
LRRK2 in Drosophila Melanogaster Model: Insights into Cellular Dysfunction and Neuroinflammation in Parkinson’s Disease

 ,
,  ,
,  , , , , and
, , , , and
Abstract
1. Introduction
2. Drosophila LRRK2 Models
2.1. Loss of Function Mutants
2.2. Gain of Function Mutants
3. Cellular Dysfunction in LRRK2 Drosophila Models
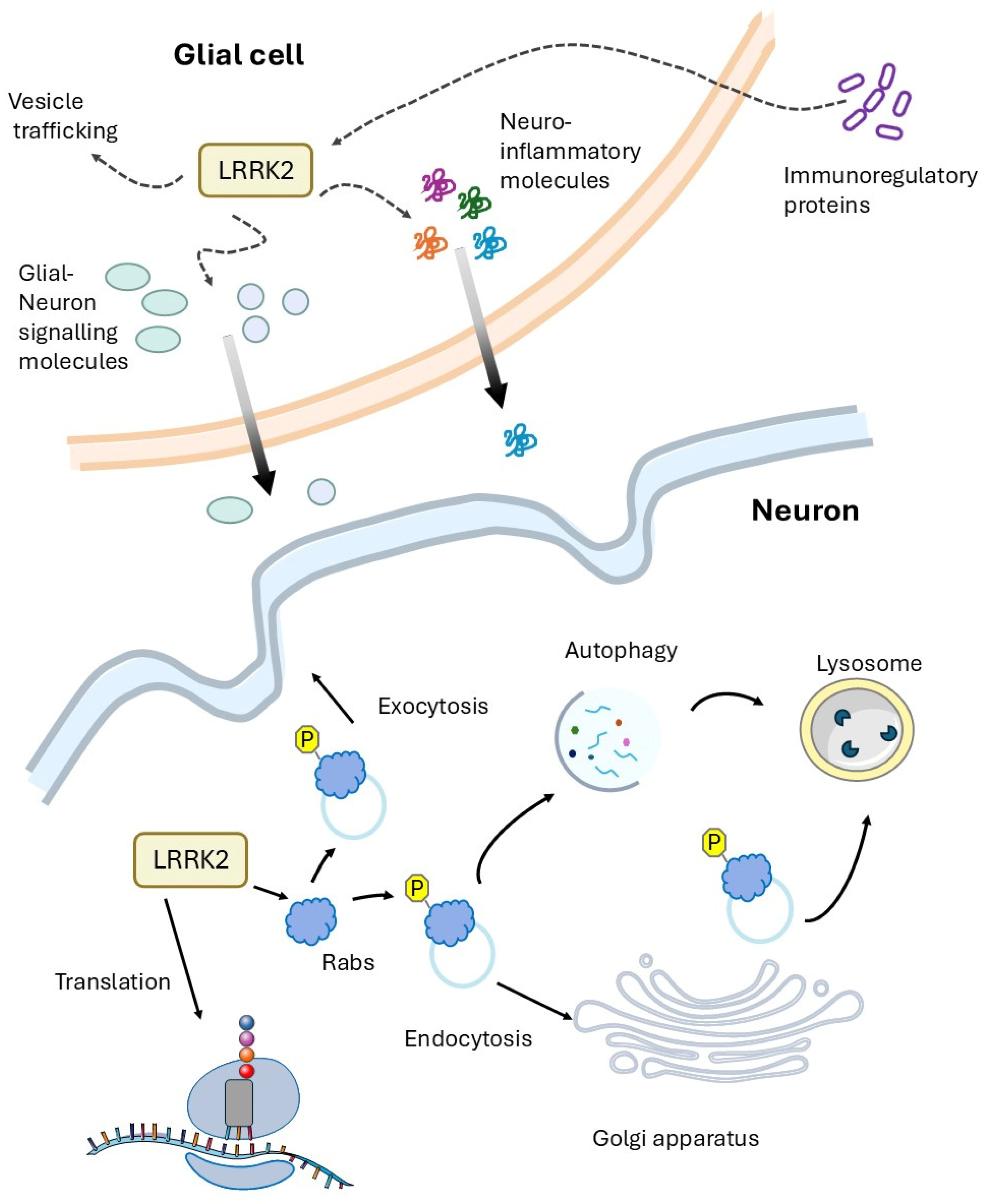
3.1. LRRK2 and Vesicle Trafficking

{kind=link}
{kind=link}
{kind=link}
| Pathway/Mechanism | Model System | Effect/Outcome | Refs. |
|---|---|---|---|
| Synaptic protein alteration | Transgenic expression in DA neurons: UAS LRRK2 R1441C/Ddc-GAL4 | Altered expression of synaptotagmin-1, syntaxin-1A, RAB3; enhanced phosphorylation of different SV proteins; LRRK2 interacts with synaptojanin-1 and EndoA. | [52] |
| Endocytosis | Lrrk loss-of-function mutants: (LrrkP1[e03680] and LrrkEX2). Ubiquity transgenic expression: UAS-LRRK2 WT or mutant/Da-GAL4 | LRRK2 phosphorylates EndoA at BAR domain; impairs synaptic vesicle endocytosis; affects macroautophagy at synapses. | [54] |
| Synaptic Function | Lrrk loss-of-function mutants: (LrrkP1[e03680]) or UAS-dLRRK-RNAi in different tissue. Transgenic expression in motor neurons or muscle of UAS-LRRK2 WT or mutants. | LRRK2 expression increased vesicle size of the readily releasable pool. LRRK2 knockdown shows opposite effect | [55] |
| Clathrin-mediated Endocytosis | Lrrk loss-of-function mutants: LrrkP1[e03680] and LrrkEX2 | dLRRK KO leads to impairments in clathrin-mediated synaptic vesicle endocytosis and neurotransmission | [54] |
| VPS35 Interaction | Transgenic expression in dopaminergic neurons or eyes: UAS-LRRK2 mut or UAS-dVPS35-VPS26 lines/Ddc-GAL4 or GMR-GAL4. dLRRK2−/− flies crossed with different dVPS35+/− and dVPS35−/− genetic backgrounds or expressing UAS-dVPS35 WT or mutant in different tissues. | VPS35 expression protects from LRRK2 pathology: improves locomotor deficits and lifespan. LRRK2 and VPS35 cooperate in endosomal pathway. | [61,62] |
| Lysosomal Function | Lrrk loss-of-function mutants: LrrkP1[e03680]. Transgenic expression in follicle cells: UAS-dLRRK G1914S/CY2-GAL4 | LRRK2 interacts with RAB7 and affects lysosome clustering | [64] |
| Autophagy | Lrrk loss-of-function mutants: LrrkP1[e03680] and LrrkEX2. Ubiquity transgenic expression: UAS-LRRK2 WT or mutant/Da-GAL4 Lrrk loss-of-function mutants: LrrkP1[e03680] mutant. Transgenic expression in motor neurons or eyes: UAS-hLRRK2-G2019S/D42-GAL4 or GMR-GAL4 | EndoA phosphorylation by LRRK2 is crucial for autophagy EndoB is required for autophagosome formation Loss of EndoB blocks G2019S-induced autophagy | [54,65] |
| ER Function | Transgenic expression in DA neurons: UAS-hLRRK2-G2019S/TH-GAL4 | LRRK2 affects ER-mitochondria contact. LRRK2 impacts calcium homeostasis | [69] |
3.2. LRRK2 and Protein Translation
