
Integrin-Linked Kinase (ILK) Promotes Mitochondrial Dysfunction by Decreasing CPT1A Expression in a Folic Acid-Based Model of Kidney Disease
 , , and
, , and
Abstract
1. Introduction
2. Results
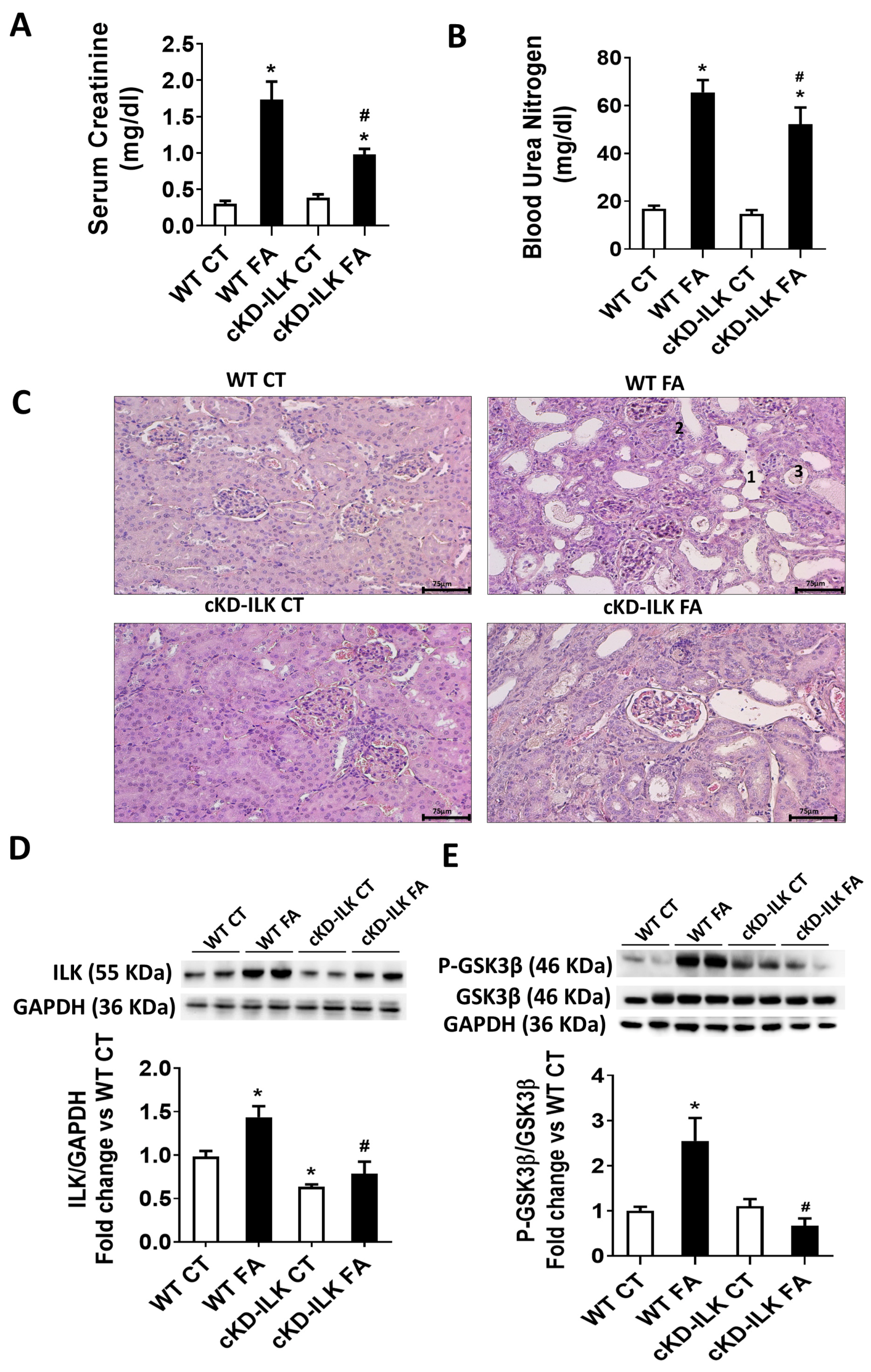
2.1. Renal ILK Depletion Prevents Folic Acid (FA)-Induced Renal Injury on Mice
2.2. Mitochondrial Dysfunction and Autophagy in FA-Induced Renal Damage Is Mediated by ILK Overexpression
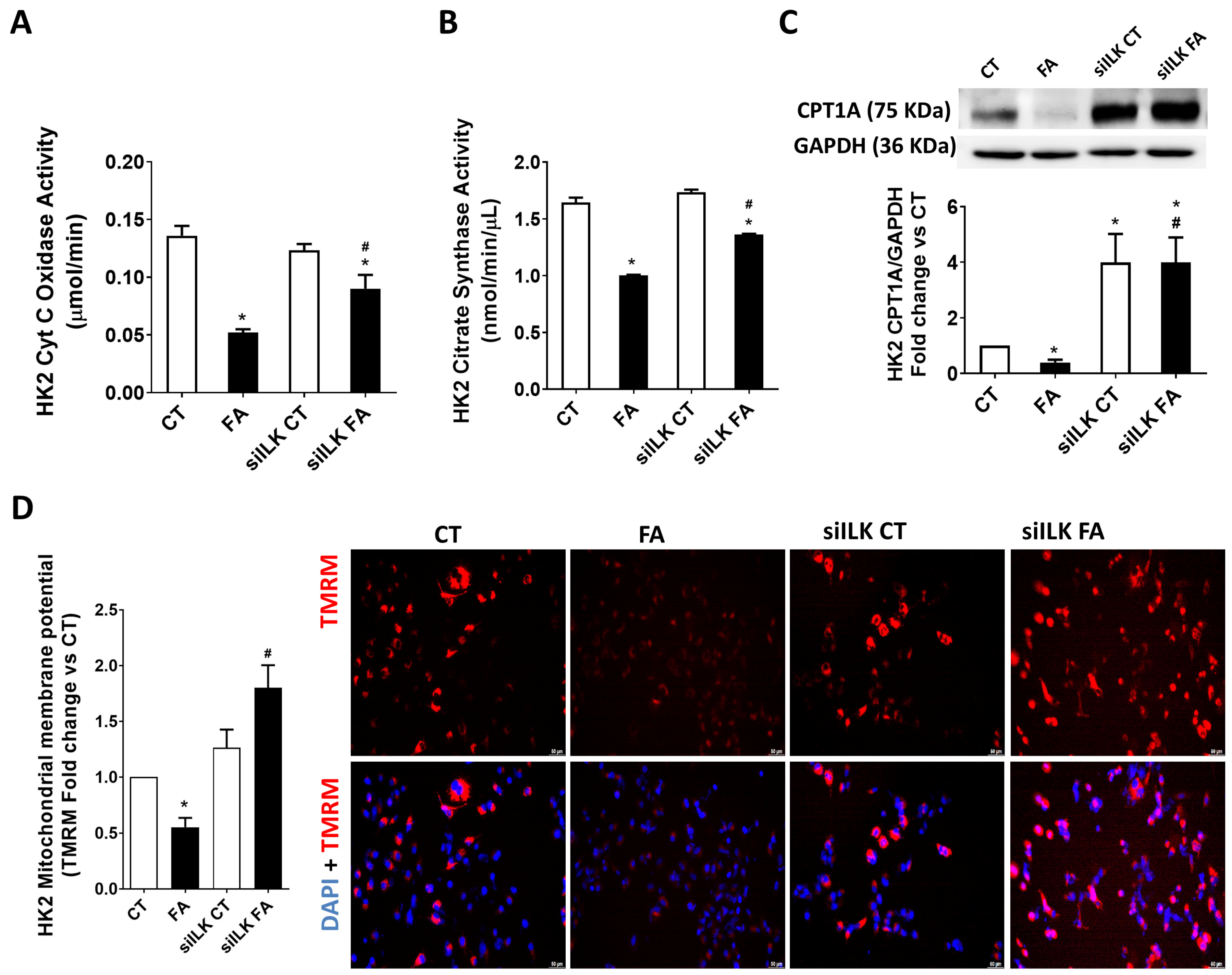
2.3. ILK Overexpression Promotes Fibrosis and Mitochondrial Dysfunction in an FA-Based Damage Model on Cultured HK2 Cells
2.4. Role of ILK/GSK3β/C/EBPβ Axis During the Transcriptional Modification of CPT1A in an FA-Based Damage Model on Cultured HK2 Cells
3. Discussion
4. Materials and Methods
4.1. Animal Model of FA-Based AKI-to-CKD Renal Damage
4.2. Serum Creatinine and Blood Urea Nitrogen Determination
4.3. Renal Histopathological Analysis
4.4. Cell Culture and In Vitro FA Treatment
4.5. Renal Mitochondrial Extracts and Determinations of Cytochrome C Oxidase and Citrate Synthase Activities
4.6. Mitochondrial Membrane Potential Assay
4.7. Western Blots
4.8. Reverse Transcription–Quantitative Polymerase Chain Reaction (RT-qPCR)
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palomba, H.; Castro, I.; Yu, L.; Burdmann, E.A. The duration of acute kidney injury after cardiac surgery increases the risk of long-term chronic kidney disease. J. Nephrol. 2017, 30, 567–572. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Górska, A.; Mazur, A.J. Integrin-linked kinase (ILK): The known vs. the unknown and perspectives. Cell. Mol. Life Sci. 2022, 79, 100. [Google Scholar] [CrossRef] [PubMed]
- Van Weeren, P.C.; De Bruyn, K.M.; De Vries-Smits, A.M.; Van Lint, J.; Burgering, B.M. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J. Biol. Chem. 1998, 273, 13150–13156. [Google Scholar] [CrossRef]
