
The Role of Inflammation in the Pathogenesis of Comorbidity of Chronic Obstructive Pulmonary Disease and Pulmonary Tuberculosis



Abstract
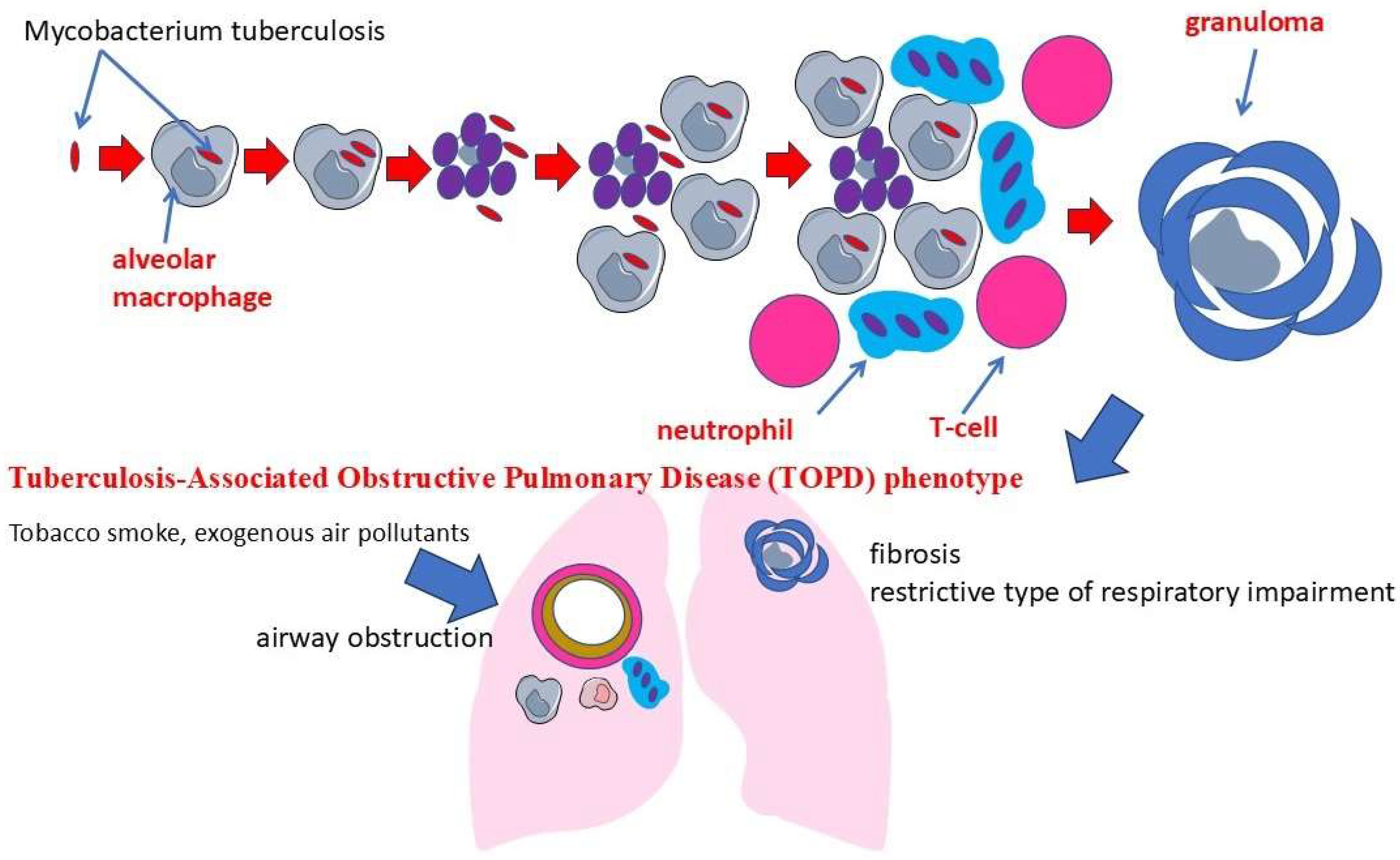
1. Introduction
2. Cytokines and Chemokines in the Pathogenesis of COPD and Tuberculosis Comorbidity: The Role of Inflammatory Mediators and Their Clinical Significance
3. Mechanisms of Cellular Inflammation in COPD and Tuberculosis Co-Morbidity: Contribution of Neutrophils, Macrophages and Dendritic Cells
3.1. The Role of Neutrophils
3.2. The Role of Macrophages
3.3. The Role of Dendritic Cells
4. Innate Immunity in COPD and Tuberculosis: Activation of Toll-like and Nod-like Receptors and Their Influence on Inflammation
5. Adaptive Immunity in COPD and Tuberculosis: The Role of T-Lymphocytes, B-Lymphocytes and Their Interactions
6. Signaling Pathways in the Pathogenesis of COPD and Tuberculosis: Influence of NF-κB, MAPK and JAK/STAT on Inflammation and Tissue Damage
7. Epigenetic Regulation of Inflammation in COPD and Tuberculosis: Role of DNA Methylation, MicroRNA and Histone Modification
8. Therapeutic Perspectives on the Study of Immune Mechanisms
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| COPD | chronic obstructive pulmonary disease |
| DCs | dendritic cells |
| IFN-γ | interferon gamma |
| IL | interleukin |
| NLRs | node-like receptors |
| PD-L1 | programmed cell death ligand 1 |
| TGF-β | transforming growth factor beta |
| Th | T-helper |
| TLRs | toll-like receptors |
| TNF-α | tumor necrosis factor-alpha |
| TOPD | Tuberculosis-Associated Obstructive Pulmonary Disease |
| Treg | regulatory T cells |
References
- Jing, C.; Zheng, H.; Wang, X.; Wang, Y.; Zhao, Y.; Liu, S.; Zhao, J.; Du, Q. Disease Burden of Tuberculosis and Post-Tuberculosis in Inner Mongolia, China, 2016–2018—Based on the Disease Burden of Post-TB Caused by COPD. BMC Infect. Dis. 2023, 23, 406. [Google Scholar] [CrossRef] [PubMed]
- Litvinjenko, S.; Magwood, O.; Wu, S.; Wei, X. Burden of Tuberculosis among Vulnerable Populations Worldwide: An Overview of Systematic Reviews. Lancet Infect. Dis. 2023, 23, 1395–1407. [Google Scholar] [CrossRef]
- Safiri, S.; Carson-Chahhoud, K.; Noori, M.; Nejadghaderi, S.A.; Sullman, M.J.M.; Ahmadian Heris, J.; Ansarin, K.; Mansournia, M.A.; Collins, G.S.; Kolahi, A.-A.; et al. Burden of Chronic Obstructive Pulmonary Disease and Its Attributable Risk Factors in 204 Countries and Territories, 1990-2019: Results from the Global Burden of Disease Study 2019. BMJ 2022, 378, e069679. [Google Scholar] [CrossRef] [PubMed]
- Nizov, A.A.; Ermachkova, A.N.; Abrosimov, V.N.; Ponomareva, I.B. Complex Assessment of the Degree of Chronic Obstructive Pulmonary Disease Copd Severity on Out-Patient Visit. I.P. Pavlov. Russ. Med. Biol. Her. 2019, 27, 59–65. [Google Scholar] [CrossRef]
- Prasiska, D.I.; Chapagain, D.D.; Osei, K.M.; Rajaguru, V.; Kang, S.J.; Kim, T.H.; Lee, S.G.; Han, W. Non-Communicable Comorbidities in Pulmonary Tuberculosis and Healthcare Utilization: A Cross-Sectional Study of 2021 Indonesian National Health Insurance Data. Arch. Public Health 2024, 82, 127. [Google Scholar] [CrossRef] [PubMed]
- Chakaya, J.; Khan, M.; Ntoumi, F.; Aklillu, E.; Fatima, R.; Mwaba, P.; Kapata, N.; Mfinanga, S.; Hasnain, S.E.; Katoto, P.D.M.C.; et al. Global Tuberculosis Report 2020—Reflections on the Global TB Burden, Treatment and Prevention Efforts. Int. J. Infect. Dis. 2021, 113, S7–S12. [Google Scholar] [CrossRef]
- Kahnert, K.; Jörres, R.A.; Behr, J.; Welte, T. The Diagnosis and Treatment of COPD and Its Comorbidities. Dtsch. Ärzteblatt Int. 2023, 120, 434–444. [Google Scholar] [CrossRef]
- Kotlyarov, S. Analysis of Differentially Expressed Genes and Signaling Pathways Involved in Atherosclerosis and Chronic Obstructive Pulmonary Disease. Biomol. Concepts 2022, 13, 34–54. [Google Scholar] [CrossRef]
- Matsunaga, K.; Harada, M.; Suizu, J.; Oishi, K.; Asami-Noyama, M.; Hirano, T. Comorbid Conditions in Chronic Obstructive Pulmonary Disease: Potential Therapeutic Targets for Unmet Needs. J. Clin. Med. 2020, 9, 3078. [Google Scholar] [CrossRef]
- Brassington, K.; Selemidis, S.; Bozinovski, S.; Vlahos, R. Chronic Obstructive Pulmonary Disease and Atherosclerosis: Common Mechanisms and Novel Therapeutics. Clin. Sci. 2022, 136, 405–423. [Google Scholar] [CrossRef]
- Goudis, C.A. Chronic Obstructive Pulmonary Disease and Atrial Fibrillation: An Unknown Relationship. J. Cardiol. 2017, 69, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Allwood, B.; Sun, Y. Advances in the Awareness of Tuberculosis-Associated Chronic Obstructive Pulmonary Disease. Chin. Med. J. Pulm. Crit. Care Med. 2024, 2, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Kamenar, K.; Hossen, S.; Gupte, A.N.; Siddharthan, T.; Pollard, S.; Chowdhury, M.; Rubinstein, A.L.; Irazola, V.E.; Gutierrez, L.; Miranda, J.J.; et al. Previous Tuberculosis Disease as a Risk Factor for Chronic Obstructive Pulmonary Disease: A Cross-Sectional Analysis of Multicountry, Population-Based Studies. Thorax 2022, 77, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Allwood, B.; Sun, Y. Post-Tuberculosis Lung Disease and Chronic Obstructive Pulmonary Disease. Chin. Med. J. 2023, 136, 1923–1928. [Google Scholar] [CrossRef]
- Xing, Z.; Sun, T.; Janssens, J.-P.; Chai, D.; Liu, W.; Tong, Y.; Wang, Y.; Ma, Y.; Pan, M.; Cui, J.; et al. Airflow Obstruction and Small Airway Dysfunction Following Pulmonary Tuberculosis: A Cross-Sectional Survey. Thorax 2023, 78, 274–280. [Google Scholar] [CrossRef]
- Amaral, A.F.S.; Coton, S.; Kato, B.; Tan, W.C.; Studnicka, M.; Janson, C.; Gislason, T.; Mannino, D.; Bateman, E.D.; Buist, S.; et al. Tuberculosis Associates with Both Airflow Obstruction and Low Lung Function: BOLD Results. Eur. Respir. J. 2015, 46, 1104–1112. [Google Scholar] [CrossRef]
- Hnizdo, E.; Singh, T.; Churchyard, G. Chronic Pulmonary Function Impairment Caused by Initial and Recurrent Pulmonary Tuberculosis Following Treatment. Thorax 2000, 55, 32–38. [Google Scholar] [CrossRef]
- Plit, M.L.; Anderson, R.; Van Rensburg, C.E.; Page-Shipp, L.; Blott, J.A.; Fresen, J.L.; Feldman, C. Influence of Antimicrobial Chemotherapy on Spirometric Parameters and Pro-Inflammatory Indices in Severe Pulmonary Tuberculosis. Eur. Respir. J. 1998, 12, 351–356. [Google Scholar] [CrossRef]
- Yakar, H.I.; Gunen, H.; Pehlivan, E.; Aydogan, S. The Role of Tuberculosis in COPD. Int. J. Chron. Obs. Pulmon Dis. 2017, 12, 323–329. [Google Scholar] [CrossRef]
- Lee, J.H.; Chang, J.H. Lung Function in Patients with Chronic Airflow Obstruction Due to Tuberculous Destroyed Lung. Respir. Med. 2003, 97, 1237–1242. [Google Scholar] [CrossRef]
- Karakasidis, E.; Kotsiou, O.S.; Gourgoulianis, K.I. Lung and Gut Microbiome in COPD. J. Pers. Med. 2023, 13, 804. [Google Scholar] [CrossRef] [PubMed]
- Alabi, F.O.; Alkhateeb, H.A.; DeBarros, K.M.; Barletti Benel, P.S.; Sanchez-Martez, R.L.; Zeper, M.L.; Ismail, R.A.; Umeh, F.; Medina-Villanueva, N. The Heterogeneity of COPD Patients in a Community-Based Practice and the Inadequacy of the Global Initiative for Chronic Obstructive Lung Disease Criteria: A Real-World Experience. Chronic Obstr. Pulm Dis. 2021, 8, 396–407. [Google Scholar] [CrossRef]
- Cavaillès, A.; Brinchault-Rabin, G.; Dixmier, A.; Goupil, F.; Gut-Gobert, C.; Marchand-Adam, S.; Meurice, J.-C.; Morel, H.; Person-Tacnet, C.; Leroyer, C.; et al. Comorbidities of COPD. Eur. Respir. Rev. 2013, 22, 454–475. [Google Scholar] [CrossRef]
- Zavala, M.J.; Becker, G.L.; Blount, R.J. Interrelationships between Tuberculosis and Chronic Obstructive Pulmonary Disease. Curr. Opin. Pulm. Med. 2023, 29, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Lee, Y.S.; Min, K.H.; Hur, G.Y.; Lee, S.Y.; Kang, K.H.; Rhee, C.K.; Park, S.J.; Shim, J.J. Difference in Systemic Inflammation and Predictors of Acute Exacerbation between Smoking-Associated COPD and Tuberculosis-Associated COPD. Int. J. Chron. Obs. Pulmon Dis. 2018, 13, 3381–3387. [Google Scholar] [CrossRef]
- Kotlyarov, S. The Role of Smoking in the Mechanisms of Development of Chronic Obstructive Pulmonary Disease and Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 8725. [Google Scholar] [CrossRef]
- Kotlyarov, S. Involvement of the Innate Immune System in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2022, 23, 985. [Google Scholar] [CrossRef]
- Aghasafari, P.; George, U.; Pidaparti, R. A Review of Inflammatory Mechanism in Airway Diseases. Inflamm. Res. 2019, 68, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Alderwick, L.J.; Harrison, J.; Lloyd, G.S.; Birch, H.L. The Mycobacterial Cell Wall—Peptidoglycan and Arabinogalactan. Cold Spring Harb. Perspect. Med. 2015, 5, a021113. [Google Scholar] [CrossRef]
- Brennan, P.J. Structure, Function, and Biogenesis of the Cell Wall of Mycobacterium Tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