| Process/Pathway | Model System | Effect/Outcome | Refs. |
|---|---|---|---|
| LRRK2 phosphorylates 4E-BP | Transgenic expression in neurons or only in DA neurons: UAS-dLRRK WT or Y1383C or I1915T/Elav-GAL4 or Ddc-GAL4 | Increased protein translation | [41] |
| LRRK2 phosphorylates ribosomal protein s15 | Lrrk loss-of-function mutants: LrrkP1[e03680] Transgenic expression in DA neurons UAS-hLRRK2 WT or mutants/Ddc-GAL4 | Increased cap-dependent and cap-independent translation Bulk increase in protein synthesis | [70] |
| G2019S mutation & amino acid diet | Transgenic expression in neurons or only in DA neurons UAS-hLRRK2-G2019S/Ddc-GAL4 or elav-GAL4 | Both amino acid restriction and supplementation attenuate LRRK2 neurodegeneration. Low amino acid diet reduces aberrant LRRK2-dependent protein synthesis. | [66] |
| LRRK2 impacts the translation of specific mRNAs | Lrrk loss-of-function mutants: LrrkP1[e03680] Transgenic expression in muscle UAS hLRRK2/G14-GAL4 | Affects translation of mRNAs with complex 5′-UTRs. Furin 1 was identified as main translational target. | [55] |
| S6 kinase as gene modifier | Transgenic UAS-hLRRK2-G2019S in dopaminergic neurons or muscle (Ddc-Gal4 or 24B-GAL4) | RNAi for S6 kinase ameliorates G2019S phenotype | [73] |
3.3. LRRK2 Pathology and Therapeutic Drugs
| Compound | Therapeutic Target | Model System | Effects | Refs. |
|---|---|---|---|---|
| LRRK2 kinase inhibitors | LRRK2 pathway | dLRRK RNAi in DA neurons UAS-Lrrk-RNAi/TH-GAL4 Different LRRK2 transgenes: UAS-LRRK2 isoforms/TH or Ddc-GAL4 | Reduces loss of dopaminergic neurons. Protects against paraquat toxic effects. Improves locomotor impairment. Reduces oxidative stress. | [76,77,78,79] |
| Lovastatin (FDA-approved drug as oral antilipemic agent) | Increases the GSK3β (Ser9) phosphorylation modulating the Akt/Nrf signalling pathway | Transgenic expression in DA neurons: UAS-LRRK2-G2019S/Ddc-GAL4 | Restores motor disability. Prevents dopaminergic neuron loss. | [44] |
| AICAr | ARE-mediated mRNA decay | Transgenic expression in DA neurons: UAS-LRRK2-G2019S/Ddc-GAL4 | Suppresses LRRK2 expression. Rescues neurodegeneration. Reduces neuroinflammation. | [79] |
| AdoCbl | LRRK2 | Transgenic expression in DA neurons UAS-LRRK2-G2019S/TH-GAL4 Lrrk loss-of-function mutants LrrkP1[e03680] | Binds directly LRRK2. Disrupts LRRK2 dimerization. Shows neuroprotective effects. | [82] |
| EGCG (Green tea derivative) | AMP-activated protein kinase (AMPK) | Transgenic expression in neurons, DA neurons or muscle: UAS-hLRRK2-G2019S/Elav-GAL4, ddc-GAL4 or 24B-GAL4 | Ameliorates DA neuron loss. Improves mitochondrial function. | [83] |
| Levetiracetam (FDA-approved antiseizure drug) | SV2A and other neuronal targets | Ubiquity transgenic expression: UAS-LRRK2 R1441C/Actin-GAL4 | Ameliorates DA neuron loss. Improves motor functions. | [48] |
