
Changes in Cells Associated with Insulin Resistance

Abstract
1. Introduction
2. Characteristics of Insulin
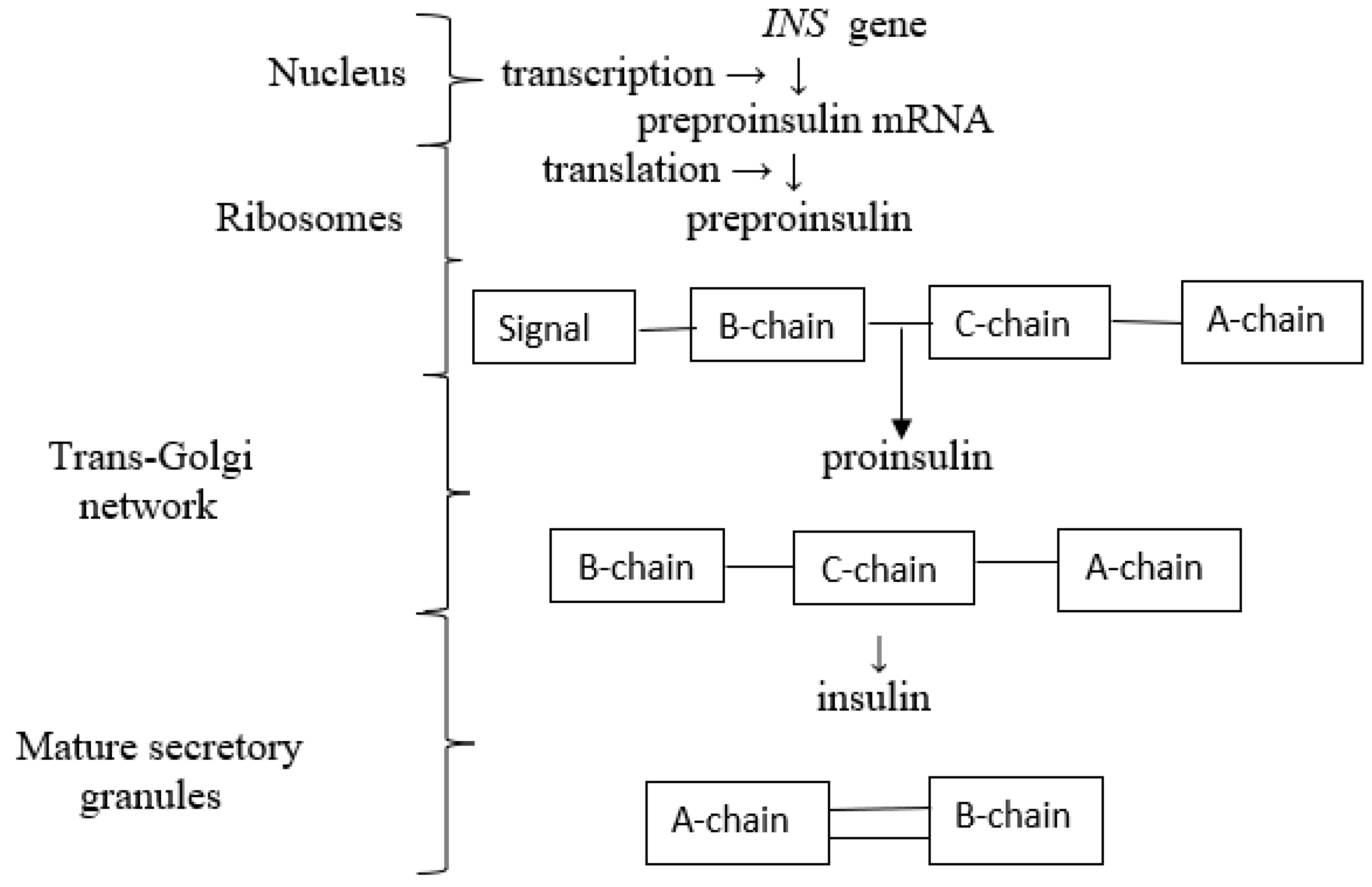
2.1. Biosynthesis and Secretion of Insulin
Biosynthesis of Insulin
3. Intracellular Insulin Signaling Pathways
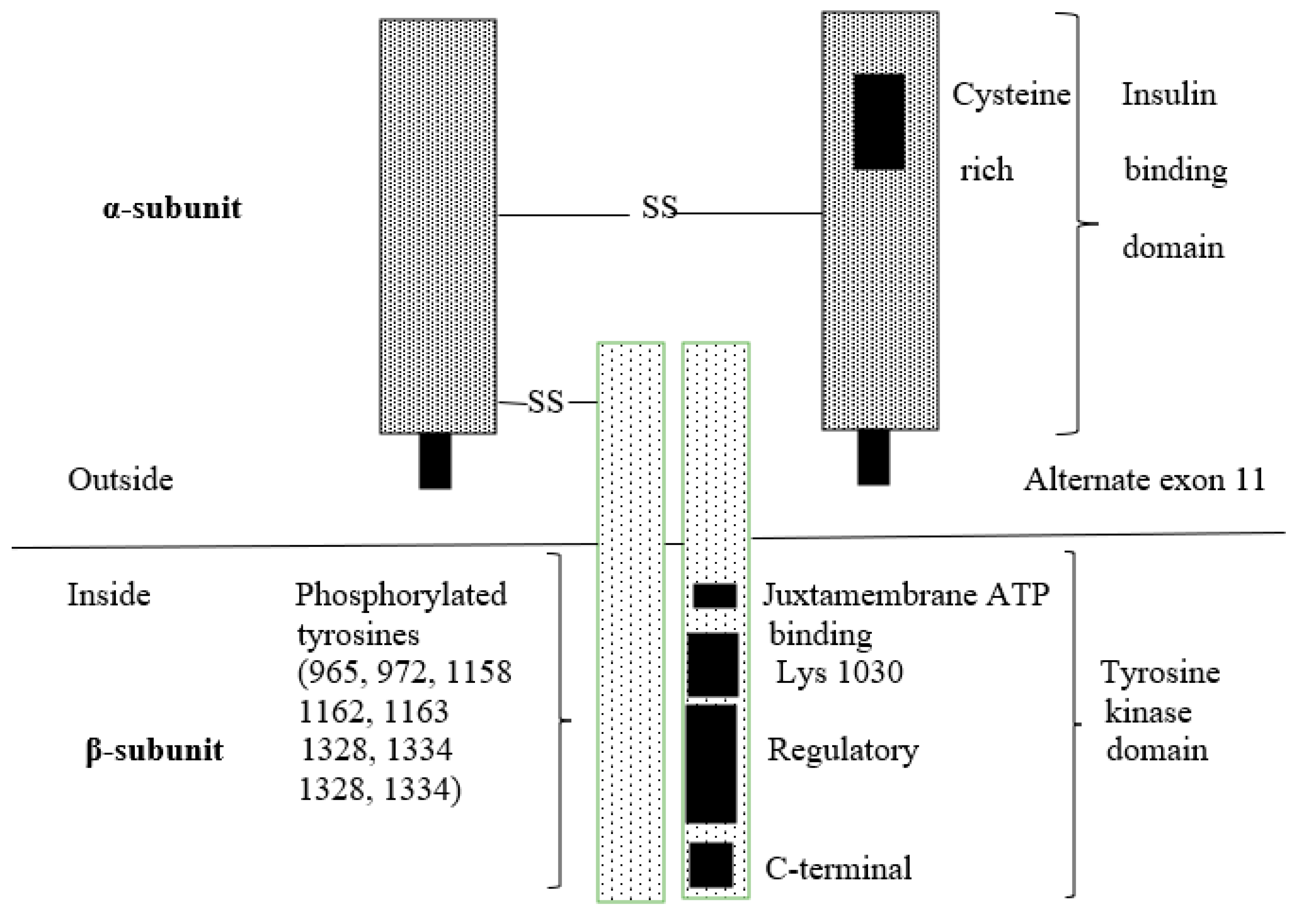
3.1. Insulin Receptor
3.2. Insulin Receptor Substrates
3.3. The PI3K/AKT Signaling Pathway
3.4. The MAPK Signaling Pathway
4. Role of Insulin in Selected Cells, Tissue, Organs
4.1. Insulin Signaling Pathway and Its Role in the Liver
4.2. Insulin Signaling Pathway and Its Role in Muscle
4.3. Insulin Signaling Pathway and Its Role in White Adipose Tissue
4.4. Insulin Signaling Pathway and Its Role in Other Organs and Cells
5. Insulin Resistance
5.1. Severe Insulin Resistance Syndromes
5.1.1. Pathogenesis of Severe Insulin Resistance
5.1.2. Defects in the Insulin Gene
5.1.3. Defects in the Insulin Receptor
5.1.4. Lipodystrophies
5.1.5. Other Severe Insulin Resistance Syndromes
5.2. Mechanisms of Insulin Resistance
5.2.1. Insulin Resistance in Select Tissues and Organs
5.2.2. The Glucose-Fatty Acid Cycle (the Randle Cycle)
5.2.3. Hexosamine Biosynthesis Pathway
5.2.4. Ectopic Lipid Accumulation
5.2.5. Diacylglycerol
5.2.6. Ceramide
5.2.7. Endoplasmic Reticulum Stress
5.2.8. Inflammation
5.2.9. Mitochondrial Dysfunction and ROS Formation
5.3. Selective Insulin Resistance
6. Therapeutic Strategies for Insulin Resistance
7. Insulin Resistance Causes Cancer: True or False? Is Insulin Our Friend or Foe?
8. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Hausler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signaling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Park, S.-Y.; Choi, C.S. Insulin resistance: From mechanisms to therapeutic strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 4, a009191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, C.; Sun, W.; Ji, H.; Lian, F. The crucial role of mechanism of insulin resistance in metabolic syndrome. Fron. Endocrinol. 2023, 14, 1149239. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.-G. Role of insulin in health and disease: An update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef]
- Zimmet, P.; Albert, K.G.; Shaw, J. Global and social implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef]
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Chawla, A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 2013, 339, 172–177. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metabol. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Csajbok, E.A.; Tamas, G. Cerebral cortex: A target and source of insulin? Diabetologia 2016, 59, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.; Lincoln, B.; Rhodes, C.I. The biosynthesis of the subtilisin-related proprotein convertase PC3, but not that of the PC2 convertase, is regulated by glucose in parallel to proinsulin biosynthesis in rat pancreatic islets. J. Biol. Chem. 1993, 268, 4276–4280. [Google Scholar] [CrossRef] [PubMed]
- Andrali, S.S.; Sampley, M.L.; Vanderford, N.L.; Ozcan, S. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem. J. 2008, 415, 1–10. [Google Scholar] [CrossRef]
- Vasilievic, J.; Tarkko, J.M.; Knoch, K.P.; Solimena, M. The making of insulin in health and disease. Diabetologia 2020, 63, 1982–1989. [Google Scholar]
- Kaufman, B.A.; Li, C.; Soleimanpour, S.A. Mitochondrial regulation of beta-cell function. Maintaining the momentum for insulin resistance. Mol. Asp. Med. 2015, 42, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Artner, I.; Stein, R. Transcriptional regulation of insulin gene expression. In Pancreatic Beta Cell in Health and Disease; Springer: Tokyo, Japan, 2008; pp. 13–30. [Google Scholar]