- García-Jérez, A.; Luengo, A.; Carracedo, J.; Ramírez-Chamond, R.; Rodriguez-Puyol, D.; Rodriguez-Puyol, M.; Calleros, L. Effect of uraemia on endothelial cell damage is mediated by the integrin-linked kinase pathway. J. Physiol. 2015, 593, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Cano-Peñalver, J.L.; Griera, M.; García-Jérez, A.; Hatem-Vaquero, M.; Ruiz-Torres, M.P.; Rodríguez-Puyol, D.; De Frutos, S.; Rodríguez-Puyol, M. Renal integrin-linked kinase depletion induces kidney cGMP axis upregulation: Consequences on basal and acutely damaged renal function. Mol. Med. 2015, 21, 873–885. [Google Scholar] [CrossRef]
- De Frutos, S.; Luengo, A.; García-Jérez, A.; Hatem-Vaquero, M.; Griera, M.; O’Valle, F.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; Calleros, L. Chronic kidney disease induced by an adenine-rich diet upregulates integrin-linked kinase (ILK), and its depletion prevents the disease progression. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Campillo, S.; Bohorquez, L.; Gutiérrez-Calabrés, E.; García-Ayuso, D.; Miguel, V.; Griera, M.; Calle, Y.; de Frutos, S.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; et al. Indoxyl sulfate- and P-cresol-induced monocyte adhesion and migration is mediated by integrin-linked kinase-dependent podosome formation. Exp. Mol. Med. 2022, 54, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Campillo, S.; Gutiérrez-Calabrés, E.; García-Miranda, S.; Griera, M.; Fernández Rodríguez, L.; de Frutos, S.; Rodríguez-Puyol, D.; Calleros, L. Integrin-linked kinase mRNA expression in circulating mononuclear cells as a biomarker of kidney and vascular damage in experimental chronic kidney disease. Cell Commun. Signal. 2024, 22, 264. [Google Scholar] [CrossRef] [PubMed]
- de Paulo Castro Teixeira, V.S.M.; Li, M.; Anders, H.J.; Cohen, C.D.; Edenhofer, I.; Calvaresi, N.; Merkle, M.; Rastaldi, M.P.; Kretzler, M. Functional consequences of integrin-linked kinase activation in podocyte damage. Kidney Int. 2005, 67, 514–523. [Google Scholar] [CrossRef]
- Kagami, S.; Shimizu, M.; Kondo, S.; Kitamura, A.; Urushihara, M.; Takamatsu, M.; Yamaji, S.; Ishigatsubo, Y.; Kawachi, H.; Shimizu, F. Up-regulation of integrin-linked kinase activity in rat mesangioproliferative glomerulonephritis. Life Sci. 2006, 78, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhu, S.; Huang, H.; He, J.; Tsuji, K.; Jin, W.W.; Xie, D.; Ham, O.; Capen, D.E.; Lu, W.; et al. Integrin-Linked Kinase Deficiency in Collecting Duct Principal Cell Promotes Necroptosis of Principal Cell and Contributes to Kidney Inflammation and Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 2073–2090. [Google Scholar] [CrossRef]
