
Coumarin Derivative Hybrids: Novel Dual Inhibitors Targeting Acetylcholinesterase and Monoamine Oxidases for Alzheimer’s Therapy




Abstract
1. Introduction
2. Results and Discussion
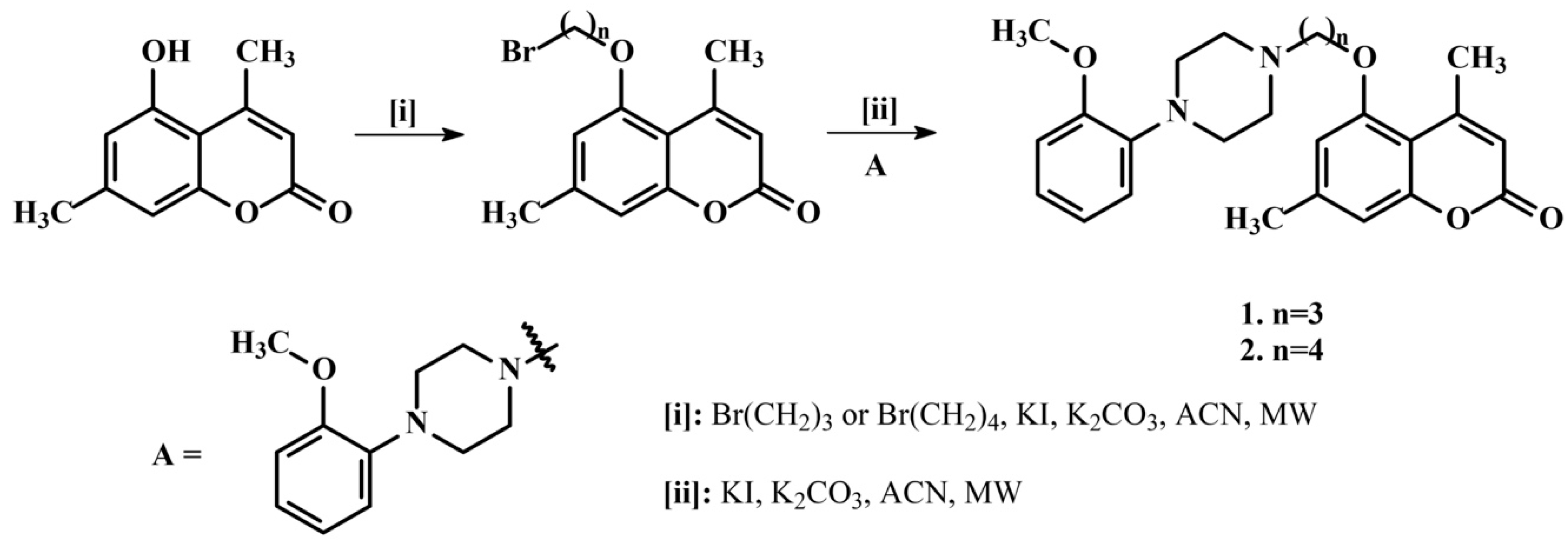
2.1. Synthetic Coumarins
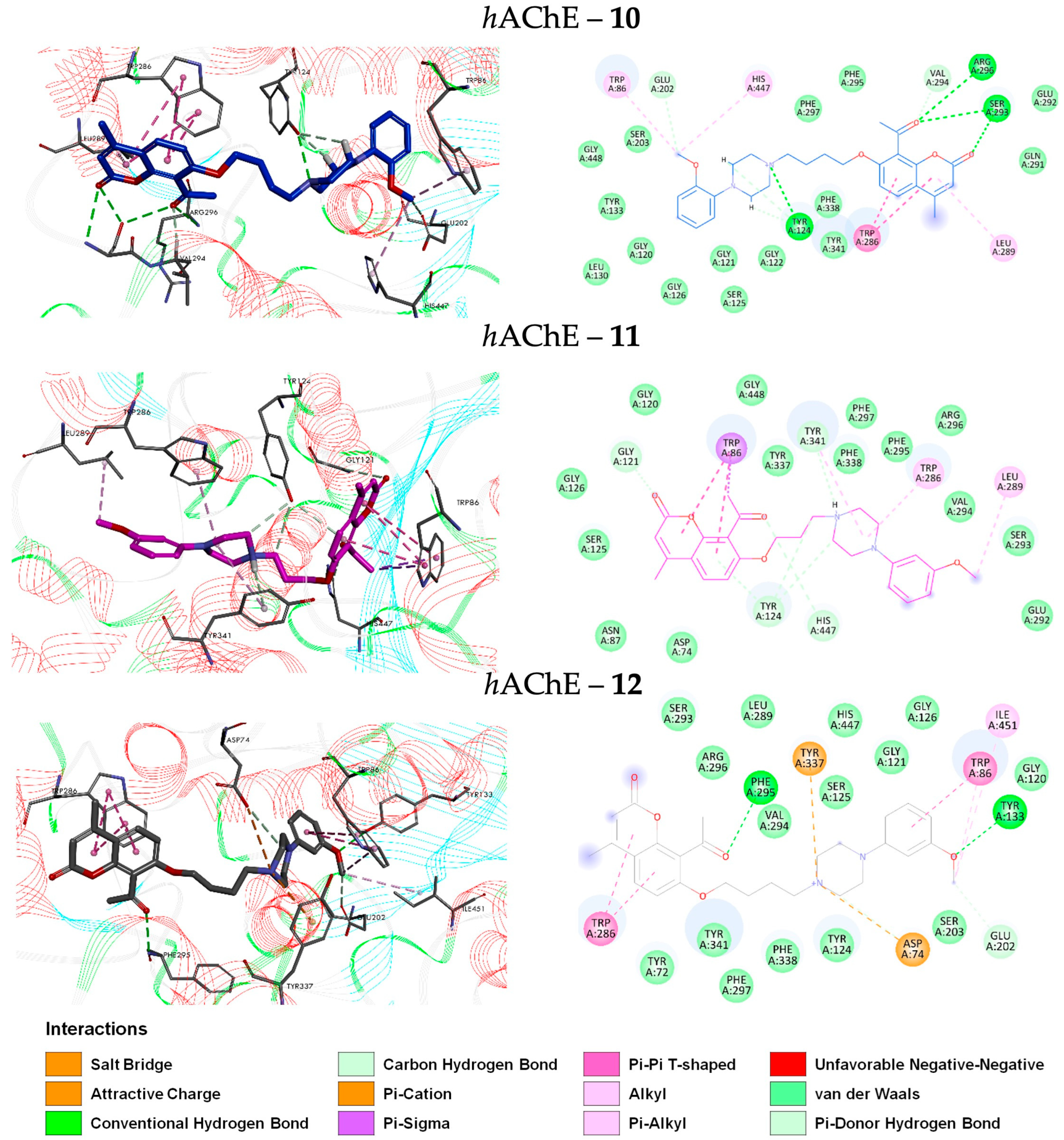