- Hutton, J.C. Insulin secretion granule biogenesis and the proinsulin-processing endopeptidases. Diabetologia 1994, 37 (Suppl. 2), S48–S56. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Runngerr-Brändle, E.; Just, I.; Jonas, J.C.; Aktories, K.; Wallheim, C.B. Effect of actin filaments by Clostridium botulinum C2 toxin on insulin secretion in HIT-T15 cells and pancreatic islets. Mol. Biol. Cell 1994, 5, 1199–1213. [Google Scholar] [CrossRef]
- Rorsman, P.; Rengström, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 2003, 46, 1029–1045. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Thurmond, D.C. Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet β cells. Exp. Mol. Med. 2013, 45, e37. [Google Scholar] [CrossRef]
- Gandasi, N.R.; Yim, P.; Riz, M.; Chibalina, M.V.; Cortese, G.; Lund, P.-E.; Matveev, V.; Rorsman, P.; Sherman, A.; Pedersen, M.G.; et al. Ca2+ channel clustering with insulin-containing granules is distributed in type 2 diabetes. J. Clin. Investig. 2017, 127, 2353–2364. [Google Scholar] [CrossRef]
- Wang, Z.; Thurmond, D.C. Mechanisms of biphasic insulin-granule exocytosis—Role of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 2009, 122, 893–903. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin receptor isoforms in physiology and diseases: An updated view. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Denley, A.; Wallace, J.C.; Cosgrove, L.J.; Forbes, B.E. The insulin receptor isoforms exon 11 (IR-A) in cancer and other diseases. A review. Horm. Metabol. Res. 2003, 35, 778–785. [Google Scholar]
- Escribano, O.; Beneit, N.; Rubio-Longás, C.; López-Pastor, A.R.; Gómez-Hernández, A. The role of insulin receptor isoforms in diabetes and its metabolism and vascular complications. J. Diab. Res. 2017, 2017, 1403206. [Google Scholar]
- Vella, V.; Milluzo, A.; Scalisi, N.M.; Vigneri, P.; Sciacca, L. Insulin receptor isoforms in cancer. Int. J. Med. 2018, 19, 3615. [Google Scholar] [CrossRef] [PubMed]
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of insulin/IGF hybrid receptors: Contributions of the insulin receptor 1,2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J. 2007, 403, 603–613. [Google Scholar] [CrossRef]
- Mantzoros, C.; Serdy, S. Insulin Action. In UpToDate Online Medical Text, Topic 1789, version 14.0; Wolters Kluwer, MEDI MEDIA: Waltham, MA, USA, 2023. [Google Scholar]
- Li, M.; Chi, X.; Wang, Y.; Seterrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- White, M.E. Regulating insulin signaling and β-cell function through IRS proteins. Can. J. Physiol. Pharmacol. 2006, 84, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M. The insulin receptor substrate (IRS) proteins: At the intersection of metabolism and cancer. Cell Cycle 2011, 10, 1750–1756. [Google Scholar] [CrossRef]
- Youngren, J.F. Regulation of insulin receptor function. Cell Mol. Life Sci. 2007, 64, 873–891. [Google Scholar] [CrossRef]
- Yee, L.D.; Mortimer, J.E.; Natarajan, R.; Dietze, E.C.; Seewaldt, V.L. Metabolic health, insulin, and breast cancer: Why oncologists should care about insulin. Front. Endocrinol. 2020, 11, 58. [Google Scholar] [CrossRef]
- Angeldi, A.M.; Filippaios, A.; Mantzoros, C.S. Severe insulin resistance syndromes. J. Clin. Investig. 2021, 131, e142245. [Google Scholar] [CrossRef]
- Vigneri, R.; Sciacca, L.; Vigneri, P. Rethinking the relationship between insulin and cancer. Trends Endocrinol. Metab. 2020, 31, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, K.; Tavares, C.D.; Rines, A.G.; Puigsener, P. Molecular pathophysiology of hepatic glucose production. Mol. Asp. Med. 2015, 46, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors: Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.C.; Coops, K.D.; Guo, S.; Li, Y.; Kollipara, R.; DePinho, R.A.; White, M.F. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptative nutrient homeostasis and endocrine growth regulation. Cell Metabol. 2008, 8, 65–76. [Google Scholar] [CrossRef]