- Mohammed Rizwan Babu Sait Decoding the Biosynthesis and Transport of α-Glucans in Mycobacterium Tuberculosis; Heinrich Heine University Düsseldorf: Düsseldorf, Germany, 2020.
- Nataraj, V.; Varela, C.; Javid, A.; Singh, A.; Besra, G.S.; Bhatt, A. Mycolic Acids: Deciphering and Targeting the A Chilles’ Heel of the Tubercle Bacillus. Mol. Microbiol. 2015, 98, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Singh, P.; Kolloli, A.; Shi, L.; Bushkin, Y.; Tyagi, S.; Subbian, S. Immunometabolism of Phagocytes During Mycobacterium Tuberculosis Infection. Front. Mol. Biosci. 2019, 6, 105. [Google Scholar] [CrossRef]
- Liu, C.H.; Liu, H.; Ge, B. Innate Immunity in Tuberculosis: Host Defense vs Pathogen Evasion. Cell Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef]
- Smith, I. Mycobacterium Tuberculosis Pathogenesis and Molecular Determinants of Virulence. Clin. Microbiol. Rev. 2003, 16, 463–496. [Google Scholar] [CrossRef] [PubMed]
- Ashenafi, S.; Brighenti, S. Reinventing the Human Tuberculosis (TB) Granuloma: Learning from the Cancer Field. Front. Immunol. 2022, 13, 1059725. [Google Scholar] [CrossRef]
- Parbhoo, T.; Mouton, J.M.; Sampson, S.L. Phenotypic Adaptation of Mycobacterium Tuberculosis to Host-Associated Stressors That Induce Persister Formation. Front. Cell. Infect. Microbiol. 2022, 12, 956607. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka-Chrebelska, K.H.; Mukherjee, D.; Maryanchik, S.V.; Rudzinska-Radecka, M. Biological and Genetic Mechanisms of COPD, Its Diagnosis, Treatment, and Relationship with Lung Cancer. Biomedicines 2023, 11, 448. [Google Scholar] [CrossRef]
- Stek, C.; Allwood, B.; Walker, N.F.; Wilkinson, R.J.; Lynen, L.; Meintjes, G. The Immune Mechanisms of Lung Parenchymal Damage in Tuberculosis and the Role of Host-Directed Therapy. Front. Microbiol. 2018, 9, 2603. [Google Scholar] [CrossRef]
- Sershen, C.L.; Salim, T.; May, E.E. Investigating the Comorbidity of COPD and Tuberculosis, a Computational Study. Front. Syst. Biol. 2023, 3, 940097. [Google Scholar] [CrossRef]
- Hu, S.; Yu, Q.; Liu, F.; Gong, F. A Novel Inflammatory Indicator for Tuberculosis-Associated Obstructive Pulmonary Disease (TOPD): The Systemic Inflammatory Response Index (SIRI). J. Inflamm. Res. 2024, 17, 4219–4228. [Google Scholar] [CrossRef]
- Siddharthan, T.; Gupte, A.; Barnes, P.J. Chronic Obstructive Pulmonary Disease Endotypes in Low- and Middle-Income Country Settings: Precision Medicine for All. Am. J. Respir. Crit. Care Med. 2020, 202, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Dai, Y.; Chang, J.; Xiang, P.; Liang, Z.; Yin, Y.; Shen, Y.; Wang, R.; Qiongda, B.; Chu, H.; et al. The Clinical Characteristics, Treatment and Prognosis of Tuberculosis-Associated Chronic Obstructive Pulmonary Disease: A Protocol for a Multicenter Prospective Cohort Study in China. COPD 2024, 19, 2097–2107. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim. Et. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Raby, K.L.; Michaeloudes, C.; Tonkin, J.; Chung, K.F.; Bhavsar, P.K. Mechanisms of Airway Epithelial Injury and Abnormal Repair in Asthma and COPD. Front. Immunol. 2023, 14, 1201658. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Verma, S.K.; Kumar, S.; Ahmad, M.K.; Nischal, A.; Singh, S.K.; Dixit, R.K. Correlation of Severity of Chronic Obstructive Pulmonary Disease with Potential Biomarkers. Immunol. Lett. 2018, 196, 1–10. [Google Scholar] [CrossRef]
- Nosik, M.; Ryzhov, K.; Kudryavtseva, A.; Kuimova, U.; Kravtchenko, A.; Sobkin, A.; Zverev, V.; Svitich, O. Decreased IL-1 β Secretion as a Potential Predictor of Tuberculosis Recurrence in Individuals Diagnosed with HIV. Biomedicines 2024, 12, 954. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.C.Y.; Hong, J.; Huang, C.; Yang, P.; Liao, S.K.; Chang, K.S.S. Increased TNF-α, IL-1β and IL-6 Levels in the Bronchoalveolar Lavage Fluid with the Upregulation of Their mRNA in Macrophages Lavaged from Patients with Active Pulmonary Tuberculosis. Tuber. Lung Dis. 1999, 79, 279–285. [Google Scholar] [CrossRef]
- Akira, S.; Hirano, T.; Taga, T.; Kishimoto, T. Biology of Multifunctional Cytokines: IL 6 and Related Molecules (IL 1 and TNF). FASEB J. 1990, 4, 2860–2867. [Google Scholar] [CrossRef]
- Barnes, P.J.; Celli, B.R. Systemic Manifestations and Comorbidities of COPD. Eur. Respir. J. 2009, 33, 1165–1185. [Google Scholar] [CrossRef]
- Choy, E.; Rose-John, S. Interleukin-6 as a Multifunctional Regulator: Inflammation, Immune Response, and Fibrosis. J. Scleroderma Relat. Disord. 2017, 2, S1–S5. [Google Scholar] [CrossRef]
- Dienz, O.; Rincon, M. The Effects of IL-6 on CD4 T Cell Responses. Clin. Immunol. 2009, 130, 27–33. [Google Scholar] [CrossRef]
- Ritter, K.; Rousseau, J.; Hölscher, C. The Role of Gp130 Cytokines in Tuberculosis. Cells 2020, 9, 2695. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, S.; Reyes-Aldasoro, C.C.; Candel, S.; Renshaw, S.A.; Mulero, V.; Calado, A. Cxcl8 (IL-8) Mediates Neutrophil Recruitment and Behavior in the Zebrafish Inflammatory Response. J. Immunol. 2013, 190, 4349–4359. [Google Scholar] [CrossRef]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic Obstructive Pulmonary Disease: Molecular and Cellularmechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef]
- Chung, K.F. Inflammatory Mediators in Chronic Obstructive Pulmonary Disease. Curr. Drug Targets Inflamm. Allergy 2005, 4, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.F. Chapter 17—Airway Mucus Hypersecretion in Asthma and COPD: Not the Same? In Asthma and COPD, 2nd ed.; Barnes, P.J., Drazen, J.M., Rennard, S.I., Thomson, N.C., Eds.; Academic Press: Oxford, UK, 2009; pp. 211–223. ISBN 978-0-12-374001-4. [Google Scholar]
- Ameixa, C.; Friedland, J.S. Interleukin-8 Secretion from Mycobacterium Tuberculosis-Infected Monocytes Is Regulated by Protein Tyrosine Kinases but Not by ERK1/2 or P38 Mitogen-Activated Protein Kinases. Infect. Immun. 2002, 70, 4743–4746. [Google Scholar] [CrossRef]
- Cavalcante-Silva, L.H.A.; Almeida, F.S.; Andrade, A.G.D.; Comberlang, F.C.; Cardoso, L.L.; Vanderley, S.E.R.; Keesen, T.S.L. Mycobacterium Tuberculosis in a Trap: The Role of Neutrophil Extracellular Traps in Tuberculosis. IJMS 2023, 24, 11385. [Google Scholar] [CrossRef] [PubMed]
- Makena, P.; Kikalova, T.; Prasad, G.L.; Baxter, S.A. Oxidative Stress and Lung Fibrosis: Towards an Adverse Outcome Pathway. IJMS 2023, 24, 12490. [Google Scholar] [CrossRef]
- Tsitoura, D.C.; Rothman, P.B. Enhancement of MEK/ERK Signaling Promotes Glucocorticoid Resistance in CD4+ T Cells. J. Clin. Invest. 2004, 113, 619–627. [Google Scholar] [CrossRef]
- Silva, D.A.A.d.; Silva, M.V.d.; Barros, C.C.O.; Alexandre, P.B.D.; Timóteo, R.P.; Catarino, J.S.; Sales-Campos, H.; Machado, J.R.; Rodrigues, D.B.R.; Oliveira, C.J.; et al. TNF-α Blockade Impairs in Vitro Tuberculous Granuloma Formation and down Modulate Th1, Th17 and Treg Cytokines. PLoS ONE 2018, 13, e0194430. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Bean, A.G.D.; Demangel, C.; France, M.P.; Briscoe, H.; Britton, W.J. TNF Regulates Chemokine Induction Essential for Cell Recruitment, Granuloma Formation, and Clearance of Mycobacterial Infection. J. Immunol. 2002, 168, 4620–4627. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; O’Dee, D.; Hamilton, T.; Nau, G.J. Cytokines Involved in Interferon-Gamma Production by Human Macrophages. J. Innate Immun. 2010, 2, 56–65. [Google Scholar] [CrossRef]
- Southworth, T.; Metryka, A.; Lea, S.; Farrow, S.; Plumb, J.; Singh, D. IFN-γ Synergistically Enhances LPS Signalling in Alveolar Macrophages from COPD Patients and Controls by Corticosteroid-resistant STAT1 Activation. Br. J. Pharmacol. 2012, 166, 2070–2083. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Young, H.A.; Rosenberg, A.S. An Overview of Cytokines and Cytokine Antagonists as Therapeutic Agents. Ann. N. Y Acad. Sci. 2009, 1182, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, X.; Huang, C.; Lin, Y.; Dai, Q. Change of Serum Inflammatory Cytokines Levels in Patients With Chronic Obstructive Pulmonary Disease, Pneumonia and Lung Cancer. Technol. Cancer Res. Treat. 2020, 19, 1533033820951807. [Google Scholar] [CrossRef]
- Carreto-Binaghi, L.E.; Sartillo-Mendoza, L.G.; Muñoz-Torrico, M.; Guzmán-Beltrán, S.; Carranza, C.; Torres, M.; González, Y.; Juárez, E. Serum Pro-Inflammatory Biomarkers Associated with Improvement in Quality of Life in Pulmonary Tuberculosis. Front. Immunol. 2023, 14, 1241121. [Google Scholar] [CrossRef]
- Yadav, A.K.; Gu, W.; Zhang, T.; Xu, X.; Yu, L. Current Perspectives on Biological Therapy for COPD. COPD J. Chronic Obstr. Pulm. Dis. 2023, 20, 197–209. [Google Scholar] [CrossRef]