2.2. Molecular Docking of Coumarin Derivatives with hAChE
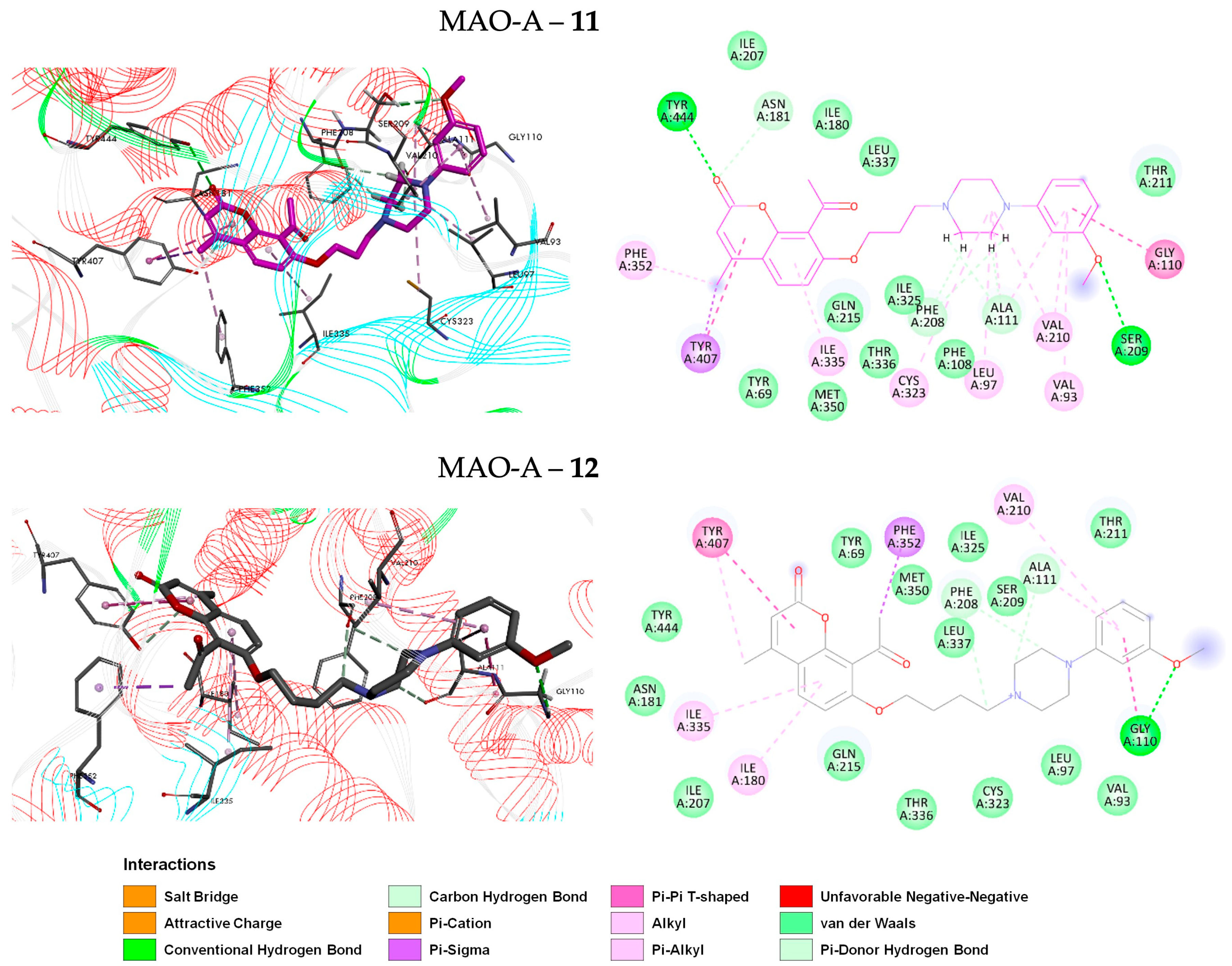
2.3. Molecular Docking of Coumarin Derivatives with hMAOs
2.4. Biological Profile of Coumarin Derivatives as Potential Drugs for Treatment of AD
2.4.1. Evaluation of hAChE Inhibitory Activity