- Abdulla, H.; Smith, K.; Atheron, P.J.; Idris, I. Role of insulin in the regulation of human skeletal muscle protein synthesis and breakdown: A systematic review and meta-analysis. Diabetologia 2016, 59, 44–55. [Google Scholar] [CrossRef]
- Choi, Y.H.; Park, S.; Hockmans, S.; Zmuda-Trzebiatowska, E.; Svennelid, F.; Haluzik, M.; Gavrilova, O.; Ahmad, F.; Pepin, L.; Napolitano, M.; et al. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J. Clin. Investig. 2006, 116, 3240–3245. [Google Scholar] [CrossRef]
- Woods, S.C.; Seeley, R.J.; Baskin, D.G.; Schwartz, M.W. Insulin and the blood-brain barrier. Curr. Pharm. Des. 2003, 9, 795–800. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J. Insulin signal transduction through protein kinase cascades. Mol. Cell Biochem. 1998, 182, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Filippi, B.M.; Abraham, M.A.; Yue, J.T.Y.; Lam, T.K.T. Insulin and glucagon signaling in the central nervous system. Rev. Endocr. Metab. Disord. 2013, 14, 365–375. [Google Scholar] [CrossRef]
- Garcia-Cáceres, C.; Quarta, C.; Vareba, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocitic insulin signaling couples brain glucose uptake with nutrient availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Woods, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Zabolotny, J.M.; Huang, H.; Lee, H.; Kim, Y.-B. Insulin in the nervous system in the mind: Functions in metabolism, memory, and mood. Mol. Metab. 2016, 5, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmidt, M. Differential sensitivity of men and women to anorexigenic and memory-improving effect of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344. [Google Scholar] [CrossRef]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brains. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef]
- Ramnanan, C.J.; Edgerton, D.S.; Cherrington, A.D. Evidence against a physiologic role for acute changes in CNS insulin action in the rapid regulation and hepatic glucose production. Cell Metab. 2012, 15, 656–664. [Google Scholar] [CrossRef]
- Ramnanan, C.J.; Kraft, G.; Smith, M.S.; Farmer, B.; Neal, D.; Williams, P.E.; Lautz, M.; Farmer, T.; Donohue, E.P.; Cherringtomn, A.D.; et al. Interaction between the central and peripheral effects of insulin in controlling hepatic glucose metabolism in the conscious dog. Diabetes 2013, 62, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Asplin, C.M.; Paquette, T.L.; Palmer, J.P. In vivo inhibition of glucagon secretion by paracrine beta cell activity in man. J. Clin. Investig. 1981, 68, 314–318. [Google Scholar] [CrossRef]
- Singh, S.; Sharma, R.; Kumari, M.; Tiwari, S. Insulin receptors in the kidneys in health and disease. World J. Nephrol. 2019, 8, 11–22. [Google Scholar] [CrossRef]
- Akhtar, M.; Taha, N.M.; Nauman, A.; Mujeeb, I.B.; Al-Nabet, A. Diabetic kidney disease: Past and present. Adv. Anat. Pathol. 2020, 27, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Gatica, R.; Bertinat, R.; Silva, P.; Carpio, D.; Ramirez, M.J.; Slebe, J.C.; San Martin, R.; Nualart, F.; Campistol, J.M.; Caelles, C.; et al. Altered expression and localization of insulin receptor in proximal tubule cells from human and rat diabetic kidneys. J. Cell Biochem. 2013, 114, 639–649. [Google Scholar] [CrossRef]
- Tiwari, S.; Halagappa, V.K.; Riazi, S.; Hu, X.; Ecelbarger, C.A. Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J. Am. Soc. Nephrol. 2007, 18, 2661–2671. [Google Scholar] [CrossRef]
- Csibi, A.; Communi, D.; Muller, N.; Bottari, S.P. Angiotensin II inhibits insulin-stimulated GLUT4 translocation and Akt activation through tyrosine nitration-dependent mechanism. PLoS ONE 2010, 5, e10070. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, B.O.; Erlandsson, M.C.; Tengvall, P.; Palmquist, A.; Ransjo, M.; Bokarewa, M.I.; Westerlund, A. Effects of implant-delivered insulin on bone formation in osteoporotic rats. J. Biomed. Mater. Res. A 2018, 106, 2472–2480. [Google Scholar] [CrossRef] [PubMed]