4. Is the PD LRRK2-Related Disease a Cell Autonomous Disease?
5. Discussion
6. Materials and Methods
6.1. Drosophila Lines
6.2. Evaluation of mRNA Expression by RT-PCR
6.3. Western Blot Analysis
6.4. Whole-Mount Immunostaining of the Adult Drosophila Brains
6.5. Climbing Assay
6.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, Genotype, and Worldwide Genetic Penetrance of LRRK2-Associated Parkinson’s Disease: A Case-Control Study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [PubMed]
- El Otmani, H.; Daghi, M.; Tahiri Jouti, N.; Lesage, S. An Overview of the Worldwide Distribution of LRRK2 Mutations in Parkinson’s Disease. Neurodegener. Dis. Manag. 2023, 13, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Sengupta, M.; Trojanowski, J.Q.; Lee, V.M.Y. Alzheimer’s Disease Tau Is a Prominent Pathology in LRRK2 Parkinson’s Disease. Acta Neuropathol. Commun. 2019, 7, 183. [Google Scholar] [CrossRef]
- Marin, I.; van Egmond, W.N.; van Haastert, P.J. The Roco Protein Family: A Functional Perspective. FASEB J. 2008, 22, 3103–3110. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Cookson, M.R. LRRK2 Recruitment, Activity, and Function in Organelles. FEBS J. 2022, 289, 6871–6890. [Google Scholar] [CrossRef]
- Taymans, J.M. Regulation of LRRK2 by Phosphatases. Adv. Neurobiol. 2017, 14, 145–160. [Google Scholar] [CrossRef]
- Cogo, S.; Ho, F.Y.; Tosoni, E.; Tomkins, J.E.; Tessari, I.; Iannotta, L.; Montine, T.J.; Manzoni, C.; Lewis, P.A.; Bubacco, L.; et al. The Roc Domain of LRRK2 as a Hub for Protein-Protein Interactions: A Focus on PAK6 and Its Impact on RAB Phosphorylation. Brain Res. 2022, 1778, 147781. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the Gene Containing Mutations That Cause PARK8-Linked Parkinson’s Disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Panicker, N.; Ge, P.; Dawson, V.L.; Dawson, T.M. The Cell Biology of Parkinson’s Disease. J. Cell Biol. 2021, 220, e202012095. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.; Outeiro, T.F. The Role of LRRK2 in Cell Signalling. Biochem. Soc. Trans. 2019, 47, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. LRRK2 Phosphorylation of Rab GTPases in Parkinson’s Disease. FEBS Lett. 2022, 597, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics Reveals That Parkinson’s Disease Kinase LRRK2 Regulates a Subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef]
- Zhang, F.R.; Huang, W.; Chen, S.M.; Sun, L.D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.X.; Yang, H.T.; Yang, R.D.; et al. Genomewide Association Study of Leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef]
- Härtlova, A.; Herbst, S.; Peltier, J.; Rodgers, A.; Bilkei-Gorzo, O.; Fearns, A.; Dill, B.D.; Lee, H.; Flynn, R.; Cowley, S.A.; et al. LRRK2 Is a Negative Regulator of Mycobacterium Tuberculosis Phagosome Maturation in Macrophages. EMBO J. 2018, 37, e98694. [Google Scholar] [CrossRef]
- Zhang, M.; Yao, C.; Cai, J.; Liu, S.; Liu, X.-N.; Chen, Y.; Wang, S.; Ji, P.; Pan, M.; Kang, Z.; et al. LRRK2 Is Involved in the Pathogenesis of System Lupus Erythematosus through Promoting Pathogenic Antibody Production. J. Transl. Med. 2019, 17, 37. [Google Scholar] [CrossRef]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.-Y.; Chuang, L.-S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional Variants in the LRRK2 Gene Confer Shared Effects on Risk for Crohn’s Disease and Parkinson’s Disease. Sci. Transl. Med. 2018, 10, eaai7795. [Google Scholar] [CrossRef]
- Gillardon, F.; Schmid, R.; Draheim, H. Parkinson’s Disease-Linked Leucine-Rich Repeat Kinase 2(R1441G) Mutation Increases Proinflammatory Cytokine Release from Activated Primary Microglial Cells and Resultant Neurotoxicity. Neuroscience 2012, 208, 21–48. [Google Scholar] [CrossRef]
- Russo, I.; Di Benedetto, G.; Kaganovich, A.; Ding, J.; Mercatelli, D.; Morari, M.; Cookson, M.R.; Bubacco, L.; Greggio, E. Leucine-Rich Repeat Kinase 2 Controls Protein Kinase A Activation State through Phosphodiesterase 4. J. Neuroinflamm. 2018, 15, 297. [Google Scholar] [CrossRef]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic Alterations in Parkinson’s Disease Astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef] [PubMed]