2.4.2. Evaluation of hMAOs Inhibitory Activity
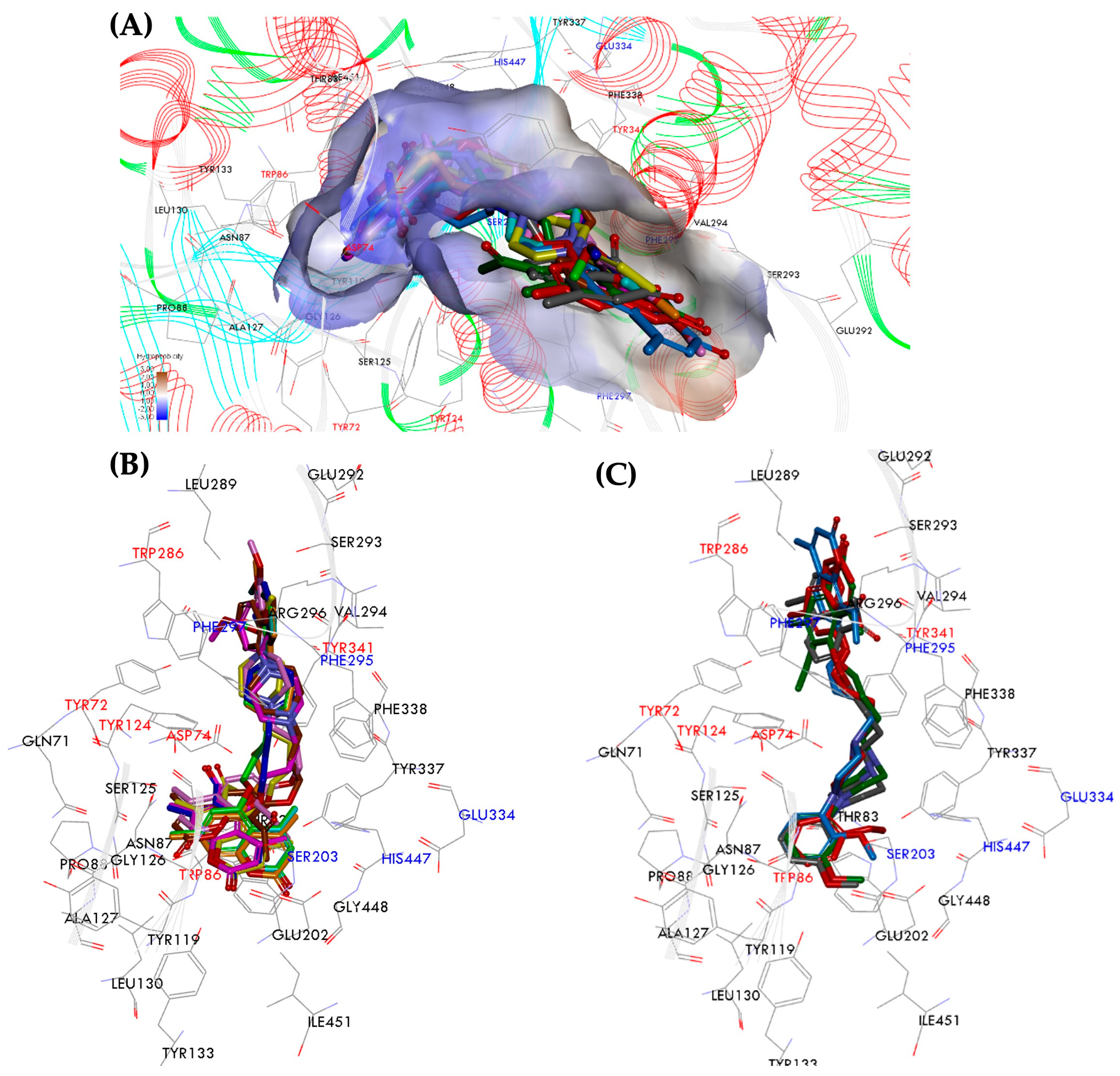
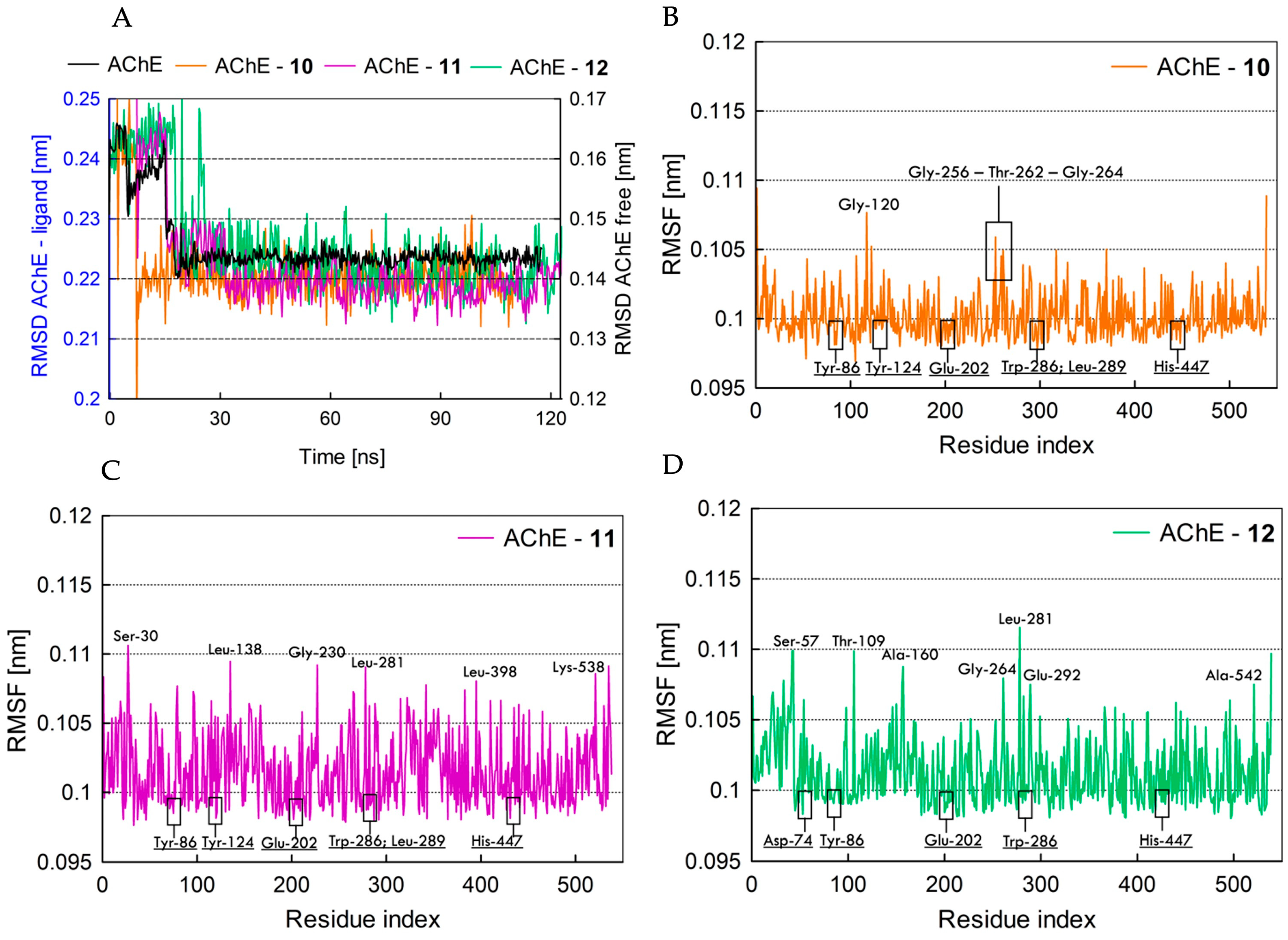
2.5. Molecular Dynamics Analysis of hAChE Inhibitors
2.5.1. Pose Analysis and Binding Mode of hAChE Inhibitors 10–12