- Li, M.; Zhou, H.; Di, J.; Yang, M.; Jia, F. ILK participates in renal interstitial fibrosis by altering the phenotype of renal tubular epithelial cells via TGF-β1/smad pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Cao, J.; Zhang, X.; Yin, D.; Xu, D.; Lu, G. EPA attenuates epithelial-mesenchymal transition and fibrosis through the TGF-β1/Smad3/ILK pathway in renal tubular epithelial HK-2 cells by up-regulating miR-541. Int. J. Clin. Exp. Pathol. 2019, 12, 2516–2525. [Google Scholar] [PubMed]
- Howard, C.; Tao, S.; Yang, H.C.; Fogo, A.B.; Woodgett, J.R.; Harris, R.C.; Rao, R. Specific deletion of glycogen synthase kinase-3β in the renal proximal tubule protects against acute nephrotoxic injury in mice. Kidney Int. 2012, 82, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Ge, Y.; Zhuang, S.; Dworkin, L.D.; Liu, Z.; Gong, R. Inhibition of glycogen synthase kinase-3β prevents NSAID-induced acute kidney injury. Kidney Int. 2012, 81, 662–673. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lianos, E.A.; Ma, J.; Lin, P.H. Autophagy, innate immunity, and tissue repair in acute kidney injury. Int. J. Mol. Sci. 2016, 17, 662. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pan, J.; Li, H.; Li, G.; Liu, B.; Tang, X.; Liu, X.; He, Z.; Peng, Z.; Zhang, H.; et al. DsbA-L interacts with VDAC1 in mitochondrion-mediated tubular cell apoptosis and contributes to the progression of acute kidney disease. eBioMedicine 2022, 76, 103859. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef] [PubMed]
- Stallons, L.J.; Whitaker, R.M.; Schnellmann, R.G. Suppressed mitochondrial biogenesis in folic acid-induced acute kidney injury and early fibrosis. Toxicol. Lett. 2014, 224, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Afshinnia, F.; Rajendiran, T.M.; Soni, T.; Byun, J.; Wernisch, S.; Sas, K.M.; Hawkins, J.; Bellovich, K.; Gipson, D.; Michailidis, G.; et al. Impaired β-Oxidation and Altered Complex Lipid Fatty Acid Partitioning with Advancing CKD. J. Am. Soc. Nephrol. 2018, 29, 295–306. [Google Scholar] [CrossRef]