- Caesar, M.; Felk, S.; Zach, S.; Brønstad, G.; Aasly, J.O.; Gasser, T.; Gillardon, F. Changes in Matrix Metalloprotease Activity and Progranulin Levels May Contribute to the Pathophysiological Function of Mutant Leucine-Rich Repeat Kinase 2. Glia 2014, 62, 1075–1092. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Pajarillo, E.; Rizor, A.; Son, D.-S.; Lee, J.; Aschner, M.; Lee, E. LRRK2 Kinase Plays a Critical Role in Manganese-Induced Inflammation and Apoptosis in Microglia. PLoS ONE 2019, 14, e0210248. [Google Scholar] [CrossRef]
- Russo, I.; Kaganovich, A.; Ding, J.; Landeck, N.; Mamais, A.; Varanita, T.; Biosa, A.; Tessari, I.; Bubacco, L.; Greggio, E.; et al. Transcriptome Analysis of LRRK2 Knock-out Microglia Cells Reveals Alterations of Inflammatory- and Oxidative Stress-Related Pathways upon Treatment with α-Synuclein Fibrils. Neurobiol. Dis. 2019, 129, 67–78. [Google Scholar] [CrossRef]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; DeSilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 Inhibition Attenuates Microglial Inflammatory Responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef]
- Munoz, L.; Kavanagh, M.E.; Phoa, A.F.; Heng, B.; Dzamko, N.; Chen, E.-J.; Doddareddy, M.R.; Guillemin, G.J.; Kassiou, M. Optimisation of LRRK2 Inhibitors and Assessment of Functional Efficacy in Cell-Based Models of Neuroinflammation. Eur. J. Med. Chem. 2015, 95, 29–34. [Google Scholar] [CrossRef]
- Russo, I.; Berti, G.; Plotegher, N.; Bernardo, G.; Filograna, R.; Bubacco, L.; Greggio, E. Leucine-Rich Repeat Kinase 2 Positively Regulates Inflammation and down-Regulates NF-κB P50 Signaling in Cultured Microglia Cells. J. Neuroinflamm. 2015, 12, 230. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammation and Neurodegeneration in Parkinson’s Disease. Parkinsonism Relat. Disord. 2004, 10 (Suppl. S1), S3–S7. [Google Scholar] [CrossRef]
- Leńska-Mieciek, M.; Madetko-Alster, N.; Alster, P.; Królicki, L.; Fiszer, U.; Koziorowski, D. Inflammation in Multiple System Atrophy. Front. Immunol. 2023, 14, 1214677. [Google Scholar] [CrossRef]
- Thakur, J.; Godad, A. Deciphering the Role of Neuropeptides as Biomarkers for Early Diagnosis of Parkinson’s Disease. Life Sci. 2025, 363, 123376. [Google Scholar] [CrossRef]
- Alster, P.; Madetko-Alster, N.; Otto-Ślusarczyk, D.; Migda, A.; Migda, B.; Struga, M.; Friedman, A. Role of Orexin in Pathogenesis of Neurodegenerative Parkinsonisms. Neurol. Neurochir. Pol. 2023, 57, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Domenicale, C.; Magnabosco, S.; Morari, M. Modeling Parkinson’s Disease in LRRK2 Rodents. Neuronal Signal. 2023, 7, NS20220040. [Google Scholar] [CrossRef] [PubMed]
- Seegobin, S.P.; Heaton, G.R.; Liang, D.; Choi, I.; Blanca Ramirez, M.; Tang, B.; Yue, Z. Progress in LRRK2-Associated Parkinson’s Disease Animal Models. Front. Neurosci. 2020, 14, 674. [Google Scholar] [CrossRef] [PubMed]
- Jeibmann, A.; Paulus, W. Drosophila Melanogaster as a Model Organism of Brain Diseases. Int. J. Mol. Sci. 2009, 10, 407–440. [Google Scholar] [CrossRef]
- Venken, K.J.T.; Simpson, J.H.; Bellen, H.J. Genetic Manipulation of Genes and Cells in the Nervous System of the Fruit Fly. Neuron 2011, 72, 202–230. [Google Scholar] [CrossRef]
- Dorkenwald, S.; McKellar, C.E.; Macrina, T.; Kemnitz, N.; Lee, K.; Lu, R.; Wu, J.; Popovych, S.; Mitchell, E.; Nehoran, B.; et al. FlyWire: Online Community for Whole-Brain Connectomics. Nat. Methods 2022, 19, 119–128. [Google Scholar] [CrossRef]
- Dorkenwald, S.; Matsliah, A.; Sterling, A.R.; Schlegel, P.; Yu, S.-C.; McKellar, C.E.; Lin, A.; Costa, M.; Eichler, K.; Yin, Y.; et al. Neuronal Wiring Diagram of an Adult Brain. Nature 2024, 634, 124–138. [Google Scholar] [CrossRef]
- Lin, A.; Yang, R.; Dorkenwald, S.; Matsliah, A.; Sterling, A.R.; Schlegel, P.; Yu, S.-C.; McKellar, C.E.; Costa, M.; Eichler, K.; et al. Network Statistics of the Whole-Brain Connectome of Drosophila. Nature 2024, 634, 153–165. [Google Scholar] [CrossRef]
- Wang, D.; Tang, B.; Zhao, G.; Pan, Q.; Xia, K.; Bodmer, R.; Zhang, Z. Dispensable Role of Drosophila Ortholog of LRRK2 Kinase Activity in Survival of Dopaminergic Neurons. Mol. Neurodegener. 2008, 3, 3. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, W.; Lee, S.; Chung, J. Loss of LRRK2/PARK8 Induces Degeneration of Dopaminergic Neurons in Drosophila. Biochem. Biophys. Res. Commun. 2007, 358, 534–539. [Google Scholar] [CrossRef]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 Affects the Maintenance of Dopaminergic Neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef]