2.5.2. Fundamental Molecular Dynamics Simulation Analysis of hAChE Inhibitors 10–12
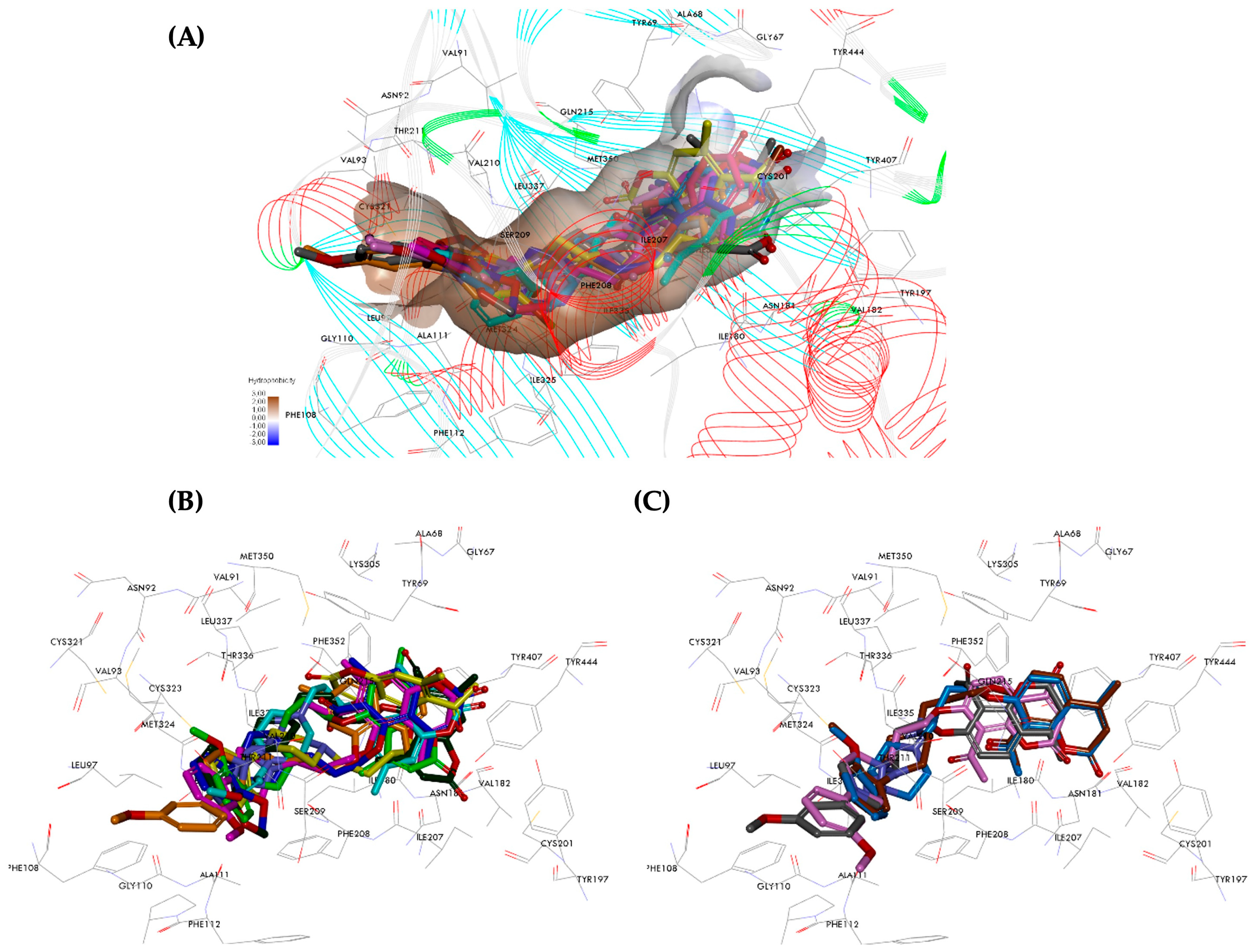
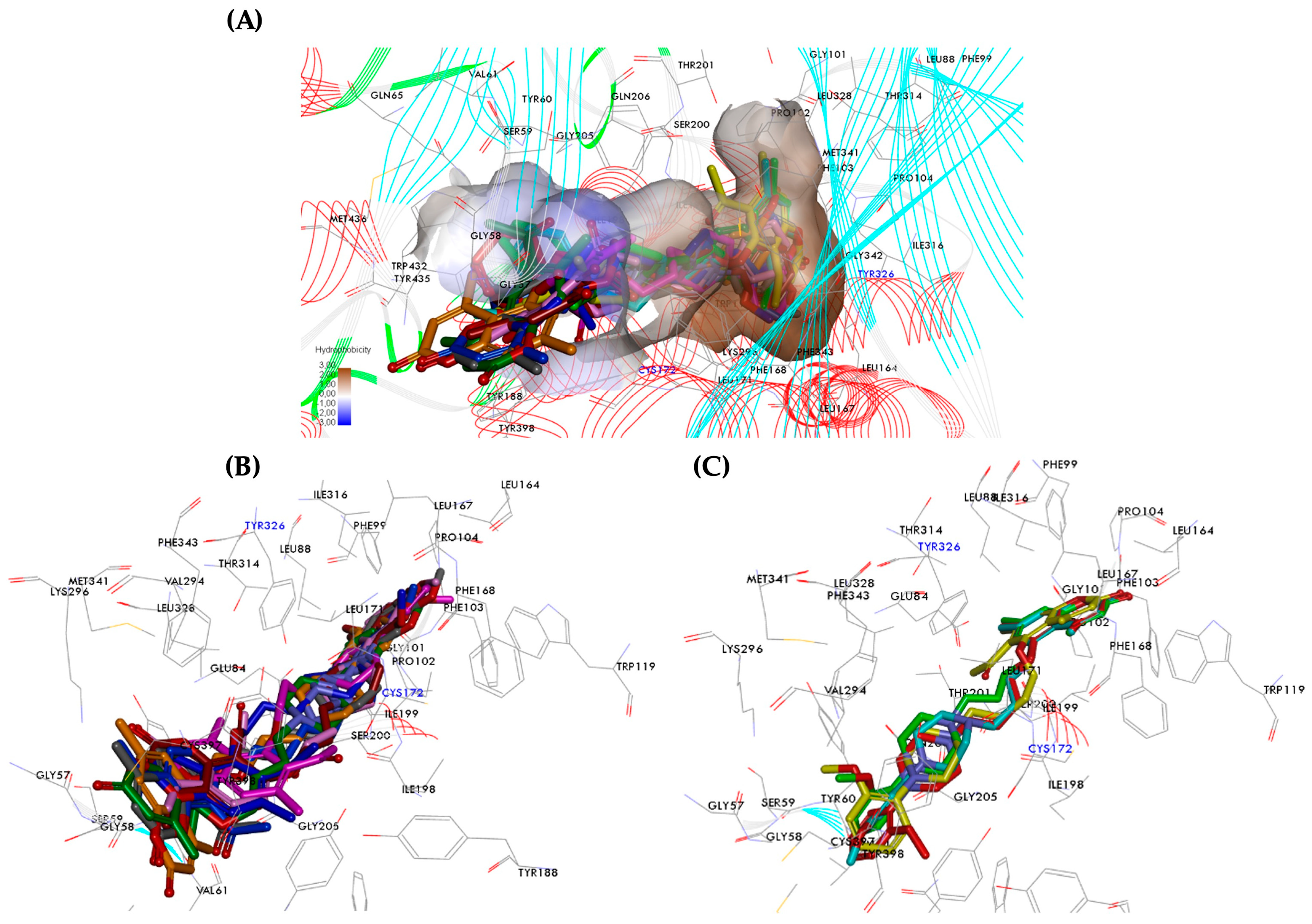
2.6. Molecular Dynamics Analysis of hMAOs Inhibitors
2.6.1. Pose Analysis and Binding Mode of hMAO-A Inhibitors 11 and 12
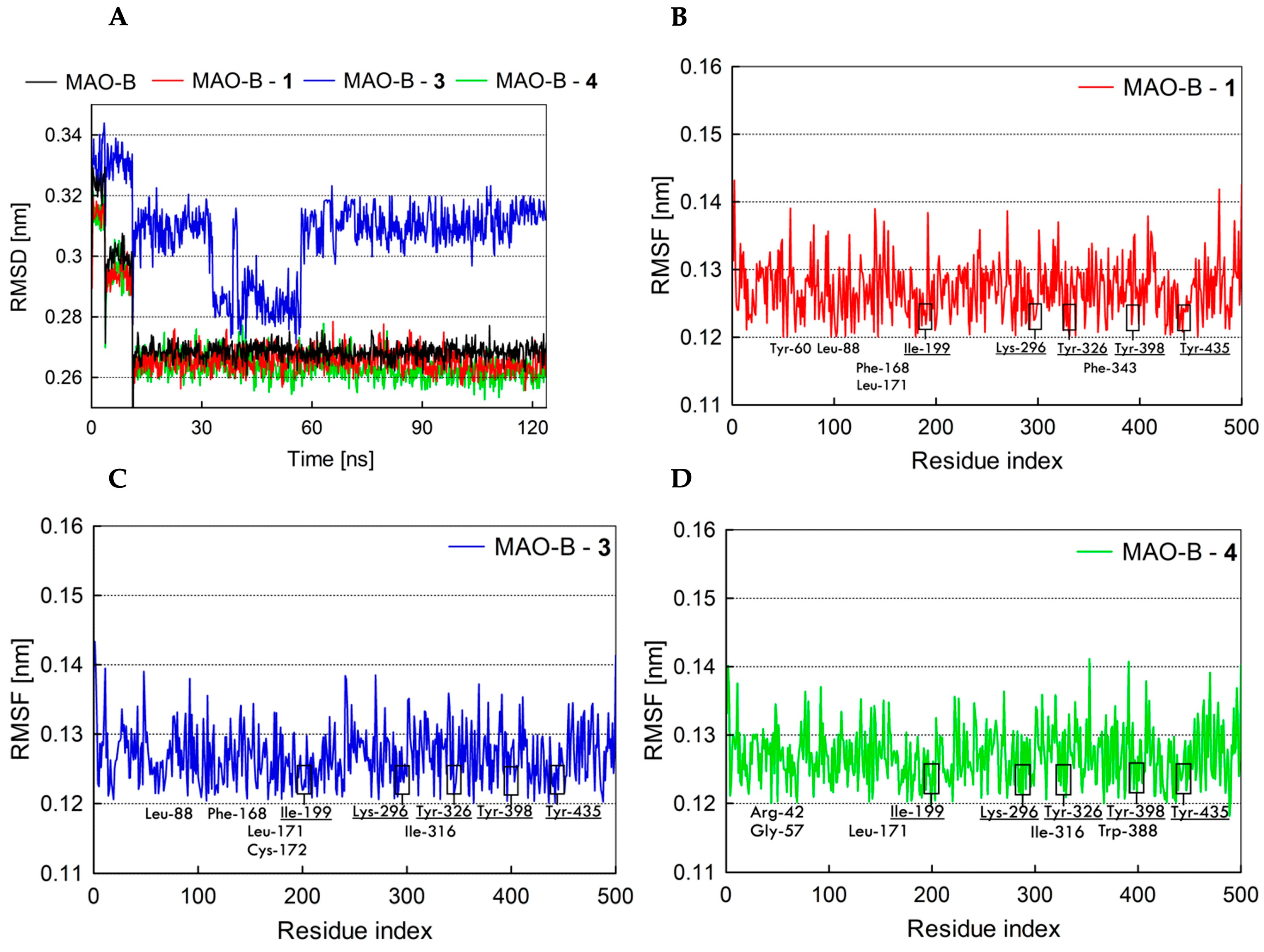
2.6.2. Pose Analysis and Binding Mode of hMAO-B Inhibitor 1, 3 and 4
2.7. Fundamental Molecular Dynamics Simulation Analysis of MAO’s Inhibitors
3. Materials and Methods
3.1. Experimental Section
3.1.1. Chemical Compounds
3.1.2. General Procedure for the Preparation of Compounds 1–12
- 1.