- Appleton, L.K.; Hanania, N.A.; Adrish, M. Personalized COPD Care: The Future of Precision-Based Therapies. J. Clin. Med. 2024, 13, 6339. [Google Scholar] [CrossRef]
- Venkatesan, P. GOLD COPD Report: 2025 Update. Lancet Respir. Med. 2025, 13, e7–e8. [Google Scholar] [CrossRef]
- Affairs, E. A Randomized, Double-Blind, Placebo Controlled, Exploratory Study to Assess the Safety and Efficacy of Multiple Doses of ACZ885 in Chronic Obstructive Pulmonary Disease (COPD) Patients. 2011. Available online: https://clinicaltrials.gov/ (accessed on 15 February 2025).
- Dentener, M.A.; Creutzberg, E.C.; Pennings, H.-J.; Rijkers, G.T.; Mercken, E.; Wouters, E.F.M. Effect of Infliximab on Local and Systemic Inflammation in Chronic Obstructive Pulmonary Disease: A Pilot Study. Respiration 2008, 76, 275–282. [Google Scholar] [CrossRef]
- van der Vaart, H.; Koëter, G.H.; Postma, D.S.; Kauffman, H.F.; ten Hacken, N.H.T. First Study of Infliximab Treatment in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2005, 172, 465–469. [Google Scholar] [CrossRef]
- Rennard, S.I.; Flavin, S.K.; Agarwal, P.K.; Lo, K.H.; Barnathan, E.S. Long-Term Safety Study of Infliximab in Moderate-to-Severe Chronic Obstructive Pulmonary Disease. Respir. Med. 2013, 107, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Aaron, S.D.; Vandemheen, K.L.; Maltais, F.; Field, S.K.; Sin, D.D.; Bourbeau, J.; Marciniuk, D.D.; FitzGerald, J.M.; Nair, P.; Mallick, R. TNFα Antagonists for Acute Exacerbations of COPD: A Randomised Double-Blind Controlled Trial. Thorax 2013, 68, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Mahler, D.A.; Huang, S.; Tabrizi, M.; Bell, G.M. Efficacy and Safety of a Monoclonal Antibody Recognizing Interleukin-8 in COPD: A Pilot Study. Chest 2004, 126, 926–934. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Alhamlan, F.S.; Al-Qahtani, A.A. Pro-Inflammatory and Anti-Inflammatory Interleukins in Infectious Diseases: A Comprehensive Review. Trop. Med. Infect. Dis. 2024, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Norazmi, M.-N. Pattern Recognition Receptors and Cytokines in Mycobacterium Tuberculosis Infection—The Double-Edged Sword? BioMed Res. Int. 2013, 2013, 179174. [Google Scholar] [CrossRef]
- Ashenafi, S.; Aderaye, G.; Bekele, A.; Zewdie, M.; Aseffa, G.; Hoang, A.T.N.; Carow, B.; Habtamu, M.; Wijkander, M.; Rottenberg, M.; et al. Progression of Clinical Tuberculosis Is Associated with a Th2 Immune Response Signature in Combination with Elevated Levels of SOCS3. Clin. Immunol. 2014, 151, 84–99. [Google Scholar] [CrossRef]
- Pyrillou, K.; Burzynski, L.C.; Clarke, M.C.H. Alternative Pathways of IL-1 Activation, and Its Role in Health and Disease. Front. Immunol. 2020, 11, 613170. [Google Scholar] [CrossRef]
- Winchell, C.G.; Mishra, B.B.; Phuah, J.Y.; Saqib, M.; Nelson, S.J.; Maiello, P.; Causgrove, C.M.; Ameel, C.L.; Stein, B.; Borish, H.J.; et al. Evaluation of IL-1 Blockade as an Adjunct to Linezolid Therapy for Tuberculosis in Mice and Macaques. Front. Immunol. 2020, 11, 891. [Google Scholar] [CrossRef]
- Turner, R.D.; Chiu, C.; Churchyard, G.J.; Esmail, H.; Lewinsohn, D.M.; Gandhi, N.R.; Fennelly, K.P. Tuberculosis Infectiousness and Host Susceptibility. J. Infect. Dis. 2017, 216, S636–S643. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, P.; Sada-Ovalle, I.; Nishimura, T.; Anderson, A.C.; Kuchroo, V.K.; Remold, H.G.; Behar, S.M. IL-1β Promotes Antimicrobial Immunity in Macrophages by Regulating TNFR Signaling and Caspase-3 Activation. J. Immunol. 2013, 190, 4196–4204. [Google Scholar] [CrossRef]
- Martinez, A.N.; Mehra, S.; Kaushal, D. Role of Interleukin 6 in Innate Immunity to Mycobacterium Tuberculosis Infection. J. Infect. Dis. 2013, 207, 1253–1261. [Google Scholar] [CrossRef]
- Druszczyńska, M.; Godkowicz, M.; Kulesza, J.; Wawrocki, S.; Fol, M. Cytokine Receptors—Regulators of Antimycobacterial Immune Response. IJMS 2022, 23, 1112. [Google Scholar] [CrossRef] [PubMed]
- Romero-Adrian, T.B. Role of Cytokines and Other Factors Involved in the Mycobacterium Tuberculosis Infection. WJI 2015, 5, 16–50. [Google Scholar] [CrossRef]
- Boni, F.G.; Hamdi, I.; Koundi, L.M.; Shrestha, K.; Xie, J. Cytokine Storm in Tuberculosis and IL-6 Involvement. Infect. Genet. Evol. 2022, 97, 105166. [Google Scholar] [CrossRef]
- Averbakh, M.M.; Ergeshow, A. Interaction between Mycobacterium Tuberculosis and Human Host: Role of Cytokines in Pathogenesis and Treatment Monitoring. In Tuberculosis; Kayembe, J.-M.N., Ed.; InTech: London, UK, 2018; ISBN 978-1-78923-781-8. [Google Scholar]
- Ritter, K.; Rousseau, J.; Hölscher, C. Interleukin-27 in Tuberculosis: A Sheep in Wolf’s Clothing? Front. Immunol. 2022, 12, 810602. [Google Scholar] [CrossRef]
- Cavalli, G.; Colafrancesco, S.; Emmi, G.; Imazio, M.; Lopalco, G.; Maggio, M.C.; Sota, J.; Dinarello, C.A. Interleukin 1α: A Comprehensive Review on the Role of IL-1α in the Pathogenesis and Treatment of Autoimmune and Inflammatory Diseases. Autoimmun. Rev. 2021, 20, 102763. [Google Scholar] [CrossRef]
- Bonecini-Almeida, M.G.; Ho, J.L.; Boéchat, N.; Huard, R.C.; Chitale, S.; Doo, H.; Geng, J.; Rego, L.; Lazzarini, L.C.O.; Kritski, A.L.; et al. Down-Modulation of Lung Immune Responses by Interleukin-10 and Transforming Growth Factor Beta (TGF-Beta) and Analysis of TGF-Beta Receptors I and II in Active Tuberculosis. Infect. Immun. 2004, 72, 2628–2634. [Google Scholar] [CrossRef]
- Sellami, M.; Fazaa, A.; Cheikh, M.; Miladi, S.; Ouenniche, K.; Ennaifer, R.; Ben Abdelghani, K.; Laatar, A. Screening for Latent Tuberculosis Infection Prior to Biologic Therapy in Patients with Chronic Immune-Mediated Inflammatory Diseases (IMID): Interferon-Gamma Release Assay (IGRA) versus Tuberculin Skin Test (TST). Egypt. Rheumatol. 2019, 41, 225–230. [Google Scholar] [CrossRef]
- Rodríguez-Jiménez, P.; Mir-Viladrich, I.; Chicharro, P.; Solano-López, G.; López-Longo, F.J.; Taxonera, C.; Sánchez-Martínez, P.; Martínez-Lacasa, X.; García-Gasalla, M.; Dorca, J.; et al. Prevention and Treatment of Tuberculosis Infection in Candidates for Biologic Therapy: A Multidisciplinary Consensus Statement Adapted to the Dermatology Patient. Actas Dermo-Sifiliográficas (Engl. Ed. ) 2018, 109, 584–601. [Google Scholar] [CrossRef]
- Kramnik, I.; Beamer, G. Mouse Models of Human TB Pathology: Roles in the Analysis of Necrosis and the Development of Host-Directed Therapies. Semin. Immunopathol. 2016, 38, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Aogáin, M.M.; Jaggi, T.K.; Chotirmall, S.H. The Airway Microbiome: Present and Future Applications. Arch. De. Bronconeumol. 2022, 58, 8–10. [Google Scholar] [CrossRef]
- Lin, L.; Yi, X.; Liu, H.; Meng, R.; Li, S.; Liu, X.; Yang, J.; Xu, Y.; Li, C.; Wang, Y.; et al. The Airway Microbiome Mediates the Interaction between Environmental Exposure and Respiratory Health in Humans. Nat. Med. 2023, 29, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Sethi, S.; Murphy, T.; Nariya, S.; Boushey, H.A.; Lynch, S.V. Airway Microbiome Dynamics in Exacerbations of Chronic Obstructive Pulmonary Disease. J. Clin. Microbiol. 2014, 52, 2813–2823. [Google Scholar] [CrossRef]
- Kayongo, A.; Robertson, N.M.; Siddharthan, T.; Ntayi, M.L.; Ndawula, J.C.; Sande, O.J.; Bagaya, B.S.; Kirenga, B.; Mayanja-Kizza, H.; Joloba, M.L.; et al. Airway Microbiome-Immune Crosstalk in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2023, 13, 1085551. [Google Scholar] [CrossRef]
- Sin, D.D. Chronic Obstructive Pulmonary Disease and the Airway Microbiome: What Respirologists Need to Know. Tuberc. Respir. Dis. 2023, 86, 166–175. [Google Scholar] [CrossRef]
- Luo, L.; Tang, J.; Du, X.; Li, N. Chronic Obstructive Pulmonary Disease and the Airway Microbiome: A Review for Clinicians. Respir. Med. 2024, 225, 107586. [Google Scholar] [CrossRef]
- Chung, K.F.; Adcock, I.M. Multifaceted Mechanisms in COPD: Inflammation, Immunity, and Tissue Repair and Destruction. Eur. Respir. J. 2008, 31, 1334–1356. [Google Scholar] [CrossRef]
- Jasper, A.E.; McIver, W.J.; Sapey, E.; Walton, G.M. Understanding the Role of Neutrophils in Chronic Inflammatory Airway Disease. F1000Res 2019, 8, F1000 Faculty Rev-557. [Google Scholar] [CrossRef]
- Teng, T.-S.; Ji, A.; Ji, X.-Y.; Li, Y.-Z. Neutrophils and Immunity: From Bactericidal Action to Being Conquered. J. Immunol. Res. 2017, 2017, 9671604. [Google Scholar] [CrossRef] [PubMed]
- Faurschou, M.; Borregaard, N. Neutrophil Granules and Secretory Vesicles in Inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.A.; Boskovic, D.S. Neutrophil Extracellular DNA Traps in Response to Infection or Inflammation, and the Roles of Platelet Interactions. Int. J. Mol. Sci. 2024, 25, 3025. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.R.; Azcutia, V.; Newton, G.; Alcaide, P.; Luscinskas, F.W. Emerging Mechanisms of Neutrophil Recruitment across Endothelium. Trends Immunol. 2011, 32, 461–469. [Google Scholar] [CrossRef]