- Fulzele, K.; Riddle, R.C.; DiGirolamo, D.J.; Cao, X.; Wan, C.; Chen, D.; Faugere, M.C.; Aja, S.; Hussain, M.A.; Bruning, J.C.; et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 2010, 142, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Thracilkill, K.M.; Lumpkin, C.K., Jr.; Bunn, R.C.; Kemp, S.F.; Fowlkes, J.L. Is insulin an anabolic agent in bone? Dissecting the diabetic bone for clues. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E735–E745. [Google Scholar] [CrossRef]
- Xiao, Y.; Woo, W.M.; Nagao, K.; Li, W.; Terunuma, A.; Mukouyama, Y.S.; Oro, A.E.; Vogel, J.C.; Brownell, I. Perivascular hair follicle stem cells associated with a venule annulus. J. Investig. Dermatol. 2013, 133, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Van Spyk, E.N.; Pham, K.; Geyfman, M.; Kumar, V.; Takahashi, J.S.; Andersen, B. The circadian clock in skin: Implications for adult stem cells, tissue regeneration, cancer, aging and immunity. J. Biol. Rhytm. 2015, 30, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Baselga Torres, E.; Torres-Pradilla, M. Cutaneous manifestations in children with diabetes mellitus and obesity. Actas Dermosifiliogr. 2014, 105, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Megna, M.; Monfrecola, G. Insulin resistance and skin diseases. Sci. World J. 2015, 2015, 479354. [Google Scholar] [CrossRef] [PubMed]
- Kahana, M.; Grossman, E.; Feinstein, A.; Ronnen, M.; Cohen, M.; Millet, M.S. Skin tags: A cutaneous marker for diabetes mellitus. Acta Derm. Venerol. 1987, 67, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kataria, M.A.; Saini, V.; Yadav, A. Role of leptin and adiponectin in insulin resistance. Clin. Chim. Acta 2013, 417, 80–84. [Google Scholar] [CrossRef]
- Abdel Hay, R.M.; Rashed, L.A. Association between the leptin gene 2548G/A polymorphism, the plasma leptin and the metabolic syndrome with psoriasis. Exp. Dermatol. 2011, 20, 715–719. [Google Scholar] [CrossRef]
- Kitabachi, A.E.; Umpierrez, G.E.; Miles, J.M.; Fisher, J.N. Hyperglycemic crisis in adult patients with diabetes. Diabetes Care 2009, 32, 1335–1343. [Google Scholar] [CrossRef]
- Keller, U.; Gerber, P.P.; Stauffacher, W. Fatty acid-independent inhibition of hepatic ketone body production by insulin in humans. Am. J. Physiol. 1988, 254, E694–E699. [Google Scholar] [CrossRef]
- Keller, U.; Lustenberger, M.; Stauffacher, W. Effect of insulin on ketone body clearance studied by a ketone body “clamp” technique in normal men. Diabetologia 1988, 31, 24–29. [Google Scholar] [CrossRef]
- Jefferson, L.S. Lilly Lecture 1979: Role of insulin in the regulation of protein synthesis. Diabetes 1980, 29, 487–496. [Google Scholar] [CrossRef]
- Flakoll, P.J.; Kulaylat, M.; Frexes-Steed, M.; Hourani, H.; Brown, L.L.; Hill, J.O.; Abumrad, N.N. Amino acid augment insulin’s suppression of whole body proteolysis. Am. J. Physiol. 1989, 257, E839–E847. [Google Scholar] [CrossRef]
- Barrett, E.J.; Eggleston, E.M.; Inyard, A.C.; Wang, H.; Li, G.; Chai, W.; Liu, O.Z. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia 2009, 52, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Nystrom, F.H.; Ravichandran, L.V.; Cong, L.N.; Kirby, M.; Mostowski, H.; Quon, M.J. Roles of insulin receptor, PI3-kinase, and Akt in insulin signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation 2000, 101, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.A.; Dawson, D.; Clark, A.D.H.; Lindner, J.R.; Rattigan, S.; Clark, M.G.; Barrett, E.J. Skeletal muscle microvascular recruitment by physiological hyperinsulinemia precedes increases in total blood flow. Diabetes 2002, 51, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.A.; Richards, S.M.; Keske, M.A.; Rattigan, S. Local NOS inhibition impairs vascular and metabolic actions of insulin in rat hindleg muscle in vivo. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E745–E750. [Google Scholar] [CrossRef] [PubMed]