- 4,7-dimethyl-5-{3-[4-(2-methoxyphenyl)piperazin-1-yl]propoxy}coumarin (1)
- 2.
- 4,7-dimethyl-5-{4-[4-(2-methoxyphenyl)piperazin-1-yl]buthoxy}coumarin (2)
3.2. Biological Assays
3.2.1. In Vitro Inhibition Studies on hAChE
3.2.2. In Vitro Inhibition Studies on hMAOs
3.3. Theoretical Methodology
3.3.1. Preparation of the Target Biomacromolecules
3.3.2. Preparation of Ligands
3.3.3. Molecular Structures and Initial System Configurations
3.3.4. Molecular Dynamics Simulations and Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xing, W.; Fu, Y.; Shi, Z.; Lu, D.; Zhang, H.; Hu, Y. Discovery of novel 2,6-disubstituted pyridazinone derivatives as acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2013, 63, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiang, X.; Luo, L.; Yang, X.; Xiao, G.; Liu, Q.; Ai, J.; Tan, Z.; Deng, Y. Aurone Mannich base derivatives as promising multifunctional agents with acetylcholinesterase inhibition, anti-beta-amyloid aggragation and neuroprotective properties for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 126, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Ashford, J.W. Treatment of Alzheimer’s Disease: The Legacy of the Cholinergic Hypothesis, Neuroplasticity, and Future Directions. J. Alzheimers Dis. 2015, 47, 149–156. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Guzior, N.; Bajda, M.; Skrok, M.; Kurpiewska, K.; Lewinski, K.; Brus, B.; Pislar, A.; Kos, J.; Gobec, S.; Malawska, B. Development of multifunctional, heterodimeric isoindoline-1,3-dione derivatives as cholinesterase and beta-amyloid aggregation inhibitors with neuroprotective properties. Eur. J. Med. Chem. 2015, 92, 738–749. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Harel, M.; Sonoda, L.K.; Silman, I.; Sussman, J.L.; Rosenberry, T.L. Crystal structure of thioflavin T bound to the peripheral site of Torpedo californica acetylcholinesterase reveals how thioflavin T acts as a sensitive fluorescent reporter of ligand binding to the acylation site. J. Am. Chem. Soc. 2008, 130, 7856–7861. [Google Scholar] [CrossRef]
- Mesulam, M.M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Birks, J.; Grimley Evans, J.; Iakovidou, V.; Tsolaki, M. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2009, 15, CD001191. [Google Scholar] [CrossRef]
- Racchi, M.; Mazzucchelli, M.; Porrello, E.; Lanni, C.; Govoni, S. Acetylcholinesterase inhibitors: Novel activities of old molecules. Pharmacol. Res. 2004, 50, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Leon, R.; Garcia, A.G.; Marco-Contelles, J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Gil-Bea, F.J.; Diez-Ariza, M.; Chen, C.P.; Francis, P.T.; Lasheras, B.; Ramirez, M.J. Cholinergic-serotonergic imbalance contributes to cognitive and behavioral symptoms in Alzheimer’s disease. Neuropsychologia 2005, 43, 442–449. [Google Scholar] [CrossRef]
- Terry, A.V., Jr.; Buccafusco, J.J.; Wilson, C. Cognitive dysfunction in neuropsychiatric disorders: Selected serotonin receptor subtypes as therapeutic targets. Behav. Brain Res. 2008, 195, 30–38. [Google Scholar] [CrossRef]
- Youdim, M.B.; Riederer, P. Monoamine oxidase B; a misunderstood and misjudged enzyme. Pharmacol. Res. Commun. 1988, 20 (Suppl. S4), 9–14. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Inoue, M.; Hromadkova, L.; Teranishi, Y.; Yamamoto, N.G.; Wiehager, B.; Bogdanovic, N.; Winblad, B.; Sandebring-Matton, A.; Frykman, S.; et al. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with gamma-secretase and regulates neuronal amyloid beta-peptide levels. Alzheimers Res. Ther. 2017, 9, 57. [Google Scholar] [CrossRef]
- Bautista-Aguilera, O.M.; Samadi, A.; Chioua, M.; Nikolic, K.; Filipic, S.; Agbaba, D.; Soriano, E.; de Andres, L.; Rodriguez-Franco, M.I.; Alcaro, S.; et al. N-Methyl-N-((1-methyl-5-(3-(1-(2-methylbenzyl)piperidin-4-yl)propoxy)-1H-indol-2-yl)methyl)prop-2-yn-1-amine, a new cholinesterase and monoamine oxidase dual inhibitor. J. Med. Chem. 2014, 57, 10455–10463. [Google Scholar] [CrossRef]
- Pisani, L.; Farina, R.; Catto, M.; Iacobazzi, R.M.; Nicolotti, O.; Cellamare, S.; Mangiatordi, G.F.; Denora, N.; Soto-Otero, R.; Siragusa, L.; et al. Exploring Basic Tail Modifications of Coumarin-Based Dual Acetylcholinesterase-Monoamine Oxidase B Inhibitors: Identification of Water-Soluble, Brain-Permeant Neuroprotective Multitarget Agents. J. Med. Chem. 2016, 59, 6791–6806. [Google Scholar] [CrossRef]
- Joubert, J.; Foka, G.B.; Repsold, B.P.; Oliver, D.W.; Kapp, E.; Malan, S.F. Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 125, 853–864. [Google Scholar] [CrossRef]
- Zhang, P.; Xu, S.; Zhu, Z.; Xu, J. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Hilgert, M.; Noldner, M.; Chatterjee, S.S.; Klein, J. KA-672 inhibits rat brain acetylcholinesterase in vitro but not in vivo. Neurosci. Lett. 1999, 263, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Peng, Q.; Shao, J.; Liu, X.; Huang, Z.; Pu, X.; Ma, L.; Li, Y.M.; Chan, A.S.; Gu, L. Synthesis and biological evaluation of functionalized coumarins as acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2005, 40, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, X.B.; Wang, T.; Kong, L.Y. Design, synthesis, and acetylcholinesterase inhibitory activity of novel coumarin analogues. Bioorg. Med. Chem. 2008, 16, 8011–8021. [Google Scholar] [CrossRef] [PubMed]