- Tain, L.S.; Mortiboys, H.; Tao, R.N.; Ziviani, E.; Bandmann, O.; Whitworth, A.J. Rapamycin Activation of 4E-BP Prevents Parkinsonian Dopaminergic Neuron Loss. Nat. Neurosci. 2009, 12, 1129–1135. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Yu, Y.; Li, X.; Wang, T.; Jiang, H.; Ren, Q.; Jiao, Y.; Sawa, A.; Moran, T.; et al. A Drosophila Model for LRRK2-Linked Parkinsonism. Proc. Natl. Acad. Sci. USA 2008, 105, 2693–2698. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Lin, H.-I.; Chen, M.-L.; Lai, T.-T.; Cao, L.-P.; Farrer, M.J.; Wu, R.-M.; Chien, C.-T. Lovastatin Protects Neurite Degeneration in LRRK2-G2019S Parkinsonism through Activating the Akt/Nrf Pathway and Inhibiting GSK3β Activity. Hum. Mol. Genet. 2016, 25, 1965–1978. [Google Scholar] [CrossRef]
- Ng, C.-H.; Mok, S.Z.S.; Koh, C.; Ouyang, X.; Fivaz, M.L.; Tan, E.-K.; Dawson, V.L.; Dawson, T.M.; Yu, F.; Lim, K.-L. Parkin Protects against LRRK2 G2019S Mutant-Induced Dopaminergic Neurodegeneration in Drosophila. J. Neurosci. 2009, 29, 11257–11262. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yuan, C.; Chen, R.; Dawson, T.M.; Dawson, V.L. ArfGAP1 Is a GTPase Activating Protein for LRRK2: Reciprocal Regulation of ArfGAP1 by LRRK2. J. Neurosci. 2012, 32, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Maksoud, E.; Liao, E.H.; Haghighi, A.P. A Neuron-Glial Trans-Signaling Cascade Mediates LRRK2-Induced Neurodegeneration. Cell Rep. 2019, 26, 1774–1786.e4. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Galleri, G.; Rassu, A.; Ciampelli, C.; Bernardoni, R.; Galioto, M.; Albani, D.; Crosio, C.; Iaccarino, C. Evaluation of Neuroinflammatory Contribution to Neurodegeneration in LRRK2 Drosophila Models. Biomedicines 2024, 12, 1555. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.; Yi, S.; Zhang, S.; Ho, M.S. A LRRK2/dLRRK-Mediated Lysosomal Pathway That Contributes to Glial Cell Death and DA Neuron Survival. Traffic 2022, 23, 506–520. [Google Scholar] [CrossRef]
- Fellgett, A.; Middleton, C.A.; Munns, J.; Ugbode, C.; Jaciuch, D.; Wilson, L.G.; Chawla, S.; Elliott, C.J.H. Multiple Pathways of LRRK2-G2019S/Rab10 Interaction in Dopaminergic Neurons. J. Park. Dis. 2021, 11, 1805–1820. [Google Scholar] [CrossRef]
- Ciampelli, C.; Galleri, G.; Puggioni, S.; Fais, M.; Iannotta, L.; Galioto, M.; Becciu, M.; Greggio, E.; Bernardoni, R.; Crosio, C.; et al. Inhibition of the Exocyst Complex Attenuates the LRRK2 Pathological Effects. Int. J. Mol. Sci. 2023, 24, 12656. [Google Scholar] [CrossRef]
- Islam, M.S.; Nolte, H.; Jacob, W.; Ziegler, A.B.; Pütz, S.; Grosjean, Y.; Szczepanowska, K.; Trifunovic, A.; Braun, T.; Heumann, H.; et al. Human R1441C LRRK2 Regulates the Synaptic Vesicle Proteome and Phosphoproteome in a Drosophila Model of Parkinson’s Disease. Hum. Mol. Genet. 2016, 25, 5365–5382. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.Y.; Li, X.; Wang, J.; Powell, J.; Wang, Q.; Zhang, Y.; Chen, Z.; Wicinski, B.; Hof, P.; Ryan, T.A.; et al. Parkinson’s Disease-Associated LRRK2 Hyperactive Kinase Mutant Disrupts Synaptic Vesicle Trafficking in Ventral Midbrain Neurons. J. Neurosci. 2017, 37, 11366–11376. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 Controls an EndoA Phosphorylation Cycle in Synaptic Endocytosis. Neuron 2012, 75, 1008–1021. [Google Scholar] [CrossRef]
- Penney, J.; Tsurudome, K.; Liao, E.H.; Kauwe, G.; Gray, L.; Yanagiya, A.; Calderon, M.R.; Sonenberg, N.; Haghighi, A.P. LRRK2 Regulates Retrograde Synaptic Compensation at the Drosophila Neuromuscular Junction. Nat. Commun. 2016, 7, 12188. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Bautista-Gomez, J.; Higgins, D.A.; Yu, J.; Xiong, Y. Dysregulation of the AP2M1 Phosphorylation Cycle by LRRK2 Impairs Endocytosis and Leads to Dopaminergic Neurodegeneration. Sci. Signal. 2021, 14, eabg3555. [Google Scholar] [CrossRef]
- Heaton, G.R.; Landeck, N.; Mamais, A.; Nalls, M.A.; Nixon-Abell, J.; Kumaran, R.; Beilina, A.; Pellegrini, L.; Li, Y.; International Parkinson Disease Genomics Consortium (IPDGC); et al. Sequential Screening Nominates the Parkinson’s Disease Associated Kinase LRRK2 as a Regulator of Clathrin-Mediated Endocytosis. Neurobiol. Dis. 2020, 141, 104948. [Google Scholar] [CrossRef]
- Mir, R.; Tonelli, F.; Lis, P.; Macartney, T.; Polinski, N.K.; Martinez, T.N.; Chou, M.-Y.; Howden, A.J.M.; König, T.; Hotzy, C.; et al. The Parkinson’s Disease VPS35[D620N] Mutation Enhances LRRK2-Mediated Rab Protein Phosphorylation in Mouse and Human. Biochem. J. 2018, 475, 1861–1883. [Google Scholar] [CrossRef]