- Mantzoros, C.S.; Flier, J.S. Insulin resistance: The clinical spectrum. In Advances in Endocrinology and Metabolism; Mazzaferi, E., Ed.; St. Louis Mosby-Year Book; Springer: Berlin/Heidelberg, Germany, 1995; Volume 6, pp. 193–232. [Google Scholar]
- Semple, R.K.; Savage, D.B.; Cohran, E.K.; Gorden, P.; O’Rahilly, S. Genetic syndromes of severe insulin resistance. Endocr. Rev. 2011, 32, 498–514. [Google Scholar] [CrossRef] [PubMed]
- Parker, V.E.; Semple, R.K. Genetics in endocrinology: Genetic forms of severe insulin resistance: What endocrinologists should know. Eur. J. Endocrinol. 2013, 169, R71–R80. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.J.; Huang, K.; Xu, B.; Hu, S.-Q.; Wang, S.; Chu, Y.-C.; Katsoyannis, P.G.; Weiss, M.A. Diabetes-associated mutation in human insulin: Crystal structure and photo-cross-linking studies of A-chain variant insulin Wakayama. Biochemistry 2005, 44, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Ataul Islam, M.; Bhayye, S.; Adeniyi, A.A.; Soliman, M.E.S.; Pillay, T.S. Diabetes mellitus caused by mutations in human insulin: Analysis of impaired receptor binding of insulins Wakayama, Los Angeles, and Chicago using pharmacoinformatics. J. Biomol. Struct. Dyn. 2017, 35, 724–737. [Google Scholar] [CrossRef]
- Carroll, R.J.; Hammer, R.E.; Chan, S.J.; Swift, H.H.; Rubenstein, A.H.; Steiner, D.F. A mutant human proinsulin is secreted from islets of Langerhans in increased amounts via an upregulated pathway. Proc. Natl. Acad. Sci. USA 1988, 85, 8943–8947. [Google Scholar] [CrossRef]
- Hovnik, T.; Bratnič, N.; Trebusăk-Podkrajsĕk, K.; Kovač, J.; Paro, D.; Podnar, T.; Bratina, N.; Battelino, T. Severe progressive obstructive cardiomyopathy and renal tubular dysfunction in Donohue syndrome with decreased insulin receptor autophosphorylation due to a novel INSR mutation. Eur. J. Pediatr. 2013, 172, 1125–1129. [Google Scholar] [CrossRef]
- Zamanfa, D.; Mohamadi, F.; Moskapil, S.R. Rabson Mendenhall syndrome; a case report and review of literature. Int. J. Environ. Chem. 2020, 4, 13–19. [Google Scholar] [CrossRef]
- Longo, N.; Wang, Y.; Pasquali, M. Progressive decline in insulin levels in Rabson-Mendenhall syndrome. J. Clin. Endocrinol. Metab. 1999, 84, 2623–2629. [Google Scholar] [CrossRef]
- Gosavi, S.; Sangamesh, S.; Rao, A.S.; Patel, S.; Hodigere, V.C. Insulin, insulin everywhere: A rare case report of Rabson-Mendenhall syndrome. Cureus 2021, 13, e13126. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine Genetics Home Reference. Type A Insulin Resistance Syndrome. 2019. Available online: https://ghr.nlm.nih.gov//condition/type-a-insulin-resistance-syndrome#statistics (accessed on 24 June 2020).
- Iwanishi, M.; Haruta, T.; Takata, Y.; Ishibashi, O.; Sasaoka, T.; Egawa, K.; Imamura, T.; Naitou, K.; Itazu, T.; Kobayashi, M. A mutation (Trp1193→Leu1193) in the tyrosine kinase domain of the insulin receptor associated with type A syndrome of insulin resistance. Diabetologia 1993, 36, 414–422. [Google Scholar] [CrossRef]
- Takasawa, K.; Tsuji-Hosokawa, A.; Takishima, S.; Wada, Y.; Nagasaki, K.; Dateki, S.; Numakura, C.; Hijikata, A.; Shirai, T.; Kashimada, K.; et al. Clinical characteristics of adolescent cases with Type A insulin resistance syndrome caused by heterozygous mutations in the β-subunit of the insulin receptor (INSR) gene. J. Diabetes 2019, 11, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chen, C.; Fang, T.; Chen, D.; Chen, K.; Quan, H. Type A insulin resistance syndrome misdiagnosed as polycystic ovary syndrome: A case report. J. Med. Case Rep. 2019, 13, 347. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Yang, J.; Wang, L.; Liu, Y.; Wang, W.; Zhu, L.; Wang, W.; Yang, J.; Chen, F. Case Report: A Chinese family of type A insulin resistance syndrome with diabetes mellitus, with a novel heterozygous missense mutation of the insulin receptor gene. Front. Endocrinol. 2022, 13, 895424. [Google Scholar] [CrossRef] [PubMed]