- Tsioumpekou, M.; Krijgsman, D.; Leusen, J.H.W.; Olofsen, P.A. The Role of Cytokines in Neutrophil Development, Tissue Homing, Function and Plasticity in Health and Disease. Cells 2023, 12, 1981. [Google Scholar] [CrossRef]
- Bordon, J.; Aliberti, S.; Fernandez-Botran, R.; Uriarte, S.M.; Rane, M.J.; Duvvuri, P.; Peyrani, P.; Morlacchi, L.C.; Blasi, F.; Ramirez, J.A. Understanding the Roles of Cytokines and Neutrophil Activity and Neutrophil Apoptosis in the Protective versus Deleterious Inflammatory Response in Pneumonia. Int. J. Infect. Dis. 2013, 17, e76–e83. [Google Scholar] [CrossRef]
- Ham, J.; Kim, J.; Ko, Y.G.; Kim, H.Y. The Dynamic Contribution of Neutrophils in the Chronic Respiratory Diseases. Allergy Asthma Immunol. Res. 2022, 14, 361–378. [Google Scholar] [CrossRef]
- Kraus, R.F.; Gruber, M.A. Neutrophils—From Bone Marrow to First-Line Defense of the Innate Immune System. Front. Immunol. 2021, 12, 767175. [Google Scholar] [CrossRef]
- Prince, L.R.; Allen, L.; Jones, E.C.; Hellewell, P.G.; Dower, S.K.; Whyte, M.K.B.; Sabroe, I. The Role of Interleukin-1beta in Direct and Toll-like Receptor 4-Mediated Neutrophil Activation and Survival. Am. J. Pathol. 2004, 165, 1819–1826. [Google Scholar] [CrossRef]
- Owen, C. Roles for Proteinases in the Pathogenesis of Chronic Obstructive Pulmonary Disease. COPD 2008, 3, 253–268. [Google Scholar] [CrossRef]
- Song, Z.; Bhattacharya, S.; Clemens, R.A.; Dinauer, M.C. Molecular Regulation of Neutrophil Swarming in Health and Disease: Lessons from the Phagocyte Oxidase. iScience 2023, 26, 108034. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, C.A.; Glassman, I.; Nguyen, K.H.; Parthasarathy, A.; Venketaraman, V. Neutrophils in Mycobacterium Tuberculosis. Vaccines 2023, 11, 631. [Google Scholar] [CrossRef] [PubMed]
- Hult, C.; Mattila, J.T.; Gideon, H.P.; Linderman, J.J.; Kirschner, D.E. Neutrophil Dynamics Affect Mycobacterium Tuberculosis Granuloma Outcomes and Dissemination. Front. Immunol. 2021, 12, 712457. [Google Scholar] [CrossRef]
- Ravimohan, S.; Kornfeld, H.; Weissman, D.; Bisson, G.P. Tuberculosis and Lung Damage: From Epidemiology to Pathophysiology. Eur. Respir. Rev. 2018, 27, 170077. [Google Scholar] [CrossRef]
- Liu, J.; Pang, Z.; Wang, G.; Guan, X.; Fang, K.; Wang, Z.; Wang, F. Advanced Role of Neutrophils in Common Respiratory Diseases. J. Immunol. Res. 2017, 2017, 6710278. [Google Scholar] [CrossRef] [PubMed]
- Muefong, C.N.; Sutherland, J.S. Neutrophils in Tuberculosis-Associated Inflammation and Lung Pathology. Front. Immunol. 2020, 11, 962. [Google Scholar] [CrossRef]
- Nikonova, A.A.; Khaitov, M.R.; Khaitov, R.M. Characteristics and Role of Macrophages in Pathogenesis of Acute and Chronic Lung Diseases. Med. Immunol. 2017, 19, 657–672. [Google Scholar] [CrossRef]
- Redente, E.F.; Higgins, D.M.; Dwyer-Nield, L.D.; Orme, I.M.; Gonzalez-Juarrero, M.; Malkinson, A.M. Differential Polarization of Alveolar Macrophages and Bone Marrow-Derived Monocytes Following Chemically and Pathogen-Induced Chronic Lung Inflammation. J. Leukoc. Biol. 2010, 88, 159–168. [Google Scholar] [CrossRef]
- Barrios, M.T.H.; Rojas, M.T.; Carvajal, E.J.; Díaz, E.S. Molecular Mechanisms of the Immune Response in Human Pulmonary Tuberculosis. Rev. Del. Inst. Nac. De. Enfermedades Respir. 2005, 18, 327–336. [Google Scholar]
- Marakalala, M.J.; Martinez, F.O.; Plüddemann, A.; Gordon, S. Macrophage Heterogeneity in the Immunopathogenesis of Tuberculosis. Front. Microbiol. 2018, 9, 1028. [Google Scholar] [CrossRef]
- Yu, J.; Tang, S. Advances in the Role of Macrophage Polarization in Tuberculosis. Chin. J. Clin. Infect. Dis. 2019, 12, 229–235. [Google Scholar] [CrossRef]
- Hussain Bhat, K.; Mukhopadhyay, S. Macrophage Takeover and the Host-Bacilli Interplay during Tuberculosis. Future Microbiol. 2015, 10, 853–872. [Google Scholar] [CrossRef]
- Sapey, E.; Stockley, R.A. The Neutrophil and Its Special Role in Chronic Obstructive Pulmonary Disease. In Asthma and COPD: Basic Mechanisms and Clinical Management; Academic Press: Oxford, UK, 2008; pp. 173–191. [Google Scholar]
- Porto, B.N.; Stein, R.T. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front. Immunol. 2016, 7, 311. [Google Scholar] [CrossRef]
- Borkute, R.R.; Woelke, S.; Pei, G.; Dorhoi, A. Neutrophils in Tuberculosis: Cell Biology, Cellular Networking and Multitasking in Host Defense. Int. J. Mol. Sci. 2021, 22, 4801. [Google Scholar] [CrossRef]
- Marakalala, M.J.; Fisher, K.L.; Rajkumar-Bhugeloo, K.; Moodley, D.; Mpotje, T.; Ramsuran, D.; Ndung’u, T. Investigating Neutrophil Cell Death in TB Pathogenesis. Gates Open Res. 2021, 5, 175. [Google Scholar] [CrossRef]
- Muefong, C.N.; Owolabi, O.; Donkor, S.; Charalambous, S.; Mendy, J.; Sey, I.C.M.; Bakuli, A.; Rachow, A.; Geldmacher, C.; Sutherland, J.S. Major Neutrophil-Derived Soluble Mediators Associate With Baseline Lung Pathology and Post-Treatment Recovery in Tuberculosis Patients. Front. Immunol. 2021, 12, 740933. [Google Scholar] [CrossRef]
- Chellappan, D.K.; Yee, L.W.; Xuan, K.Y.; Kunalan, K.; Rou, L.C.; Jean, L.S.; Ying, L.Y.; Wie, L.X.; Chellian, J.; Mehta, M.; et al. Targeting Neutrophils Using Novel Drug Delivery Systems in Chronic Respiratory Diseases. Drug Dev. Res. 2020, 81, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Shin, S.J. Pathological and Protective Roles of Dendritic Cells in Mycobacterium Tuberculosis Infection: Interaction between Host Immune Responses and Pathogen Evasion. Front. Cell. Infect. Microbiol. 2022, 12, 891878. [Google Scholar] [CrossRef]
- Freeman, C.M.; Curtis, J.L.; Chensue, S.W. CC Chemokine Receptor 5 and CXC Chemokine Receptor 6 Expression by Lung CD8+ Cells Correlates with Chronic Obstructive Pulmonary Disease Severity. Am. J. Pathol. 2007, 171, 767–776. [Google Scholar] [CrossRef]
- Vassallo, R.; Walters, P.R.; Lamont, J.; Kottom, T.J.; Yi, E.S.; Limper, A.H. Cigarette Smoke Promotes Dendritic Cell Accumulation in COPD; a Lung Tissue Research Consortium Study. Respir. Res. 2010, 11, 45. [Google Scholar] [CrossRef]
- Lu, Y.-B.; Xiao, D.-Q.; Liang, K.-D.; Zhang, J.-A.; Wang, W.-D.; Yu, S.-Y.; Zheng, B.-Y.; Gao, Y.-C.; Dai, Y.-C.; Jia, Y.; et al. Profiling Dendritic Cell Subsets in the Patients with Active Pulmonary Tuberculosis. Mol. Immunol. 2017, 91, 86–96. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in Immunoregulation and Therapeutics. Sig Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Akata, K.; van Eeden, S.F. Lung Macrophage Functional Properties in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2020, 21, 853. [Google Scholar] [CrossRef]
- Ross, E.A.; Devitt, A.; Johnson, J.R. Macrophages: The Good, the Bad, and the Gluttony. Front. Immunol. 2021, 12, 708186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of Inflammatory Cells in Airway Remodeling in COPD. COPD 2018, 13, 3341–3348. [Google Scholar] [CrossRef]
- Maphasa, R.E.; Meyer, M.; Dube, A. The Macrophage Response to Mycobacterium Tuberculosis and Opportunities for Autophagy Inducing Nanomedicines for Tuberculosis Therapy. Front. Cell Infect. Microbiol. 2020, 10, 618414. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J.; Lin, P.L. Macrophages and Control of Granulomatous Inflammation in Tuberculosis. Mucosal Immunol. 2011, 4, 271–278. [Google Scholar] [CrossRef]
- Chai, Q.; Zhang, Y.; Liu, C.H. Mycobacterium Tuberculosis: An Adaptable Pathogen Associated With Multiple Human Diseases. Front. Cell Infect. Microbiol. 2018, 8, 158. [Google Scholar] [CrossRef]
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 Macrophages and Their Overlaps - Myth or Reality? Clin. Sci. 2023, 137, 1067–1093. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their Role, Activation and Polarization in Pulmonary Diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 Polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic Reprogramming in Macrophage Responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Finicelli, M.; Digilio, F.A.; Galderisi, U.; Peluso, G. The Emerging Role of Macrophages in Chronic Obstructive Pulmonary Disease: The Potential Impact of Oxidative Stress and Extracellular Vesicle on Macrophage Polarization and Function. Antioxidants 2022, 11, 464. [Google Scholar] [CrossRef] [PubMed]
- Bo, H.; Moure, U.A.E.; Yang, Y.; Pan, J.; Li, L.; Wang, M.; Ke, X.; Cui, H. Mycobacterium Tuberculosis-Macrophage Interaction: Molecular Updates. Front. Cell Infect. Microbiol. 2023, 13, 1062963. [Google Scholar] [CrossRef] [PubMed]
- Hussell, T.; Bell, T.J. Alveolar Macrophages: Plasticity in a Tissue-Specific Context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef]
- Labonte, A.C.; Tosello-Trampont, A.-C.; Hahn, Y.S. The Role of Macrophage Polarization in Infectious and Inflammatory Diseases. Mol. Cells 2014, 37, 275–285. [Google Scholar] [CrossRef]