- Kadgien, C.A.; Kamesh, A.; Milnerwood, A.J. Endosomal Traffic and Glutamate Synapse Activity Are Increased in VPS35 D620N Mutant Knock-in Mouse Neurons, and Resistant to LRRK2 Kinase Inhibition. Mol. Brain 2021, 14, 143. [Google Scholar] [CrossRef]
- McCarron, K.R.; Elcocks, H.; Mortiboys, H.; Urbé, S.; Clague, M.J. The Parkinson’s Disease Related Mutant VPS35 (D620N) Amplifies the LRRK2 Response to Endolysosomal Stress. Biochem. J. 2024, 481, 265–278. [Google Scholar] [CrossRef]
- Linhart, R.; Wong, S.A.; Cao, J.; Tran, M.; Huynh, A.; Ardrey, C.; Park, J.M.; Hsu, C.; Taha, S.; Peterson, R.; et al. Vacuolar Protein Sorting 35 (Vps35) Rescues Locomotor Deficits and Shortened Lifespan in Drosophila Expressing a Parkinson’s Disease Mutant of Leucine-Rich Repeat Kinase 2 (LRRK2). Mol. Neurodegener. 2014, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Inoshita, T.; Arano, T.; Hosaka, Y.; Meng, H.; Umezaki, Y.; Kosugi, S.; Morimoto, T.; Koike, M.; Chang, H.-Y.; Imai, Y.; et al. Vps35 in Cooperation with LRRK2 Regulates Synaptic Vesicle Endocytosis through the Endosomal Pathway in Drosophila. Hum. Mol. Genet. 2017, 26, 2933–2948. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Upadhyay, S.; Hassan, M.I. Novel Prospects in Targeting Neurodegenerative Disorders via Autophagy. Eur. J. Pharmacol. 2024, 984, 177060. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 Homolog in Rab7-Dependent Lysosomal Positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Diaz, S.; Ghimire, S.; Sanchez-Mirasierra, I.; Montecinos-Oliva, C.; Swerts, J.; Kuenen, S.; Verstreken, P.; Soukup, S.-F. Endophilin-B Regulates Autophagy during Synapse Development and Neurodegeneration. Neurobiol. Dis. 2022, 163, 105595. [Google Scholar] [CrossRef]
- Chittoor-Vinod, V.G.; Villalobos-Cantor, S.; Roshak, H.; Shea, K.; Abalde-Atristain, L.; Martin, I. Dietary Amino Acids Impact LRRK2-Induced Neurodegeneration in Parkinson’s Disease Models. J. Neurosci. 2020, 40, 6234–6249. [Google Scholar] [CrossRef]
- Cho, H.J.; Yu, J.; Xie, C.; Rudrabhatla, P.; Chen, X.; Wu, J.; Parisiadou, L.; Liu, G.; Sun, L.; Ma, B.; et al. Leucine-Rich Repeat Kinase 2 Regulates Sec16A at ER Exit Sites to Allow ER-Golgi Export. EMBO J. 2014, 33, 2314–2331. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Cookson, M.R. The Endoplasmic Reticulum Contributes to Lysosomal Tubulation/Sorting Driven by LRRK2. Mol. Biol. Cell 2022, 33, ar124. [Google Scholar] [CrossRef]
- Lee, K.-S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.-K.; Kang, H.-Y.; Lu, B. Altered ER-Mitochondria Contact Impacts Mitochondria Calcium Homeostasis and Contributes to Neurodegeneration in Vivo in Disease Models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef]
- Martin, I.; Kim, J.W.; Lee, B.D.; Kang, H.C.; Xu, J.-C.; Jia, H.; Stankowski, J.; Kim, M.-S.; Zhong, J.; Kumar, M.; et al. Ribosomal Protein S15 Phosphorylation Mediates LRRK2 Neurodegeneration in Parkinson’s Disease. Cell 2014, 157, 472–485. [Google Scholar] [CrossRef]
- Kim, J.W.; Yin, X.; Jhaldiyal, A.; Khan, M.R.; Martin, I.; Xie, Z.; Perez-Rosello, T.; Kumar, M.; Abalde-Atristain, L.; Xu, J.; et al. Defects in mRNA Translation in LRRK2-Mutant hiPSC-Derived Dopaminergic Neurons Lead to Dysregulated Calcium Homeostasis. Cell Stem Cell 2020, 27, 633–645.e7. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Yin, X.; Martin, I.; Xiong, Y.; Eacker, S.M.; Ingolia, N.T.; Dawson, T.M.; Dawson, V.L. Dysregulated mRNA Translation in the G2019S LRRK2 and LRRK2 Knock-Out Mouse Brains. eNeuro 2021, 8, ENEURO.0310-21.2021. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.P.L.; Ziyin, W.; Basil, A.H.; Lin, S.; Chen, Z.; Zhang, C.; Zeng, L.; Cai, Y.; Lim, K.-L. Identification of PP2A and S6 Kinase as Modifiers of Leucine-Rich Repeat Kinase-Induced Neurotoxicity. Neuromolecular Med. 2020, 22, 218–226. [Google Scholar] [CrossRef]
- Toh, J.; Chua, L.L.; Ho, P.; Sandanaraj, E.; Tang, C.; Wang, H.; Tan, E.K. Identification of Targets from LRRK2 Rescue Phenotypes. Cells 2021, 10, 76. [Google Scholar] [CrossRef]
- Morez, M.; Lara Ordóñez, A.J.; Melnyk, P.; Liberelle, M.; Lebègue, N.; Taymans, J.-M. Leucine-Rich Repeat Kinase 2 (LRRK2) Inhibitors for Parkinson’s Disease: A Patent Review of the Literature to Date. Expert. Opin. Ther. Pat. 2024, 34, 773–788. [Google Scholar] [CrossRef]