- Hosoe, J.; Kadowaki, H.; Miya, F.; Aizu, K.; Kawamura, T.; Miyata, I.; Satomura, K.; Ito, T.; Hara, K.; Tanaka, M.; et al. Structural basis and genotype-phenotype correlations of INSR mutations causing severe insulin resistance. Diabetes 2017, 66, 2713–2723. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Taira, M.; Hashimoto, N.; Makino, H.; Sasaki, N. Familial type C syndrome of insulin resistance and short stature with possible autosomal dominant transmission. Endocrinology 1989, 36, 349–358. [Google Scholar] [CrossRef]
- Globerman, H.; Karnieli, E. Analysis of the insulin receptor gene tyrosine kinase domain in obese patients with hyperandrogenism, insulin resistance and acanthosis nigricans (type C insulin resistance). Int. J. Obes. 1998, 22, 349–353. [Google Scholar] [CrossRef]
- Kumakura, S.; Sakamoto, Y.; Iwamoto, Y.; Matsuda, A.; Kuzuya, T. Hyperinsulinemia, acanthosis nigricans and normal insulin binding in young women—Evidence for familial occurrence of post-binding defect in insulin action. J. Japan Diab. Soc. 1988, 31, 499–504. [Google Scholar]
- Moller, D.E.; Vidal-Puig, A.; Azziz, R. Severe insulin-resistance hyperandrogenic syndromes. In Contemporary Endocrinology; Azziz, R., Nestler, D., Eds.; Androgen Excess Disorders in Women; Human Press Inc.: Totova, NJ, USA, 2007; pp. 129–138. [Google Scholar]
- Hong, J.H.; Kim, H.J.; Park, K.S.; Ku, B.J. Paradigm shift in the management of type A insulin resistance. Ann. Transl. Med. 2018, 6 (Suppl. 2), S98. [Google Scholar] [CrossRef]
- Sjöholm, A.; Pereira, M.J.; Nilsson, T.; Linde, T.; Katsogiannos, P.; Saaf, J.W.; Eriksson, J.W. Type B insulin resistance syndrome in a patient with type 1 diabetes. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020, 32478674. [Google Scholar] [CrossRef] [PubMed]
- Łebkowska, A.; Krentowska, A.; Adamska, A.; Lipińska, D.; Piasecka, B.; Kowal-Bielecka, O.; Górska, M.; Semple, R.K.; Kowalska, I. Type B insulin resistance syndrome associated with connective tissue disease and psoriasis. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020, 32755965. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.; Fernandes, V.O.; Dias de Carvalho, M.M.; Gadalha, D.D.; de Queiroz, P.C.; Montenegro, R.M., Jr. Type B insulin resistance syndrome: A systematic review. Arch. Endocrinol. Metab. 2020, 64, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Caga-anan, G.; DeCastro, I.; Bates, J.T.; Sharma, M.D. Type B insulin resistance: A rare type of diabetes mellitus. AACC Clin. Case Rep. 2016, 2, e256. [Google Scholar] [CrossRef]
- Malek, R.; Chong, A.; Lupsa, B.C.; Lungu, A.O.; Cochran, E.K.; Soos, M.A.; Semple, R.K.; Balow, J.E.; Gorden, P. Treatment of type B insulin resistance: A novel approach to reduce insulin receptor autoantibodies. J. Clin. Endocrinol. Metab. 2010, 95, 3641–3647. [Google Scholar] [CrossRef] [PubMed]
- Censi, S.; Mian, C.; Betterle, C. Insulin autoimmune syndrome: From diagnosis to clinical management. Ann. Transl. Med. 2018, 6, 335. [Google Scholar] [CrossRef] [PubMed]
- Willard, D.L.; Stevenson, M.; Steenkamp, D. Type B insulin resistance syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 4, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Puig, A.; Moller, D.E. Insulin resistance: Classification, prevalence, clinical manifestations, and diagnosis. In Androgen Excess Disorders in Women; Azziz, R., Nestler, J.E., Dewailly, D., Eds.; Lippincott Raven: Philadelphia, PA, USA, 1997; pp. 227–236. [Google Scholar]