- Zhu, J.; Qiu, Y.; Valobra, M.; Qiu, S.; Majumdar, S.; Matin, D.; De Rose, V.; Jeffery, P.K. Plasma Cells and IL-4 in Chronic Bronchitis and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2007, 175, 1125–1133. [Google Scholar] [CrossRef]
- Li, M.; Wang, M.; Wen, Y.; Zhang, H.; Zhao, G.-N.; Gao, Q. Signaling Pathways in Macrophages: Molecular Mechanisms and Therapeutic Targets. MedComm (2020) 2023, 4, e349. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, L.E.; Sheedy, F.J.; Palsson-McDermott, E.M.; Triglia, D.; O’Leary, S.M.; O’Sullivan, M.P.; O’Neill, L.A.J.; Keane, J. Cutting Edge: Mycobacterium Tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J. Immunol. 2016, 196, 2444–2449. [Google Scholar] [CrossRef]
- Mehrotra, P.; Jamwal, S.V.; Saquib, N.; Sinha, N.; Siddiqui, Z.; Manivel, V.; Chatterjee, S.; Rao, K.V.S. Pathogenicity of Mycobacterium Tuberculosis Is Expressed by Regulating Metabolic Thresholds of the Host Macrophage. PLoS Pathog. 2014, 10, e1004265. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-Directed Therapy of Tuberculosis Based on Interleukin-1 and Type I Interferon Crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef]
- Gupta, P.K.; Jahagirdar, P.; Tripathi, D.; Devarajan, P.V.; Kulkarni, S. Macrophage Targeted Polymeric Curcumin Nanoparticles Limit Intracellular Survival of Mycobacterium Tuberculosis through Induction of Autophagy and Augment Anti-TB Activity of Isoniazid in RAW 264.7 Macrophages. Front. Immunol. 2023, 14, 1233630. [Google Scholar] [CrossRef]
- Boscardin, S.B.; Dudziak, D.; Münz, C.; Rosa, D.S. Editorial: Harnessing the Participation of Dendritic Cells in Immunity and Tolerance. Front. Immunol. 2020, 11, 595841. [Google Scholar] [CrossRef]
- Condon, T.V.; Sawyer, R.T.; Fenton, M.J.; Riches, D.W.H. Lung Dendritic Cells at the Innate-Adaptive Immune Interface. J. Leukoc. Biol. 2011, 90, 883–895. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Dendritic Cell Subsets in Primary and Secondary T Cell Responses at Body Surfaces. Nat. Immunol. 2009, 10, 1237–1244. [Google Scholar] [CrossRef]
- Chapoval, S.; Dasgupta, P.; Dorsey, N.J.; Keegan, A.D. Regulation of the T Helper Cell Type 2 (Th2)/T Regulatory Cell (Treg) Balance by IL-4 and STAT6. J. Leukoc. Biol. 2010, 87, 1011–1018. [Google Scholar] [CrossRef]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef]
- Madan-Lala, R.; Sia, J.K.; King, R.; Adekambi, T.; Monin, L.; Khader, S.A.; Pulendran, B.; Rengarajan, J. Mycobacterium Tuberculosis Impairs Dendritic Cell Functions through the Serine Hydrolase Hip1. J. Immunol. 2014, 192, 4263–4272. [Google Scholar] [CrossRef] [PubMed]
- Trinath, J.; Maddur, M.S.; Kaveri, S.V.; Balaji, K.N.; Bayry, J. Mycobacterium Tuberculosis Promotes Regulatory T-Cell Expansion via Induction of Programmed Death-1 Ligand 1 (PD-L1, CD274) on Dendritic Cells. J. Infect. Dis. 2012, 205, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Skokos, D.; Nussenzweig, M.C. CD8- DCs Induce IL-12-Independent Th1 Differentiation through Delta 4 Notch-like Ligand in Response to Bacterial LPS. J. Exp. Med. 2007, 204, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Verhagen, J.; Blaser, K.; Akdis, M.; Akdis, C.A. Mechanisms of Immune Suppression by Interleukin-10 and Transforming Growth Factor-β: The Role of T Regulatory Cells. Immunology 2006, 117, 433–442. [Google Scholar] [CrossRef]
- Hasegawa, H.; Matsumoto, T. Mechanisms of Tolerance Induction by Dendritic Cells In Vivo. Front. Immunol. 2018, 9, 350. [Google Scholar] [CrossRef]
- Rao, Y.; Gai, X.; Le, Y.; Xiong, J.; Liu, Y.; Zhang, X.; Wang, J.; Cao, W.; Sun, Y. Enhanced Proinflammatory Cytokine Production and Immunometabolic Impairment of NK Cells Exposed to Mycobacterium Tuberculosis and Cigarette Smoke. Front. Cell. Infect. Microbiol. 2022, 11, 799276. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273, Table of Contents. [Google Scholar] [CrossRef]
- Le, J.; Kulatheepan, Y.; Jeyaseelan, S. Role of Toll-like Receptors and Nod-like Receptors in Acute Lung Infection. Front. Immunol. 2023, 14, 1249098. [Google Scholar] [CrossRef]
- Mishra, A.K.; Driessen, N.N.; Appelmelk, B.J.; Besra, G.S. Lipoarabinomannan and Related Glycoconjugates: Structure, Biogenesis and Role in Mycobacterium Tuberculosis Physiology and Host-Pathogen Interaction. FEMS Microbiol. Rev. 2011, 35, 1126–1157. [Google Scholar] [CrossRef]
- Perros, F.; Lambrecht, B.N.; Hammad, H. TLR4 Signalling in Pulmonary Stromal Cells Is Critical for Inflammation and Immunity in the Airways. Respir. Res. 2011, 12, 125. [Google Scholar] [CrossRef]
- Lafferty, E.I.; Qureshi, S.T.; Schnare, M. The Role of Toll-like Receptors in Acute and Chronic Lung Inflammation. J. Inflamm. 2010, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Lea, S.; Reynolds, S.; Kaur, M.; Simpson, K.; Hall, S.; Hessel, E.; Singh, D. The Effects of Repeated Toll-like Receptors 2 and 4 Stimulation in COPD Alveolar Macrophages. COPD 2018, 13, 771–780. [Google Scholar] [CrossRef]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like Receptors in Microbial Recognition and Host Defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Yao, J.; Sterling, K.; Wang, Z.; Zhang, Y.; Song, W. The Role of Inflammasomes in Human Diseases and Their Potential as Therapeutic Targets. Sig Transduct. Target. Ther. 2024, 9, 10. [Google Scholar] [CrossRef]
- Li, Y.; Qiang, R.; Cao, Z.; Wu, Q.; Wang, J.; Lyu, W. NLRP3 Inflammasomes: Dual Function in Infectious Diseases. J. Immunol. 2024, 213, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Ferwerda, G.; Girardin, S.E.; Kullberg, B.-J.; Le Bourhis, L.; De Jong, D.J.; Langenberg, D.M.L.; Van Crevel, R.; Adema, G.J.; Ottenhoff, T.H.M.; Van Der Meer, J.W.M.; et al. NOD2 and Toll-like Receptors Are Nonredundant Recognition Systems of Mycobacterium Tuberculosis. PLoS Pathog. 2005, 1, 0279–0285. [Google Scholar] [CrossRef]
- Haw, T.J.; Starkey, M.R.; Pavlidis, S.; Fricker, M.; Arthurs, A.L.; Nair, P.M.; Liu, G.; Hanish, I.; Kim, R.Y.; Foster, P.S.; et al. Toll-like Receptor 2 and 4 Have Opposing Roles in the Pathogenesis of Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L298–L317. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Q.; Sun, W.; Zhou, X.; Fu, D.; Mao, L. New Insights into the Role of NLRP3 Inflammasome in Pathogenesis and Treatment of Chronic Obstructive Pulmonary Disease. J. Inflamm. Res. 2021, 14, 4155–4168. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Z.; Zheng, Y.; Yu, Q.; Zeng, M.; Bai, L.; Yang, L.; Guo, M.; Jiang, X.; Gan, J. Inhibitors of the NLRP3 Inflammasome Pathway as Promising Therapeutic Candidates for Inflammatory Diseases (Review). Int. J. Mol. Med. 2023, 51, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, M. Associations between Genetic Polymorphisms of TLRs and Susceptibility to Tuberculosis: A Meta-Analysis. Innate Immun. 2020, 26, 75–83. [Google Scholar] [CrossRef]
- Ning, Y.; Wang, W.; Jordan, P.M.; Barth, S.A.; Hofstetter, R.K.; Xu, J.; Zhang, X.; Cai, Y.; Menge, C.; Chen, X.; et al. Mycobacterium Tuberculosis –Induced Prostaglandin J2 and 15-Deoxy-Prostaglandin J2 Inhibit Inflammatory Signals in Human M1 Macrophages via a Negative Feedback Loop. J. Immunol. 2023, 210, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Brusselle, G.G.; Joos, G.F.; Bracke, K.R. New Insights into the Immunology of Chronic Obstructive Pulmonary Disease. Lancet 2011, 378, 1015–1026. [Google Scholar] [CrossRef]
- Lee, H.-G.; Cho, M.-J.; Choi, J.-M. Bystander CD4+ T Cells: Crossroads between Innate and Adaptive Immunity. Exp. Mol. Med. 2020, 52, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Lyadova, I.V.; Panteleev, A.V. Th1 and Th17 Cells in Tuberculosis: Protection, Pathology, and Biomarkers. Mediat. Inflamm. 2015, 2015, 854507. [Google Scholar] [CrossRef]
- Barnes, P.J. The Cytokine Network in Asthma and Chronic Obstructive Pulmonary Disease. J. Clin. Invest. 2008, 118, 3546–3556. [Google Scholar] [CrossRef]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef]
- Lourenço, J.D.; Ito, J.T.; Martins, M.D.A.; Tibério, I.D.F.L.C.; Lopes, F.D.T.Q.D.S. Th17/Treg Imbalance in Chronic Obstructive Pulmonary Disease: Clinical and Experimental Evidence. Front. Immunol. 2021, 12, 804919. [Google Scholar] [CrossRef]
- Bao, K.; Reinhardt, R.L. The Differential Expression of IL-4 and IL-13 and Its Impact on Type-2 Immunity. Cytokine 2015, 75, 25–37. [Google Scholar] [CrossRef]
