10791–10803. [Google Scholar] [CrossRef]
- Mahajan, S.; Saini, A.; Chandra, V.; Nanduri, R.; Kalra, R.; Bhagyaraj, E.; Khatri, N.; Gupta, P. Nuclear Receptor Nr4a2 Promotes Alternative Polarization of Macrophages and Confers Protection in Sepsis. J. Biol. Chem. 2015, 290, 18304–18314. [Google Scholar] [CrossRef]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A Nurr1/CoREST Pathway in Microglia and Astrocytes Protects Dopaminergic Neurons from Inflammation-Induced Death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef]
- Hanna, R.N.; Shaked, I.; Hubbeling, H.G.; Punt, J.A.; Wu, R.; Herrley, E.; Zaugg, C.; Pei, H.; Geissmann, F.; Ley, K.; et al. NR4A1 (Nur77) Deletion Polarizes Macrophages Toward an Inflammatory Phenotype and Increases Atherosclerosis. Circ. Res. 2012, 110, 416–427. [Google Scholar] [CrossRef]
- Chen, H.; Yu, X.; Hu, L.; Peng, Y.; Yu, Q.; Zhou, H.; Xu, C.; Zeng, H.; Cao, Y.; Zhuang, J.; et al. Activation of Nurr1 with Amodiaquine Protected Neuron and Alleviated Neuroinflammation after Subarachnoid Hemorrhage in Rats. Oxidative Med. Cell. Longev. 2021, 2021, 6669787. [Google Scholar] [CrossRef]
- Varani, K.; Padovan, M.; Govoni, M.; Vincenzi, F.; Trotta, F.; Borea, P.A. The Role of Adenosine Receptors in Rheumatoid Arthritis. Autoimmun. Rev. 2010, 10, 61–64. [Google Scholar] [CrossRef]
- Sasaki, K.; Ishikawa, M.; Monma, K.; Takayanagi, C. Effect of Organogermanium Compound Ge-132, a Biological Response Modifier on the Rat Adjuvant Arthritis. Annu. Rep. Tohoku Coll. Pharm. 1983, 30, 143–147. [Google Scholar]
- Shigeru, A.; Miyoko, Y.; Kaori, I. Improved rheumatoid arthritis case with Ge-132 administration evaluated by clinically and immunologically using two-color flow cytometry. Jpn. J. Clin. Immunol. 1990, 13, 80–86. [Google Scholar] [CrossRef]
- Da Silva, J.; Gabriel-Costa, D.; Sudo, R.; Wang, H.; Groban, L.; Ferraz, E.; Nascimento, J.; Fraga, C.; Barreiro, E.; Zapata-Sudo, G. Adenosine A2A Receptor Agonist Prevents Cardiac Remodeling and Dysfunction in Spontaneously Hypertensive Male Rats after Myocardial Infarction. Drug Des. Dev. Ther. 2017, 11, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on Adenosine Receptors as a Potential Targeted Therapy in Human Diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Kutryb-Zajac, B.; Mierzejewska, P.; Slominska, E.M.; Smolenski, R.T. Therapeutic Perspectives of Adenosine Deaminase Inhibition in Cardiovascular Diseases. Molecules 2020, 25, 4652. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, Q.; Lv, H.; Wang, F.; Liu, R.; Zeng, N. Effect of Pulegone on the NLPR3 Inflammasome during Inflammatory Activation of THP-1 Cells. Exp. Ther. Med. 2020, 19, 1304–1312. [Google Scholar] [CrossRef]
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.R.; Flavell, R.A.; Cronstein, B.N.; Mehal, W.Z. Adenosine Is Required for Sustained Inflammasome Activation via the A2A Receptor and the HIF-1α Pathway. Nat. Commun. 2013, 4, 2909. [Google Scholar] [CrossRef]
- Zhang, T.; Fang, Z.; Linghu, K.; Liu, J.; Gan, L.; Lin, L. Small Molecule-driven SIRT3-autophagy-mediated NLRP3 Inflammasome Inhibition Ameliorates Inflammatory Crosstalk between Macrophages and Adipocytes. Br. J. Pharmacol. 2020, 177, 4645–4665. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target mRNA | Forward Primer Sequence (5′→3′) Reverse Primer Sequence (5′→3′) | Amplicon Size (bp) | Gene Reference |
|---|---|---|---|
| Tnf-α | TCAGCCTCTTCTCCTTCCTG GGCTACAGGCTTGTCACTCG | 173 | NM_000594.4 |
| Il-6 | GGATTCAATGAGGAGACTTGC GTTGGGTCAGGGGTGGTTAT | 197 | NM_001318095.2 |
| Nr4a2 | CTAACCTGCAGGCAGAACCTGAA ACATTTGTCTGAACTGCAACCA | 197 | NM_006186.4 |
| Rps18 | TTTGCGAGTACTCAACACCAACATC GAGCATATCTTCGGCCCACAC | 89 | NM_022551.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azumi, J.; Takeda, T.; Shibata, S.; Shimada, Y.; Aso, H.; Nakamura, T. The Organogermanium Compound 3-(trihydroxygermyl)propanoic Acid Exerts Anti-Inflammatory Effects via Adenosine-NR4A2 Signaling. Int. J. Mol. Sci. 2025, 26, 2449. https://doi.org/10.3390/ijms26062449
Azumi J, Takeda T, Shibata S, Shimada Y, Aso H, Nakamura T. The Organogermanium Compound 3-(trihydroxygermyl)propanoic Acid Exerts Anti-Inflammatory Effects via Adenosine-NR4A2 Signaling. International Journal of Molecular Sciences. 2025; 26(6):2449. https://doi.org/10.3390/ijms26062449
Chicago/Turabian StyleAzumi, Junya, Tomoya Takeda, Shunya Shibata, Yasuhiro Shimada, Hisashi Aso, and Takashi Nakamura. 2025. "The Organogermanium Compound 3-(trihydroxygermyl)propanoic Acid Exerts Anti-Inflammatory Effects via Adenosine-NR4A2 Signaling" International Journal of Molecular Sciences 26, no. 6: 2449. https://doi.org/10.3390/ijms26062449
APA StyleAzumi, J., Takeda, T., Shibata, S., Shimada, Y., Aso, H., & Nakamura, T. (2025). The Organogermanium Compound 3-(trihydroxygermyl)propanoic Acid Exerts Anti-Inflammatory Effects via Adenosine-NR4A2 Signaling. International Journal of Molecular Sciences, 26(6), 2449. https://doi.org/10.3390/ijms26062449