- Yuan, Q.; Lv, Y.; Ding, H.; Ke, Q.; Shi, C.; Luo, J.; Jiang, L.; Yang, J.; Zhou, Y. CPT1α maintains phenotype of tubules via mitochondrial respiration during kidney injury and repair. Cell Death Dis. 2021, 12, 792. [Google Scholar] [CrossRef] [PubMed]
- Miguel, V.; Tituaña, J.; Herrero, J.I.; Herrero, L.; Serra, D.; Cuevas, P.; Barbas, C.; Puyol, D.R.; Márquez-Expósito, L.; Ruiz-Ortega, M.; et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Investig. 2021, 131, e140695. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimer’s Dis. 2011, 2011, 505607. [Google Scholar] [CrossRef]
- Liu, Z.; Gong, R. Remote ischemic preconditioning for kidney protection: GSK3β-centric insights into the mechanism of action. Am. J. Kidney Dis. 2015, 66, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.Y.; Tin, A.; Schlosser, P.; Ko, Y.A.; Qiu, C.; Yao, C.; Joehanes, R.; Grams, M.E.; Liang, L.; Gluck, C.A.; et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 2017, 8, 1286. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.; Qian, L.; Zhou, Y.; Feng, L.; Zhang, Z.; Zheng, C.; Chen, M.; Huang, X.; Wu, X. Identification of hub genes and transcription factor-miRNA-mRNA pathways in mice and human renal ischemia-reperfusion injury. PeerJ 2021, 9, e12375. [Google Scholar] [CrossRef] [PubMed]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Lopez-Diaz, A.M.; Guerrero-Mauvecin, J.; Miguel, V.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Tubular Mitochondrial Dysfunction, Oxidative Stress, and Progression of Chronic Kidney Disease. Antioxidants 2022, 11, 1356. [Google Scholar] [CrossRef]
- Yan, L.J. Folic acid-induced animal model of kidney disease. Anim. Model. Exp. Med. 2021, 4, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Miguel, V.; Rey-Serra, C.; Tituaña, J.; Sirera, B.; Alcalde-Estévez, E.; Herrero, J.I.; Ranz, I.; Fernández, L.; Castillo, C.; Sevilla, L.; et al. Enhanced fatty acid oxidation through metformin and baicalin as therapy for COVID-19 and associated inflammatory states in lung and kidney. Redox Biol. 2023, 68, 102957. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Renal Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Zhao, X.; Li, Y.; Yu, J.; Teng, H.; Wu, S.; Wang, Y.; Zhou, H.; Li, F. Role of mitochondria in pathogenesis and therapy of renal fibrosis. Metabolism 2024, 155, 155913. [Google Scholar] [CrossRef] [PubMed]