- Arranz, A.M.; Delbroek, L.; Van Kolen, K.; Guimaraes, M.R.; Mandemakers, W.; Daneels, G.; Matta, S.; Calafate, S.; Shaban, H.; Baatsen, P.; et al. LRRK2 Functions in Synaptic Vesicle Endocytosis through a Kinase-Dependent Mechanism. J. Cell Sci. 2015, 128, 541–552. [Google Scholar] [CrossRef]
- Yang, D.; Li, T.; Liu, Z.; Arbez, N.; Yan, J.; Moran, T.H.; Ross, C.A.; Smith, W.W. LRRK2 Kinase Activity Mediates Toxic Interactions between Genetic Mutation and Oxidative Stress in a Drosophila Model: Suppression by Curcumin. Neurobiol. Dis. 2012, 47, 385–392. [Google Scholar] [CrossRef]
- Cording, A.C.; Shiaelis, N.; Petridi, S.; Middleton, C.A.; Wilson, L.G.; Elliott, C.J.H. Targeted Kinase Inhibition Relieves Slowness and Tremor in a Drosophila Model of LRRK2 Parkinson’s Disease. npj Parkinson’s Dis. 2017, 3, 34. [Google Scholar] [CrossRef]
- Quintero-Espinosa, D.A.; Jimenez-Del-Rio, M.; Velez-Pardo, C. LRRK2 Kinase Inhibitor PF-06447475 Protects Drosophila Melanogaster against Paraquat-Induced Locomotor Impairment, Life Span Reduction, and Oxidative Stress. Neurochem. Res. 2024, 49, 2440–2452. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, D.; Li, N.; Chen, S.; Hu, L.; Yu, J.; Xiong, Y. Regulation of LRRK2 mRNA Stability by ATIC and Its Substrate AICAR through ARE-Mediated mRNA Decay in Parkinson’s Disease. EMBO J. 2023, 42, e113410. [Google Scholar] [CrossRef]
- Cao, R.; Chen, C.; Wen, J.; Zhao, W.; Zhang, C.; Sun, L.; Yuan, L.; Wu, C.; Shan, L.; Xi, M.; et al. Recent Advances in Targeting Leucine-Rich Repeat Kinase 2 as a Potential Strategy for the Treatment of Parkinson’s Disease. Bioorg. Chem. 2023, 141, 106906. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, A.; Li, X.; Gomez-Llorente, Y.; Leandrou, E.; Memou, A.; Clemente, N.; Yao, C.; Afsari, F.; Zhi, L.; Pan, N.; et al. Vitamin B12 Modulates Parkinson’s Disease LRRK2 Kinase Activity through Allosteric Regulation and Confers Neuroprotection. Cell Res. 2019, 29, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.-H.; Guan, M.S.H.; Koh, C.; Ouyang, X.; Yu, F.; Tan, E.-K.; O’Neill, S.P.; Zhang, X.; Chung, J.; Lim, K.-L. AMP Kinase Activation Mitigates Dopaminergic Dysfunction and Mitochondrial Abnormalities in Drosophila Models of Parkinson’s Disease. J. Neurosci. 2012, 32, 14311–14317. [Google Scholar] [CrossRef]
- Wallings, R.L.; Herrick, M.K.; Tansey, M.G. LRRK2 at the Interface Between Peripheral and Central Immune Function in Parkinson’s. Front. Neurosci. 2020, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Hoffmann, J. The Host Defense of Drosophila Melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef]
- Hanson, M.A.; Lemaitre, B. New Insights on Drosophila Antimicrobial Peptide Function in Host Defense and Beyond. Curr. Opin. Immunol. 2020, 62, 22–30. [Google Scholar] [CrossRef]
- Kounatidis, I.; Chtarbanova, S.; Cao, Y.; Hayne, M.; Jayanth, D.; Ganetzky, B.; Ligoxygakis, P. NF-κB Immunity in the Brain Determines Fly Lifespan in Healthy Aging and Age-Related Neurodegeneration. Cell Rep. 2017, 19, 836–848. [Google Scholar] [CrossRef]
- Badinloo, M.; Nguyen, E.; Suh, W.; Alzahrani, F.; Castellanos, J.; Klichko, V.I.; Orr, W.C.; Radyuk, S.N. Overexpression of Antimicrobial Peptides Contributes to Aging through Cytotoxic Effects in Drosophila Tissues. Arch. Insect Biochem. Physiol. 2018, 98, e21464. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 as a Target for Modulating Immune System Responses. Neurobiol. Dis. 2022, 169, 105724. [Google Scholar] [CrossRef]
- Kim, M.E.; Lee, J.S. Mechanisms and Emerging Regulators of Neuroinflammation: Exploring New Therapeutic Strategies for Neurological Disorders. Curr. Issues Mol. Biol. 2024, 47, 8. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; Janelidze, S.; Surova, Y.; Widner, H.; Zetterberg, H.; Hansson, O. Cerebrospinal Fluid Concentrations of Inflammatory Markers in Parkinson’s Disease and Atypical Parkinsonian Disorders. Sci. Rep. 2018, 8, 13276. [Google Scholar] [CrossRef] [PubMed]
- Arbo, B.D.; Schimith, L.E.; Goulart Dos Santos, M.; Hort, M.A. Repositioning and Development of New Treatments for Neurodegenerative Diseases: Focus on Neuroinflammation. Eur. J. Pharmacol. 2022, 919, 174800. [Google Scholar] [CrossRef]