- Hoerr, R.; Noeldner, M. Ensaculin (KA-672 HCl): A multitransmitter approach to dementia treatment. CNS Drug Rev. 2002, 8, 143–158. [Google Scholar] [CrossRef]
- Weinstock, M. Selectivity of Cholinesterase Inhibition. CNS Drugs 1999, 12, 307–323. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef]
- Patil, P.O.; Bari, S.B.; Firke, S.D.; Deshmukh, P.K.; Donda, S.T.; Patil, D.A. A comprehensive review on synthesis and designing aspects of coumarin derivatives as monoamine oxidase inhibitors for depression and Alzheimer’s disease. Bioorg. Med. Chem. 2013, 21, 2434–2450. [Google Scholar] [CrossRef]
- Sun, Q.; Peng, D.Y.; Yang, S.G.; Zhu, X.L.; Yang, W.C.; Yang, G.F. Syntheses of coumarin-tacrine hybrids as dual-site acetylcholinesterase inhibitors and their activity against butylcholinesterase, Abeta aggregation, and beta-secretase. Bioorg. Med. Chem. 2014, 22, 4784–4791. [Google Scholar] [CrossRef]
- Pisani, L.; Farina, R.; Nicolotti, O.; Gadaleta, D.; Soto-Otero, R.; Catto, M.; Di Braccio, M.; Mendez-Alvarez, E.; Carotti, A. In silico design of novel 2H-chromen-2-one derivatives as potent and selective MAO-B inhibitors. Eur. J. Med. Chem. 2015, 89, 98–105. [Google Scholar] [CrossRef]
- Zawadowski, T.; Kossakowski, J. Synthesis of 3-methyl-5-methoxy-6-hydroxy-7-acetyl-coumaronic and 3,7-dimethyl-5-methoxy-9-H-furo [2,3-f] [1] benzopyran-9-on-2-carboxylic acids and their derivatives. Pol. J. Pharmacol. Pharm. 1980, 32, 789–792. [Google Scholar] [PubMed]
- Trykowska Konc, J.; Hejchman, E.; Kruszewska, H.; Wolska, I.; Maciejewska, D. Synthesis and pharmacological activity of O-aminoalkyl derivatives of 7-hydroxycoumarin. Eur. J. Med. Chem. 2011, 46, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, K.; Mlodzikowska, K.; Gluch-Lutwin, M.; Grybos, A.; Siwek, A. Synthesis of a new series of aryl/heteroarylpiperazinyl derivatives of 8-acetyl-7-hydroxy-4-methylcoumarin with low nanomolar 5-HT(1A) affinities. Eur. J. Med. Chem. 2017, 137, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, K.; Grzeszczuk, D.; Gluch-Lutwin, M.; Grybos, A.; Siwek, A.; Lesniak, A.; Sacharczuk, M.; Trzaskowski, B. 5-HT(1A) and 5-HT(2A) receptors affinity, docking studies and pharmacological evaluation of a series of 8-acetyl-7-hydroxy-4-methylcoumarin derivatives. Bioorg. Med. Chem. 2018, 26, 527–535. [Google Scholar] [CrossRef]
- Ostrowska, K.; Grzeszczuk, D.; Gluch-Lutwin, M.; Grybos, A.; Siwek, A.; Dobrzycki, L.; Trzaskowski, B. Development of selective agents targeting serotonin 5HT(1A) receptors with subnanomolar activities based on a coumarin core. MedChemComm 2017, 8, 1690–1696. [Google Scholar] [CrossRef]
- Ostrowska, K.; Lesniak, A.; Karczynska, U.; Jeleniewicz, P.; Gluch-Lutwin, M.; Mordyl, B.; Siwek, A.; Trzaskowski, B.; Sacharczuk, M.; Bujalska-Zadrozny, M. 6-Acetyl-5-hydroxy-4,7-dimethylcoumarin derivatives: Design, synthesis, modeling studies, 5-HT(1A), 5-HT(2A) and D(2) receptors affinity. Bioorg. Chem. 2020, 100, 103912. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-A resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef]
- Hubalek, F.; Binda, C.; Khalil, A.; Li, M.; Mattevi, A.; Castagnoli, N.; Edmondson, D.E. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J. Biol. Chem. 2005, 280, 15761–15766. [Google Scholar] [CrossRef]
- Zhao, H.; Caflisch, A. Molecular dynamics in drug design. Eur. J. Med. Chem. 2015, 91, 4–14. [Google Scholar] [CrossRef]
- Bharadwaj, K.K.; Ahmad, I.; Pati, S.; Ghosh, A.; Sarkar, T.; Rabha, B.; Patel, H.; Baishya, D.; Edinur, H.A.; Abdul Kari, Z.; et al. Potent Bioactive Compounds From Seaweed Waste to Combat Cancer Through Bioinformatics Investigation. Front. Nutr. 2022, 9, 889276. [Google Scholar] [CrossRef] [PubMed]
- Sudevan, S.T.; Oh, J.M.; Abdelgawad, M.A.; Abourehab, M.A.S.; Rangarajan, T.M.; Kumar, S.; Ahmad, I.; Patel, H.; Kim, H.; Mathew, B. Introduction of benzyloxy pharmacophore into aryl/heteroaryl chalcone motifs as a new class of monoamine oxidase B inhibitors. Sci. Rep. 2022, 12, 22404. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Catto, M.; De Palma, A.; Farina, R.; Cellamare, S.; Altomare, C.D. Discovery of Potent Dual Binding Site Acetylcholinesterase Inhibitors via Homo- and Heterodimerization of Coumarin-Based Moieties. ChemMedChem 2017, 12, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Iacobazzi, R.M.; Catto, M.; Rullo, M.; Farina, R.; Denora, N.; Cellamare, S.; Altomare, C.D. Investigating alkyl nitrates as nitric oxide releasing precursors of multitarget acetylcholinesterase-monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2019, 161, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef]
- Wang, D.; Hong, R.Y.; Guo, M.; Liu, Y.; Chen, N.; Li, X.; Kong, D.X. Novel C7-Substituted Coumarins as Selective Monoamine Oxidase Inhibitors: Discovery, Synthesis and Theoretical Simulation. Molecules 2019, 24, 4003. [Google Scholar] [CrossRef]
- Gaweska, H.; Fitzpatrick, P.F. Structures and Mechanism of the Monoamine Oxidase Family. Biomol. Concepts 2011, 2, 365–377. [Google Scholar] [CrossRef]
- Schrödinger Release 2023-1: Desmond Molecular Dynamics System, D.E.S.R., New York, NY, 2021. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2021.