- Chen, G.; Mu, Q.; Meng, Z.-J. Cigarette Smoking Contributes to Th1/Th2 Cell Dysfunction via the Cytokine Milieu in Chronic Obstructive Pulmonary Disease. COPD 2023, 18, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Carabalí-Isajar, M.L.; Rodríguez-Bejarano, O.H.; Amado, T.; Patarroyo, M.A.; Izquierdo, M.A.; Lutz, J.R.; Ocampo, M. Clinical Manifestations and Immune Response to Tuberculosis. World J. Microbiol. Biotechnol. 2023, 39, 206. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Xiao, Z. Changes in Peripheral Blood TBNK Lymphocyte Subsets and Their Association with Acute Exacerbation of Chronic Obstructive Pulmonary Disease. J. Int. Med. Res. 2023, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Ma, N.; Huang, Z. Correlation between Elderly Patients with COPD and the Impact on Immunity in Tuberculosis Patients: A Retrospective Study. Medicine 2024, 103, e40140. [Google Scholar] [CrossRef]
- Kumar, N.P.; Moideen, K.; Viswanathan, V.; Kornfeld, H.; Babu, S. Effect of Standard Tuberculosis Treatment on Naive, Memory and Regulatory T-Cell Homeostasis in Tuberculosis–Diabetes Co-Morbidity. Immunology 2016, 149, 87–97. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, J.; Tang, P.; Song, H.; Liu, X.; Huang, Z.; Zhang, X.; Wang, X.; Wu, M. Expression and Clinical Significance of B and T Lymphocyte Attenuator on CD4+ and CD8+ T Cells from Patients with Pulmonary Tuberculosis. Indian. J. Pathol. Microbiol. 2019, 62, 232–238. [Google Scholar] [CrossRef]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T Cells (Tregs) and Their Therapeutic Potential against Autoimmune Disorders—Advances and Challenges. Hum. Vaccin. Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Ma, R.; Su, H.; Jiao, K.; Liu, J. Role of Th17 Cells, Treg Cells, and Th17/Treg Imbalance in Immune Homeostasis Disorders in Patients with Chronic Obstructive Pulmonary Disease. Immun. Inflam. Amp; Dis. 2023, 11, e784. [Google Scholar] [CrossRef]
- da Silva, M.V.; Tiburcio, M.G.S.; Machado, J.R.; Silva, D.A.A.; Rodrigues, D.B.R.; Rodrigues, V.; Oliveira, C.J.F. Complexity and Controversies over the Cytokine Profiles of T Helper Cell Subpopulations in Tuberculosis. J. Immunol. Res. 2015, 2015, 639107. [Google Scholar] [CrossRef]
- Kozakiewicz, L.; Phuah, J.; Flynn, J.; Chan, J. The Role of B Cells and Humoral Immunity in Mycobacterium Tuberculosis Infection. Adv. Exp. Med. Biol. 2013, 783, 225–250. [Google Scholar] [CrossRef]
- Chan, J.; Mehta, S.; Bharrhan, S.; Chen, Y.; Achkar, J.M.; Casadevall, A.; Flynn, J. The Role of B Cells and Humoral Immunity in Mycobacterium Tuberculosis Infection. Semin. Immunol. 2014, 26, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Seys, L.J.M.; Bracke, K.R.; Owen, C.A. B Cells in Chronic Obstructive Pulmonary Disease: Moving to Center Stage. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2016, 311, L687–L695. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.H.; Wilkes, D.S. Complement System in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2014, 51, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.; Grigsby, S.J.; Philips, J.A. Immune Evasion and Provocation by Mycobacterium Tuberculosis. Nat. Rev. Microbiol. 2022, 20, 750–766. [Google Scholar] [CrossRef]
- Bhat, T.A.; Panzica, L.; Kalathil, S.G.; Thanavala, Y. Immune Dysfunction in Patients with Chronic Obstructive Pulmonary Disease. Ann. ATS 2015, 12, S169–S175. [Google Scholar] [CrossRef]
- Smith, A.J.P.; Humphries, S.E. Cytokine and Cytokine Receptor Gene Polymorphisms and Their Functionality. Cytokine Growth Factor. Rev. 2009, 20, 43–59. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF- B Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Batra, S.; Balamayooran, G.; Sahoo, M.K. Nuclear Factor-κB: A Key Regulator in Health and Disease of Lungs. Arch. Immunol. Ther. Exp. 2011, 59, 335–351. [Google Scholar] [CrossRef]
- Zaynagetdinov, R.; Sherrill, T.P.; Gleaves, L.A.; Hunt, P.; Han, W.; McLoed, A.G.; Saxon, J.A.; Tanjore, H.; Gulleman, P.M.; Young, L.R.; et al. Chronic NF-κB Activation Links COPD and Lung Cancer through Generation of an Immunosuppressive Microenvironment in the Lungs. Oncotarget 2016, 7, 5470–5482. [Google Scholar] [CrossRef]
- Bai, X.; Feldman, N.E.; Chmura, K.; Ovrutsky, A.R.; Su, W.-L.; Griffin, L.; Pyeon, D.; McGibney, M.T.; Strand, M.J.; Numata, M.; et al. Inhibition of Nuclear Factor-Kappa B Activation Decreases Survival of Mycobacterium Tuberculosis in Human Macrophages. PLoS ONE 2013, 8, e61925. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 Activation Controls Cell Proliferation and Cell Death: Is Subcellular Localization the Answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Pua, L.J.W.; Mai, C.-W.; Chung, F.F.-L.; Khoo, A.S.-B.; Leong, C.-O.; Lim, W.-M.; Hii, L.-W. Functional Roles of JNK and P38 MAPK Signaling in Nasopharyngeal Carcinoma. IJMS 2022, 23, 1108. [Google Scholar] [CrossRef]
- Huang, G.; Shi, L.Z.; Chi, H. Regulation of JNK and P38 MAPK in the Immune System: Signal Integration, Propagation and Termination. Cytokine 2009, 48, 161–169. [Google Scholar] [CrossRef]
- Ahmadi, A.; Ahrari, S.; Salimian, J.; Salehi, Z.; Karimi, M.; Emamvirdizadeh, A.; Jamalkandi, S.A.; Ghanei, M. P38 MAPK Signaling in Chronic Obstructive Pulmonary Disease Pathogenesis and Inhibitor Therapeutics. Cell Commun. Signal 2023, 21, 314. [Google Scholar] [CrossRef]
- Barnes, P.J. Cellular and Molecular Mechanisms of Asthma and COPD. Clin. Sci. 2017, 131, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT Signaling Pathway: From Bench to Clinic. Sig Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT Signaling in Immunity and Disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef]
- Ihle, J.N. STATs: Signal Transducers and Activators of Transcription. Cell 1996, 84, 331–334. [Google Scholar] [CrossRef]
- Korn, T.; Hiltensperger, M. Role of IL-6 in the Commitment of T Cell Subsets. Cytokine 2021, 146, 155654. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.; Ben-Moshe, N.B.; Hoffman, D.; Trzebanski, S.; Yehezkel, D.; Vainman, L.; Netea, M.; Avraham, R. Early and Delayed STAT1-Dependent Responses Drive Local Trained Immunity of Macrophages in the Spleen. Elife 2024, 13, RP100922. [Google Scholar] [CrossRef]
- Forrellad, M.A.; Klepp, L.I.; Gioffré, A.; Sabio y García, J.; Morbidoni, H.R.; de la Paz Santangelo, M.; Cataldi, A.A.; Bigi, F. Virulence Factors of the Mycobacterium Tuberculosis Complex. Virulence 2013, 4, 3–66. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, N.; Giang, P.H.; Basu, S.K.; Mir, F.A.; Siddiqui, I.; Sharma, P. Mycobacterium Tuberculosis 6-kDa Early Secreted Antigenic Target (ESAT-6) Protein Downregulates Lipopolysaccharide Induced c-Myc Expression by Modulating the Extracellular Signal Regulated Kinases 1/2. BMC Immunol. 2007, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and Aging: Signaling Pathways and Intervention Therapies. Sig Transduct. Target. Ther. 2023, 8, 239. [Google Scholar] [CrossRef]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.-S.; Zeng, J.; Mei, J.; Wang, P.-Y. JAK/STAT Pathway: Extracellular Signals, Diseases, Immunity, and Therapeutic Regimens. Front. Bioeng. Biotechnol. 2023, 11, 1110765. [Google Scholar] [CrossRef]
- Verres, Y.; da Silva, C.O.; Aljebawi, B.; Bodin, A.; Barreto, E.; Lagente, V.; Victoni, T. Impact of JAK/STAT Inhibitors on Human Monocyte-Derived-Macrophages Stimulated by Cigarette Smoke Extract and Lipopolysaccharide. Clin. Exp. Pharmacol. Physiol. 2022, 49, 1187–1196. [Google Scholar] [CrossRef]
- Keizer, S.; Gerritsen, R.; Jauw, Y.; Janssen, J.; Koopman, B.; Bresser, P. Fatal Tuberculosis during Treatment with Ruxolitinib. Ned. Tijdschr. Voor Geneeskd. 2015, 159, A8650. [Google Scholar]
- Tiwari, N.; Singh, A.; Singh, B.; Verma, S.P.; Tripathi, A.K. Ruxolitinib and Tuberculosis: A Case Report with Brief Review. Indian. J. Tuberc. 2022, 69, 354–358. [Google Scholar] [CrossRef]
- Neethu, C.M.; James, J.; Prabhu, R. Reactivation of a Common Infection Following Treatment with a Novel Agent for an Uncommon Disease- Ruxolitinib Associated Tuberculosis: Two Cases. J. Pharm. Sci. Res. 2017, 9, 2437–2439. [Google Scholar]
- Cantini, F.; Blandizzi, C.; Niccoli, L.; Petrone, L.; Goletti, D. Systematic Review on Tuberculosis Risk in Patients with Rheumatoid Arthritis Receiving Inhibitors of Janus Kinases. Expert. Opin. Drug Saf. 2020, 19, 861–872. [Google Scholar] [CrossRef]
- He, L.-X.; Tang, Z.-H.; Huang, Q.-S.; Li, W.-H. DNA Methylation: A Potential Biomarker of Chronic Obstructive Pulmonary Disease. Front. Cell Dev. Biol. 2020, 8, 585. [Google Scholar] [CrossRef]
- Wrede, D.; Bordak, M.; Abraham, Y.; Mehedi, M. Pulmonary Pathogen-Induced Epigenetic Modifications. Epigenomes 2023, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, K.S.; Alshehri, S.M.; Alenezi, S.K. Epigenetic Optimization in Chronic Obstructive Pulmonary Disease (COPD). In Targeting Epigenetics in Inflammatory Lung Diseases; Gupta, G., Oliver, B.G., Dua, K., Ali, M.K., Dave, P., Eds.; Springer Nature Singapore: Singapore, 2023; pp. 99–110. ISBN 978-981-9947-79-9. [Google Scholar]