- Sampson, T.R.; Tansey, M.G.; West, A.B.; Liddle, R.A. Lewy Body Diseases and the Gut. Mol. Neurodegener. 2025, 20, 14. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi Rastegar, D.; Hughes, L.P.; Perera, G.; Keshiya, S.; Zhong, S.; Gao, J.; Halliday, G.M.; Schüle, B.; Dzamko, N. Effect of LRRK2 Protein and Activity on Stimulated Cytokines in Human Monocytes and Macrophages. npj Parkinson’s Dis. 2022, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakopoulou, V.; Ivanyuk, D.; De Cicco, S.; Haq, W.; Arsić, A.; Yu, C.; Messelodi, D.; Oldrati, M.; Schöndorf, D.C.; Perez, M.-J.; et al. Interferon-γ Signaling Synergizes with LRRK2 in Neurons and Microglia Derived from Human Induced Pluripotent Stem Cells. Nat. Commun. 2020, 11, 5163. [Google Scholar] [CrossRef]
- Neyen, C.; Bretscher, A.J.; Binggeli, O.; Lemaitre, B. Methods to Study Drosophila Immunity. Methods 2014, 68, 116–128. [Google Scholar] [CrossRef]

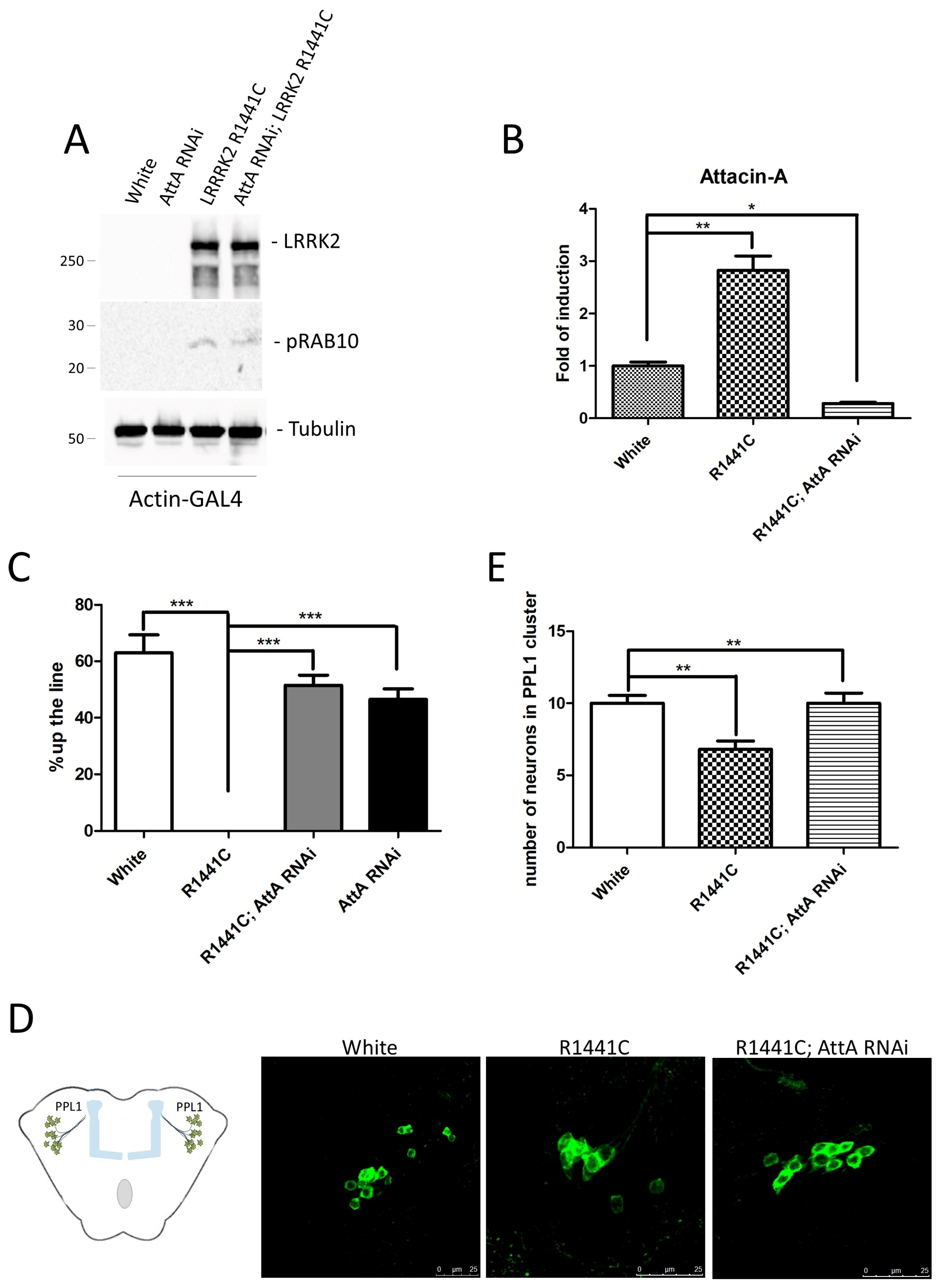
| Genetic Modification | Expression | Phenotype | Refs. |
|---|---|---|---|
| dLRRK loss-of-function: LRRKex1, generated by imprecise excision of the EP-element in the G7459 line | No expression | Severely impaired locomotor activity at 3 days after eclosion. Reduced TH-positive neurons | [40] |
| dLRRK loss-of-function P-element insertion line | No expression | Viable; reduced female fertility; increased DA content; more resistant to oxidative stress (paraquat/H2O2). No significant change in TH-positive neurons | [41] |
| Truncated dLRRK: P-element insertion in dLRRK gene causing the C-terminal kinase deletion | Endogenous truncated dLRR | More sensitive to H2O2 stress No sensitivity to paraquat, rotenone, beta-mercaptoethanol. No significant change in TH-positive neurons | [39] |
| Transgenic expression in dopaminergic neurons: UAS-dLRRK WT or Y1383C or I1915T/TH-GAL4 or Ddc-GAL4 | DA neurons | Selective loss of dopaminergic neurons Locomotor dysfunction Early mortality | [41] |
| Transgenic expression UAS-LRRK2/TH-GAL4 or Ddc-GAL4 | DA neurons | Selective loss of dopaminergic neurons Locomotor dysfunction Early mortality | [43] |
| Transgenic expression UAS-LRRK2-G2019S/TH-GAL4 or Ddc-GAL4 | DA neurons | Loss of TH-positive neurons Locomotor dysfunction (improved with L-DOPA) | [43,44,45,46] |
| Transgenic expression UAS-LRRK2-G2019S/Ddc-GAL4 | DA neurons | Loss of TH-positive neurons Neuron-glial BMP-signaling cascade is critical for mediating age-dependent neurodegeneration. | [47] |
| Transgenic expression UAS-LRRK2 WT or mutant/elav-GAL4 | Pan-neuronal | Less severe or absent phenotype compared to DA neuron expression | [43,48] |
| RNA interference UAS-dLRRK-RNAi/repo-GAL4 | Pan-glial | Locomotor deficits Glial apoptosis DA neurodegeneration | [49] |
| Transgenic expression UAS-LRRK R1441C/repo-GAL4 | Pan-glial | Locomotor deficits DA neurodegeneration | [48] |
| Transgenic expression UAS-LRRK2-G2019S/Actin-GAL4 | Ubiquitous | Loss of TH-positive neurons Locomotor deficits Increased inflammatory signals | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciampelli, C.; Galleri, G.; Galioto, M.; Mereu, P.; Pirastru, M.; Bernardoni, R.; Albani, D.; Crosio, C.; Iaccarino, C. LRRK2 in Drosophila Melanogaster Model: Insights into Cellular Dysfunction and Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2025, 26, 2093. https://doi.org/10.3390/ijms26052093
Ciampelli C, Galleri G, Galioto M, Mereu P, Pirastru M, Bernardoni R, Albani D, Crosio C, Iaccarino C. LRRK2 in Drosophila Melanogaster Model: Insights into Cellular Dysfunction and Neuroinflammation in Parkinson’s Disease. International Journal of Molecular Sciences. 2025; 26(5):2093. https://doi.org/10.3390/ijms26052093
Chicago/Turabian StyleCiampelli, Cristina, Grazia Galleri, Manuela Galioto, Paolo Mereu, Monica Pirastru, Roberto Bernardoni, Diego Albani, Claudia Crosio, and Ciro Iaccarino. 2025. "LRRK2 in Drosophila Melanogaster Model: Insights into Cellular Dysfunction and Neuroinflammation in Parkinson’s Disease" International Journal of Molecular Sciences 26, no. 5: 2093. https://doi.org/10.3390/ijms26052093
APA StyleCiampelli, C., Galleri, G., Galioto, M., Mereu, P., Pirastru, M., Bernardoni, R., Albani, D., Crosio, C., & Iaccarino, C. (2025). LRRK2 in Drosophila Melanogaster Model: Insights into Cellular Dysfunction and Neuroinflammation in Parkinson’s Disease. International Journal of Molecular Sciences, 26(5), 2093. https://doi.org/10.3390/ijms26052093