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Ahmad, I.; Jadhav, H.; Shinde, Y.; Jagtap, V.; Girase, R.; Patel, H. Optimizing Bedaquiline for cardiotoxicity by structure based virtual screening, DFT analysis and molecular dynamic simulation studies to identify selective MDR-TB inhibitors. In Silico Pharmacol. 2021, 9, 23. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Mehta, C.H.; Narayan, R.; Aithal, G.; Pandiyan, S.; Bhat, P.; Dengale, S.; Shah, A.; Nayak, U.Y.; Garg, S. Molecular simulation driven experiment for formulation of fixed dose combination of Darunavir and Ritonavir as anti-HIV nanosuspension. J. Mol. Liq. 2019, 293, 111469. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Koh, Y.; Tojo, Y.; Ghosh, A.K.; Mitsuya, H. Prediction of potency of protease inhibitors using free energy simulations with polarizable quantum mechanics-based ligand charges and a hybrid water model. J. Chem. Inf. Model. 2009, 49, 2851–2862. [Google Scholar] [CrossRef]
- Gnerre, C.; Catto, M.; Leonetti, F.; Weber, P.; Carrupt, P.A.; Altomare, C.; Carotti, A.; Testa, B. Inhibition of monoamine oxidases by functionalized coumarin derivatives: Biological activities, QSARs, and 3D-QSARs. J. Med. Chem. 2000, 43, 4747–4758. [Google Scholar] [CrossRef]
- Zolek, T.; Enyedy, E.A.; Ostrowska, K.; Posa, V.; Maciejewska, D. Drug likeness prediction of 5-hydroxy-substituted coumarins with high affinity to 5-HT(1A) and 5-HT(2A) receptors. Eur. J. Pharm. Sci. 2018, 115, 25–36. [Google Scholar] [CrossRef]
- Zolek, T.; Domotor, O.; Ostrowska, K.; Enyedy, E.A.; Maciejewska, D. Evaluation of blood-brain barrier penetration and examination of binding to human serum albumin of 7-O-arylpiperazinylcoumarins as potential antipsychotic agents. Bioorg. Chem. 2019, 84, 211–225. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | hAChE [a,b] | hMAO-A [a,b] | hMAO-B [a,b] | ΔGbind [kcal/mol] | ||
|---|---|---|---|---|---|---|
| hAChE | hMAO-A | hMAO-B | ||||
| 1 | 61 ± 1% | 44 ± 4% | 2.18 ± 0.48 | −66.89 | −52.88 | −72.32 |
| 2 | 61 ± 4% | 19 ± 1% | 33 ± 2% | −66.30 | −9.56 | −53.61 |
| 3 | 53 ± 2% | 39 ± 5% | 1.88 ± 0.45 | −65.30 | −26.74 | −75.57 |
| 4 | 56 ± 1% | 38 ± 4% | 3.18 ± 0.63 | −65.78 | −24.47 | −70.04 |
| 5 | 50 ± 6% | 22 ± 3% | 34 ± 2% | −61.09 | −10.86 | −45.51 |
| 6 | 48 ± 1% | 30 ± 5% | 43 ± 3% | −60.44 | −14.87 | −58.17 |
| 7 | 46 ± 3% | 39 ± 3% | 35 ± 3% | −60.42 | −25.85 | −45.07 |
| 8 | 54 ± 8% | 55 ± 2% | 26 ± 2% | −62.24 | −59.35 | −50.82 |
| 9 | 59 ± 7% | 32 ± 4% | 4.76 ± 0.34 | −66.25 | −17.42 | −69.95 |
| 10 | 1.52 ± 0.66 | 52 ± 4% | 24 ± 1% | −77.24 | −56.26 | −59.12 |
| 11 | 2.80 ± 0.69 | 6.97 ± 0.76 | 32 ± 1% | −76.32 | −66.27 | −55.07 |
| 12 | 4.95 ± 0.48 | 7.65 ± 0.36 | 57 ± 4% | −70.12 | −62.89 | −50.82 |
| Ensaculin | 0.36 ± 0.01 | - | - | |||
| Pargyline | - | 10.9 ± 0.6 | 2.69 ± 0.48 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żołek, T.; Purgatorio, R.; Kłopotowski, Ł.; Catto, M.; Ostrowska, K. Coumarin Derivative Hybrids: Novel Dual Inhibitors Targeting Acetylcholinesterase and Monoamine Oxidases for Alzheimer’s Therapy. Int. J. Mol. Sci. 2024, 25, 12803. https://doi.org/10.3390/ijms252312803
Żołek T, Purgatorio R, Kłopotowski Ł, Catto M, Ostrowska K. Coumarin Derivative Hybrids: Novel Dual Inhibitors Targeting Acetylcholinesterase and Monoamine Oxidases for Alzheimer’s Therapy. International Journal of Molecular Sciences. 2024; 25(23):12803. https://doi.org/10.3390/ijms252312803
Chicago/Turabian StyleŻołek, Teresa, Rosa Purgatorio, Łukasz Kłopotowski, Marco Catto, and Kinga Ostrowska. 2024. "Coumarin Derivative Hybrids: Novel Dual Inhibitors Targeting Acetylcholinesterase and Monoamine Oxidases for Alzheimer’s Therapy" International Journal of Molecular Sciences 25, no. 23: 12803. https://doi.org/10.3390/ijms252312803
APA StyleŻołek, T., Purgatorio, R., Kłopotowski, Ł., Catto, M., & Ostrowska, K. (2024). Coumarin Derivative Hybrids: Novel Dual Inhibitors Targeting Acetylcholinesterase and Monoamine Oxidases for Alzheimer’s Therapy. International Journal of Molecular Sciences, 25(23), 12803. https://doi.org/10.3390/ijms252312803