- Brown, R.J.; Araujo-Vilar, D.; To Cheung, P.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.A.; Patni, N.; Rother, K.I.; et al. The diagnosis and management of lipodystrophy syndromes: Multi-society practice guidelines. J. Clin. Endocrinol. Metab. 2016, 12, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Robbins, A.L.; Savage, D.B. The genetics of lipid storage and human lipodystrophies. Trends. Mol. Med. 2015, 7, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Chiquette, E.; Oral, E.A.; Garg, A.; Vilar, D.-A.; Dhankhar, P. Estimating the prevalence of generalized and partial lipodystrophy: Findings and challenges. Diabetes Metab. Syndr. Obes. 2017, 10, 375–383. [Google Scholar] [CrossRef]
- Tritos, N.A.; Mantzoros, C.S. Syndromes of severe insulin resistance. J. Clin. Endocrinol. Metab. 1998, 83, 3025–3030. [Google Scholar] [CrossRef]
- Uotani, S.; Yamaguchi, Y.; Yokata, A.; Yamasaki, H.; Takino, H.; Chikuba, N.; Goto, Y.; Fujishima, N.; Yano, M.; Matsumoto, K.; et al. Molecular analysis of insulin receptor gene in Werner’s syndrome. Diabetes Res. Clin. Pract. 1994, 26, 175–176. [Google Scholar] [CrossRef]
- Nolan, C.J.; Ruderman, N.B.; Kahn, S.E.; Pedersen, O.; Prentki, M. Insulin resistance as a physiological defense against metabolic stress: Implications for the management of subset of type 2 diabetes. Diabetes 2015, 64, 673–686. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Beauloye, C.; Harmancey, R.; Hue, L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am. J. Physiol. Heart C 2013, 305, H1693–H1697. [Google Scholar] [CrossRef]
- Nolan, C.J.; Ruderman, N.B.; Prentki, M. Intensive insulin for type 2 diabetes: The risk of causing harm. Lancet Diabetes Endocrinol. 2013, 1, 9–10. [Google Scholar] [CrossRef]
- Nolan, C.J.; Prentki, M. Insulin resistance and insulin hypersecretion in the metabolic syndrome and type 2 diabetes: Time for a conceptual framework shift. Diabetes Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Goodpaster, B.H.; Lee, J.S.; Kuller, L.H.; Boudreau, R.; de Rekeneire, N.; Harris, T.B.; Kritchevsky, S.; Tylavsky, F.A.; Nevit, M.; et al. Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 2009, 32, 1993–1997. [Google Scholar] [CrossRef]
- Baron, A.D.; Brechtel, G.; Wallace, P.; Edelman, S.V. Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am. J. Physiol. 1988, 255 Pt 1, E769–E774. [Google Scholar] [CrossRef]
- Kim, J.K.; Michael, M.D.; Previs, S.F.; Peroni, O.D.; Mauvais-Jarvis, F.; Neschen, S.; Kahn, B.B.; Kahn, C.R.; Shulman, G.I. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J. Clin. Investig. 2000, 105, 1791–1797. [Google Scholar] [CrossRef]
- Kim, J.K.; Zisman, A.; Fillmore, J.J.; Peroni, O.D.; Kotani, K.; Perret, P.; Zong, H.; Dong, J.; Kahn, C.R.; Kahn, B.B. Glucose toxicity and the development of diabetes in mice with muscle-specific inactivation of GLUT4. J. Clin. Investig. 2001, 108, 153–160. [Google Scholar] [CrossRef]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jørgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice revealed as essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef]
- Le Marchand-Brustel, Y.; Gremeaux, T.; Ballotti, R.; Van Obberghen, E. Insulin receptor tyrosine kinase is defective in skeletal muscle of insulin-resistant obese mice. Nature 1985, 315, 676–679. [Google Scholar] [CrossRef]
- Frojdo, S.; Vidal, H.; Pirola, L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochim. Biophys. Acta 2009, 1792, 83–92. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Z.; Hu, J.; Du, J.; Mitch, W.E. Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 2006, 147, 4160–4168. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Chen, P.-J.; Xiao, W.-H. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol. Metabol. Syndr. 2020, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.M.; Barzilai, N. Role of visceral adipose tissue in aging. BBA Gen. Subj. 2009, 1790, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Meneilly, G.S.; Elliott, T.; Tessier, D.; Hards, L.; Tildesley, H. NIDDM in the elderly. Diabetes Care 1996, 19, 1320–1325. [Google Scholar] [CrossRef] [PubMed]