- Creed, S.; McKenzie, M. Measurement of Mitochondrial Membrane Potential with the Fluorescent Dye Tetramethylrhodamine Methyl Ester (TMRM). Methods Mol. Biol. 2019, 1928, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Illescas, M.; Peñas, A.; Arenas, J.; Martín, M.A.; Ugalde, C. Regulation of Mitochondrial Function by the Actin Cytoskeleton. Front. Cell Dev. Biol. 2021, 9, 795838. [Google Scholar] [CrossRef] [PubMed]
- Trefts, E.; Hughey, C.C.; Lantier, L.; Lark, D.S.; Boyd, K.L.; Pozzi, A.; Zent, R.; Wasserman, D.H. Energy metabolism couples hepatocyte integrin-linked kinase to liver glucoregulation and postabsorptive responses of mice in an age-dependent manner. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E1118–E1135. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Gil, D.; Laidler, P.; Zarzycka, M.; Dulińska-Litewka, J. Inhibition Effect of Chloroquine and Integrin-Linked Kinase Knockdown on Translation in Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 3682. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, S.; Gao, H.; Wang, P.; Zhang, Y.; Zhang, A.; Jia, Z.; Huang, S. Ursodeoxycholic acid protects against cisplatin-induced acute kidney injury and mitochondrial dysfunction through acting on ALDH1L2. Free Radic. Biol. Med. 2020, 152, 821–837. [Google Scholar] [CrossRef]
- Ding, H.; Bai, F.; Cao, H.; Xu, J.; Fang, L.; Wu, J.; Yuan, Q.; Zhou, Y.; Sun, Q.; He, W.; et al. PDE/cAMP/Epac/C/EBP-β Signaling Cascade Regulates Mitochondria Biogenesis of Tubular Epithelial Cells in Renal Fibrosis. Antioxid. Redox Signal. 2018, 29, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Morales, P.; León-Contreras, J.C.; Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Ramírez-Rangel, A.; Hernández-Pando, R.; Pedraza-Chaverri, J.; Tapia, E. The Protective Role of Cysteine Against Mitochondrial Damage and Oxidative Stress Induced by Kidney Injury. Int. J. Mol. Sci. 2023, 24, 2887. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Group | Tubular Dilatation (%) | Tubular Atrophy (%) | Pus Casts in Tubules (%) |