- Liu, R.; Zhao, E.; Yu, H.; Yuan, C.; Abbas, M.N.; Cui, H. Methylation across the Central Dogma in Health and Diseases: New Therapeutic Strategies. Sig Transduct. Target. Ther. 2023, 8, 310. [Google Scholar] [CrossRef]
- Pan, W.; An, S.; Dai, L.; Xu, S.; Liu, D.; Wang, L.; Zhang, R.; Wang, F.; Wang, Z. Identification of Potential Differentially-Methylated/Expressed Genes in Chronic Obstructive Pulmonary Disease. COPD: J. Chronic Obstr. Pulm. Dis. 2023, 20, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Körholz, J.; Chen, L.-S.; Strauss, T.; Schuetz, C.; Dalpke, A.H. One Gene to Rule Them All—Clinical Perspectives of a Potent Suppressor of Cytokine Signaling—SOCS1. Front. Immunol. 2024, 15, 1385190. [Google Scholar] [CrossRef] [PubMed]
- Brighenti, S.; Joosten, S.A. Friends and Foes of Tuberculosis: Modulation of Protective Immunity. J. Intern. Med. 2018, 284, 125–144. [Google Scholar] [CrossRef]
- Diener, C.; Keller, A.; Meese, E. The miRNA–Target Interactions: An Underestimated Intricacy. Nucleic Acids Res. 2024, 52, 1544–1557. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G.; Labbaye, C. miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. ncRNA 2017, 3, 22. [Google Scholar] [CrossRef]
- Liu, G.; Abraham, E. MicroRNAs in Immune Response and Macrophage Polarization. ATVB 2013, 33, 170–177. [Google Scholar] [CrossRef]
- Wang, J.; Wu, M.; Wen, J.; Yang, K.; Li, M.; Zhan, X.; Feng, L.; Li, M.; Huang, X. MicroRNA-155 Induction by Mycobacterium Bovis BCG Enhances ROS Production through Targeting SHIP1. Mol. Immunol. 2014, 62, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Yang, L.; Guo, J.; Hu, H.; Li, F.; Liu, Y.; Pang, Y. miR-145-5p Is Associated with Smoke-Related Chronic Obstructive Pulmonary Disease via Targeting KLF5. Chem. -Biol. Interact. 2019, 300, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Sun, Q.; Lu, L.; Luo, F.; Zhou, L.; Liu, J.; Cao, L.; Wang, Q.; Xue, J.; Yang, Q.; et al. MicroRNA-218 Acts by Repressing TNFR1-Mediated Activation of NF-κB, Which Is Involved in MUC5AC Hyper-Production and Inflammation in Smoking-Induced Bronchiolitis of COPD. Toxicol. Lett. 2017, 280, 171–180. [Google Scholar] [CrossRef]
- Nouws, J.; Wan, F.; Finnemore, E.; Roque, W.; Kim, S.-J.; Bazan, I.; Li, C.-X.; Skold, C.M.; Dai, Q.; Yan, X.; et al. MicroRNA miR-24-3p Reduces DNA Damage Responses, Apoptosis, and Susceptibility to Chronic Obstructive Pulmonary Disease. JCI Insight 2021, 6, e134218. [Google Scholar] [CrossRef] [PubMed]
- Moattar Husseini, N.; Mohamadnia, A.; Hosseini, F.; Bahrami, N. Molecular Detection of Chlamydia Pneumoniae, Haemophilus Influenza, and Streptococcus Pneumoniae and Expression of miR-146, miR-16, and miR-221 in Patients with Chronic Obstructive Pulmonary Diseases. Biomed. Biotechnol. Res. J. (BBRJ) 2024, 8, 356–362. [Google Scholar] [CrossRef]
- Ezzie, M.E.; Crawford, M.; Cho, J.-H.; Orellana, R.; Zhang, S.; Gelinas, R.; Batte, K.; Yu, L.; Nuovo, G.; Galas, D.; et al. Gene Expression Networks in COPD: MicroRNA and mRNA Regulation. Thorax 2012, 67, 122–131. [Google Scholar] [CrossRef]
- Roffel, M.P.; Bracke, K.R.; Heijink, I.H.; Maes, T. miR-223: A Key Regulator in the Innate Immune Response in Asthma and COPD. Front. Med. 2020, 7, 196. [Google Scholar] [CrossRef]
- Fu, Y.; Yang, X.; Chen, H.; Lu, Y. Diagnostic Value of miR-145 and Its Regulatory Role in Macrophage Immune Response in Tuberculosis. Genet. Mol. Biol. 2020, 43, e20190238. [Google Scholar] [CrossRef]
- Song, Q.; Bian, Q.; Liang, T.; Zhang, Y.; Zhang, K. Identification of Immune-Related Genes and Susceptible Population of Pulmonary Tuberculosis by Constructing TF-miRNA-mRNA Regulatory Network. Tuberculosis 2021, 131, 102139. [Google Scholar] [CrossRef]
- Shepelkova, G.S.; Evstifeev, V.V.; Yeremeev, V.V. Overexpressing miR-222-3p in Cultured Mycobacterium Tuberculosis-Infected Macrophages Does Not Affect Their Bacteriostatic Activity. Russ. J. Infect. Immun. 2024, 14, 532–538. [Google Scholar] [CrossRef]
- XiXiue; ZhangChunxiao; HanWei; ZhaoHuayang; ZhangHuiqiang; JiaoJunhua MicroRNA-223 Is Upregulated in Active Tuberculosis Patients and Inhibits Apoptosis of Macrophages by Targeting FOXO3. Genet. Test. Mol. Biomark. 2015, 19, 650–656. [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Eberharter, A.; Becker, P.B. Histone Acetylation: A Switch between Repressive and Permissive Chromatin: Second in Review Series on Chromatin Dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Xuan, L.; Cao, S.; Yu, G.; Hou, Q.; Wang, H. Decreased Histone Deacetylase 2 (HDAC2) in Peripheral Blood Monocytes (PBMCs) of COPD Patients. PLoS ONE 2016, 11, e0147380. [Google Scholar] [CrossRef]
- Chandran, A.; Antony, C.; Jose, L.; Mundayoor, S.; Natarajan, K.; Kumar, R.A. Mycobacterium Tuberculosis Infection Induces HDAC1-Mediated Suppression of IL-12B Gene Expression in Macrophages. Front. Cell Infect. Microbiol. 2015, 5, 90. [Google Scholar] [CrossRef]
- Rodríguez-Carlos, A.; Jacobo-Delgado, Y.; Santos-Mena, A.O.; García-Hernández, M.H.; De Jesus-Gonzalez, L.A.; Lara-Ramirez, E.E.; Rivas-Santiago, B. Histone Deacetylase (HDAC) Inhibitors- Based Drugs Are Effective to Control Mycobacterium Tuberculosis Infection and Promote the Sensibility for Rifampicin in MDR Strain. Mem. Inst. Oswaldo Cruz 2023, 118, e230143. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tang, X.; Zhou, Z.; Huang, Q. Histone Deacetylase 6 Inhibitor Enhances Resistance to Mycobacterium Tuberculosis Infection through Innate and Adaptive Immunity in Mice. Pathog. Dis. 2018, 76, fty064. [Google Scholar] [CrossRef]
- Peng, L.; Zhong, X. Epigenetic Regulation of Drug Metabolism and Transport. Acta Pharm. Sin. B 2015, 5, 106–112. [Google Scholar] [CrossRef]
- Wu, D.-D.; Song, J.; Bartel, S.; Krauss-Etschmann, S.; Rots, M.G.; Hylkema, M.N. The Potential for Targeted Rewriting of Epigenetic Marks in COPD as a New Therapeutic Approach. Pharmacol. Ther. 2018, 182, 1–14. [Google Scholar] [CrossRef]
- Sui, J.; Qiao, W.; Xiang, X.; Luo, Y. Epigenetic Changes in Mycobacterium Tuberculosis and Its Host Provide Potential Targets or Biomarkers for Drug Discovery and Clinical Diagnosis. Pharmacol. Res. 2022, 179, 106195. [Google Scholar] [CrossRef]
- Mukherjee, S.; Dasgupta, S.; Mishra, P.K.; Chaudhury, K. Air Pollution-Induced Epigenetic Changes: Disease Development and a Possible Link with Hypersensitivity Pneumonitis. Environ. Sci. Pollut. Res. 2021, 28, 55981–56002. [Google Scholar] [CrossRef]
- Hussain, T.; Zhao, D.; Shah, S.Z.A.; Sabir, N.; Wang, J.; Liao, Y.; Song, Y.; Dong, H.; Hussain Mangi, M.; Ni, J.; et al. Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy. Cells 2019, 8, 506. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Kim, I.; Jo, E.-K. Host Immune Pathways to Mycobacterium Tuberculosis Infection. J. Bacteriol. Virol. 2024, 54, 167–190. [Google Scholar] [CrossRef]
- Olmo-Fontánez, A.M.; Torrelles, J.B. Alveolar Epithelial Cells. In Advances in Host-Directed Therapies Against Tuberculosis; Springer: Berlin/Heidelberg, Germany, 2020; pp. 247–255. [Google Scholar]
- Ouyang, Q.; Zhang, K.; Lin, D.; Feng, C.G.; Cai, Y.; Chen, X. Bazedoxifene Suppresses Intracellular Mycobacterium Tuberculosis Growth by Enhancing Autophagy. mSphere 2020, 5, e00124-20. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, L.; Meng, Z.; Qi, K.; Gao, X.; Feng, J.; Luo, J. Recognition of Mycobacterium Tuberculosis by Macrophage Toll-like Receptor and Its Role in Autophagy. Inflamm. Res. 2024, 73, 753–770. [Google Scholar] [CrossRef]
- Michalaki, C.; Albers, G.J.; Byrne, A.J. Itaconate as a Key Regulator of Respiratory Disease. Clin. Exp. Immunol. 2024, 215, 120–125. [Google Scholar] [CrossRef]
- Chandra, P.; Rajmani, R.S.; Verma, G.; Bhavesh, N.S.; Kumar, D. Targeting Drugsensitive and -Resistant Strains of Mycobacterium Tuberculosis by Inhibition of Src Family Kinases Lowers Disease Burden and Pathology. mSphere 2016, 1. [Google Scholar] [CrossRef]
- Mandal, M.; Pires, D.; Catalão, M.J.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin F in Human Macrophages Impacts Cathepsin-Driven Killing of Multidrug-Resistant Mycobacterium Tuberculosis. Microorganisms 2023, 11, 1861. [Google Scholar] [CrossRef]
- Vu, A.; Glassman, I.; Campbell, G.; Yeganyan, S.; Nguyen, J.; Shin, A.; Venketaraman, V. Host Cell Death and Modulation of Immune Response against Mycobacterium Tuberculosis Infection. Int. J. Mol. Sci. 2024, 25, 6255. [Google Scholar] [CrossRef]
- Hu, S.; He, W.; Du, X.; Huang, Y.; Fu, Y.; Yang, Y.; Hu, C.; Li, S.; Wang, Q.; Wen, Q.; et al. Vitamin B1 Helps to Limit Mycobacterium Tuberculosis Growth via Regulating Innate Immunity in a Peroxisome Proliferator-Activated Receptor-γ-Dependent Manner. Front. Immunol. 2018, 9, 1778. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, X.; Wu, Y.; Chen, X.; Feng, L.; Xie, N.; Shen, G. Nanotechnology’s Frontier in Combatting Infectious and Inflammatory Diseases: Prevention and Treatment. Sig Transduct. Target. Ther. 2024, 9, 34. [Google Scholar] [CrossRef]