|---|---|---|---|
| WT CT | 3.2 ± 0.3 | 1.2 ± 0.9 | 0 ± 0 |
| WT FA | 26.2 ± 3.2 * | 33.4 ± 0.9 * | 18.3 ± 2.9 * |
| cKD-ILK CT | 4.5 ± 0.7 | 1 ± 0.6 | 0 ± 0 |
| cKD-ILK FA | 10.9 ± 1.3 *# | 21.8 ± 1 *# | 11.4 ± 2.15 *# |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Serna-Soto, M.; Calleros, L.; Martos-Elvira, M.; Moreno-Piedra, A.; García-Villoria, S.; Griera, M.; Alcalde-Estévez, E.; Asenjo-Bueno, A.; Rodríguez-Puyol, D.; de Frutos, S.; et al. Integrin-Linked Kinase (ILK) Promotes Mitochondrial Dysfunction by Decreasing CPT1A Expression in a Folic Acid-Based Model of Kidney Disease. Int. J. Mol. Sci. 2025, 26, 1861. https://doi.org/10.3390/ijms26051861
de la Serna-Soto M, Calleros L, Martos-Elvira M, Moreno-Piedra A, García-Villoria S, Griera M, Alcalde-Estévez E, Asenjo-Bueno A, Rodríguez-Puyol D, de Frutos S, et al. Integrin-Linked Kinase (ILK) Promotes Mitochondrial Dysfunction by Decreasing CPT1A Expression in a Folic Acid-Based Model of Kidney Disease. International Journal of Molecular Sciences. 2025; 26(5):1861. https://doi.org/10.3390/ijms26051861
Chicago/Turabian Stylede la Serna-Soto, Mariano, Laura Calleros, María Martos-Elvira, Ariadna Moreno-Piedra, Sergio García-Villoria, Mercedes Griera, Elena Alcalde-Estévez, Ana Asenjo-Bueno, Diego Rodríguez-Puyol, Sergio de Frutos, and et al. 2025. "Integrin-Linked Kinase (ILK) Promotes Mitochondrial Dysfunction by Decreasing CPT1A Expression in a Folic Acid-Based Model of Kidney Disease" International Journal of Molecular Sciences 26, no. 5: 1861. https://doi.org/10.3390/ijms26051861
APA Stylede la Serna-Soto, M., Calleros, L., Martos-Elvira, M., Moreno-Piedra, A., García-Villoria, S., Griera, M., Alcalde-Estévez, E., Asenjo-Bueno, A., Rodríguez-Puyol, D., de Frutos, S., & Ruiz-Torres, M. P. (2025). Integrin-Linked Kinase (ILK) Promotes Mitochondrial Dysfunction by Decreasing CPT1A Expression in a Folic Acid-Based Model of Kidney Disease. International Journal of Molecular Sciences, 26(5), 1861. https://doi.org/10.3390/ijms26051861