- Simonson, A.W.; Umstead, T.M.; Lawanprasert, A.; Klein, B.; Almarzooqi, S.; Halstead, E.S.; Medina, S.H. Extracellular Matrix-Inspired Inhalable Aerogels for Rapid Clearance of Pulmonary Tuberculosis. Biomaterials 2021, 273, 120848. [Google Scholar] [CrossRef]
- Shen, J.; Fu, Y.; Liu, F.; Ning, B.; Jiang, X. Ursolic Acid Promotes Autophagy by Inhibiting Akt/mTOR and TNF-α/TNFR1 Signaling Pathways to Alleviate Pyroptosis and Necroptosis in Mycobacterium Tuberculosis-Infected Macrophages. Inflammation 2023, 46, 1749–1763. [Google Scholar] [CrossRef] [PubMed]
- Su, V.Y.-F.; Pan, S.-W.; Yen, Y.-F.; Feng, J.-Y.; Su, W.-J.; Chen, Y.-M. Statin Use and Impact on Tuberculosis Risk. Expert. Rev. Anti-Infect. Ther. 2021, 19, 1093–1098. [Google Scholar] [CrossRef]
- Ou, Q.; Power, R.; Griffin, M.D. Revisiting Regulatory T Cells as Modulators of Innate Immune Response and Inflammatory Diseases. Front. Immunol. 2023, 14, 1287465. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, Q.; Li, Y.; Lu, L.; Xiang, Z.; Yin, Z.; Kabelitz, D.; Wu, Y. Γδ T Cells: Origin and Fate, Subsets, Diseases and Immunotherapy. Sig Transduct. Target. Ther. 2023, 8, 434. [Google Scholar] [CrossRef]
- Abdeladhim, M.; Karnell, J.L.; Rieder, S.A. In or out of Control: Modulating Regulatory T Cell Homeostasis and Function with Immune Checkpoint Pathways. Front. Immunol. 2022, 13, 1033705. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.P.; Moideen, K.; Banurekha, V.V.; Nair, D.; Babu, S. Modulation of Th1/Tc1 and Th17/Tc17 Responses in Pulmonary Tuberculosis by IL-20 Subfamily of Cytokines. Cytokine 2018, 108, 190–196. [Google Scholar] [CrossRef]
- Nouailles, G.; Day, T.A.; Kuhlmann, S.; Loewe, D.; Dorhoi, A.; Gamradt, P.; Hurwitz, R.; Jörg, S.; Pradl, L.; Hutloff, A.; et al. Impact of Inducible Co-stimulatory Molecule (ICOS) on T-cell Responses and Protection against Mycobacterium Tuberculosis Infection. Eur. J. Immunol. 2011, 41, 981–991. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Adjei, A.A. Anti–PD-1 Antibody Treatment and the Development of Acute Pulmonary Tuberculosis. J. Thorac. Oncol. 2016, 11, 2048–2050. [Google Scholar] [CrossRef]
- Ahmed, M.; Tezera, L.B.; Elkington, P.T.; Leslie, A.J. The Paradox of Immune Checkpoint Inhibition Re-Activating Tuberculosis. Eur. Respir. J. 2022, 60, 2102512. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, L.N.; Stüve, P.; Gentilini, M.V.; Swallow, M.; Bartel, J.; Lycke, N.Y.; Barkan, D.; Martina, M.; Lujan, H.D.; Kalay, H.; et al. Targeting Mycobacterium Tuberculosis Antigens to Dendritic Cells via the DC-Specific-ICAM3-Grabbing-Nonintegrin Receptor Induces Strong T-Helper 1 Immune Responses. Front. Immunol. 2018, 9, 471. [Google Scholar] [CrossRef] [PubMed]
- du Plessis, N.; Kotze, L.A.; Leukes, V.; Walzl, G. Translational Potential of Therapeutics Targeting Regulatory Myeloid Cells in Tuberculosis. Front. Cell. Infect. Microbiol. 2018, 8, 332. [Google Scholar] [CrossRef]
- Simwela, N.V.; Johnston, L.; Bitar, P.P.; Jaecklein, E.; Altier, C.; Sassetti, C.M.; Russell, D.G. Genome-Wide Screen of Mycobacterium Tuberculosis-Infected Macrophages Revealed GID/CTLH Complex-Mediated Modulation of Bacterial Growth. Nat. Commun. 2024, 15, 9322. [Google Scholar] [CrossRef] [PubMed]
- Zein-Eddine, R.; Refrégier, G.; Cervantes, J.; Yokobori, N.K. The Future of CRISPR in Mycobacterium Tuberculosis Infection. J. Biomed. Sci. 2023, 30, 34. [Google Scholar] [CrossRef]
- Schaible, U.E.; Linnemann, L.; Redinger, N.; Patin, E.C.; Dallenga, T. Strategies to Improve Vaccine Efficacy against Tuberculosis by Targeting Innate Immunity. Front. Immunol. 2017, 8, 1755. [Google Scholar] [CrossRef]
- Kayongo, A.; Ntayi, M.L.; Olweny, G.; Kyalo, E.; Ndawula, J.; Ssengooba, W.; Kigozi, E.; Kalyesubula, R.; Munana, R.; Namaganda, J.; et al. Airway Microbiome Signature Accurately Discriminates Mycobacterium Tuberculosis Infection Status. iScience 2024, 27, 110142. [Google Scholar] [CrossRef]
- Stankovic, M.M. Lung Microbiota: From Healthy Lungs to Development of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2025, 26, 1403. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, C.; Wu, H.; Liu, J.; Ye, W.; Zhao, T.; Li, Z. The Gut Microbiota-SCFA-Inflammation Axis in Patients with AECOPD. PLoS ONE 2025, 20, e0312606. [Google Scholar] [CrossRef]
- Rana, A.K.; Kumar Saraswati, S.S.; Anang, V.; Singh, A.; Singh, A.; Verma, C.; Natarajan, K. Butyrate Induces Oxidative Burst Mediated Apoptosis via Glucose-6-Phosphate Dehydrogenase (G6PDH) in Macrophages during Mycobacterial Infection. Microbes Infect. 2024, 26, 105271. [Google Scholar] [CrossRef]
- Luo, M.; Liu, Y.; Wu, P.; Luo, D.-X.; Sun, Q.; Zheng, H.; Hu, R.; Pandol, S.J.; Li, Q.-F.; Han, Y.-P.; et al. Alternation of Gut Microbiota in Patients with Pulmonary Tuberculosis. Front. Physiol. 2017, 8, 822. [Google Scholar] [CrossRef] [PubMed]
- Lachmandas, E.; Van Den Heuvel, C.N.A.M.; Damen, M.S.M.A.; Cleophas, M.C.P.; Netea, M.G.; Van Crevel, R. Diabetes Mellitus and Increased Tuberculosis Susceptibility: The Role of Short-Chain Fatty Acids. J. Diabetes Res. 2016, 2016, 6014631. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cytokine | Medication | Study | Result | References |
|---|---|---|---|---|
| IL-1β | Canakinumab | 147 participants | No efficacy. | [73] |
| TNF-α | Infliximab | 16 patients with cachexia and moderate to severe COPD | Infliximab did not induce a marked reduction in local inflammation in cachectic patients with COPD and had little effect on systemic inflammation. | [74] |
| 22 smokers with mild to moderate COPD | No clinically significant beneficial effects of infliximab and no significant safety concerns were observed. | [75] | ||
| 234 patients with moderate to severe COPD | Twenty-six patients (total 11.1%; placebo, 9.1%; infliximab, 12.1%) died, including nine during COPD treatment. Lung cancer was the most common type of malignancy (placebo, two cases; infliximab, ten cases). | [76] | ||
| Etanercept | 81 patients with acute exacerbation of COPD | Etanercept is no more effective than prednisone in the treatment of acute exacerbations of COPD. | [77] | |
| IL-8 | ABX-IL8 | 109 patients with stable COPD. | There were no significant differences in lung function, health status, distance traveled in 6 min, or side effects between groups. | [78] |
| Aspect | Tuberculosis | COPD | References |
|---|---|---|---|
| Macrophages | |||
| Primary role | Defense against M. tuberculosis, granuloma formation, cytokine production | Chronic inflammation, tissue damage, cytokine production | [121,122,123,124,125] |
| Interaction with pathogens | M. tuberculosis evades immune response, inhibits macrophage functions | Contribute to chronic inflammation and tissue damage | [121,122,125,126] |
| Macrophage polarization | M1 (initial response) and M2 (chronic infection) | M1 and M2 phenotypes depending on stage of disease | [121,122,125] |
| Neutrophils | |||
| Main function | Initial immune response, phagocytosis M. tuberculosis | Causes pathologic changes (emphysema, mucus hypersecretion) | [127,128,129,130] |
| Tissue damage | Through pro-inflammatory mediators, networks and enzyme release | Through oxidative stress, networks and enzyme release | [120,128,131,132] |
| Dendritic cells | |||
| Main function | Enhance host defense and manage pathogen evasion | Modulates chronic inflammation and immune response | [133,134] |
| Key changes | Decreased DC numbers, altered cytokine profiles | Increased pro-inflammatory markers, altered survival and migration | [135,136] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotlyarov, S.; Oskin, D. The Role of Inflammation in the Pathogenesis of Comorbidity of Chronic Obstructive Pulmonary Disease and Pulmonary Tuberculosis. Int. J. Mol. Sci. 2025, 26, 2378. https://doi.org/10.3390/ijms26062378
Kotlyarov S, Oskin D. The Role of Inflammation in the Pathogenesis of Comorbidity of Chronic Obstructive Pulmonary Disease and Pulmonary Tuberculosis. International Journal of Molecular Sciences. 2025; 26(6):2378. https://doi.org/10.3390/ijms26062378
Chicago/Turabian StyleKotlyarov, Stanislav, and Dmitry Oskin. 2025. "The Role of Inflammation in the Pathogenesis of Comorbidity of Chronic Obstructive Pulmonary Disease and Pulmonary Tuberculosis" International Journal of Molecular Sciences 26, no. 6: 2378. https://doi.org/10.3390/ijms26062378
APA StyleKotlyarov, S., & Oskin, D. (2025). The Role of Inflammation in the Pathogenesis of Comorbidity of Chronic Obstructive Pulmonary Disease and Pulmonary Tuberculosis. International Journal of Molecular Sciences, 26(6), 2378. https://doi.org/10.3390/ijms26062378