- da Costa, J.P.; Vitorino, R.; Silva, G.M.; Vogel, C.; Duarte, A.C.; Rocha-Santos, T.A. A synopsis, on aging theories, mechanisms and future prospects. Ageing Res. Rev. 2016, 29, 90–112. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Morino, K.; Alves, T.C.; Kibbey, R.G.; Dufour, S.; Sano, S.; Yoo, P.S.; Cline, G.E.; Shulman, G.I. Effect of aging on muscle mitochondrial substrate utilization in humans. Proc. Natl. Acad. Sci. USA 2017, 112, 11300–11334. [Google Scholar] [CrossRef] [PubMed]
- Dagdeviren, S.; Jung, D.Y.; Friedline, R.H.; Noh, H.L.; Kim, J.H.; Patel, P.R.; Tsitsilianos, N.; Inashima, K.; Tran, D.A.; Hu, X.; et al. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 2017, 31, 701–710. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Lewis, G.F.; Carpentier, A.C.; Pereira, S.; Hahn, M.; Giacca, A. Direct and indirect control of hepatic glucose production by insulin. Cell Metab. 2021, 33, 709–720. [Google Scholar] [CrossRef]
- Magnusson, I.; Rothman, D.L.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J. Clin. Investig. 1992, 90, 1323–1327. [Google Scholar] [CrossRef]
- Krssak, M.; Brehm, A.; Bernroider, E.; Anderwald, C.; Nowotny, P.; Man, C.D.; Cobelli, C.; Cline, G.W.; Shulman, G.I.; Waldhäusl, W.; et al. Alterations in postprandial hepatic glycogen metabolism in Iype 2 diabetes. Diabetes 2004, 53, 3048–3056. [Google Scholar] [CrossRef]
- Basu, R.; Chandramouli, V.; Dicke, B.; Landau, B.; Rizza, R. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes 2005, 54, 1942–1948. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.D.; Clark, M.G. Role of blood flow in the regulation of muscle glucose uptake. Annu. Rev. Nutr. 1997, 17, 487–499. [Google Scholar] [CrossRef]
- Potenza, M.A.; Marasciulo, F.L.; Chieppa, D.M.; Brigiani, G.S.; Formoso, G.; Quon, M.J.; Montagnani, M. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am. J. Physiol. 2005, 289, H813–H822. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Quon, M.J. Insulin-stimulated production of nitric oxide is inhibited by wortmannin: Direct measurement in vascular endothelial cells. J. Clin. Investig. 1996, 98, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, M.; Ravichandran, I.V.; Chen, H.; Esposito, D.L.; Quon, M.J. Insulin receptor substrat-1 and phosphoinositide-kinase-1 are required for insulin-stimulating productoion of nitric oxide in endothelial cells. Mol. Endocrinol. 2002, 16, 1931–1942. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-A.; Montagnani, M.; Koh, K.K.; Quon, M.J. Reciprocal relationship between insulin resistance and endothelial dysfunction. Molecular and pathophysiological mechanisms. Circulation 2006, 113, 1888–1904. [Google Scholar] [CrossRef] [PubMed]
- Mărmol, J.M.; Carlsson, M.; Raun, S.H.; Grand, M.K.; Sørensen, J.; Lang Lehrskov, L.; Richter, E.A.; Norgaard, O.; Sylow, L. Insulin resistance in patients with cancer: A systematic review and meta-analysis. Acta Oncol. 2023, 62, 364–371. [Google Scholar] [CrossRef]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic syndrome and risk of cancer: A systematic review and meta-analysis. Diabetes Care 2012, 35, 2402–2411. [Google Scholar] [CrossRef]
- Cowey, S.; Hardy, R.W. The metabolic syndrome: A high risk state of cancer? Am. J. Pathol. 2006, 169, 1505–1522. [Google Scholar] [CrossRef]
- Kundaktepe, B.P.; Durmus, S.; Cengiz, M.; Kundaktepe, F.O.; Sozer, V.; Papila, C.; Gelisgen, R.; Uzun, H. The significance of insulin resistance in nondiabetic breast cancer patients. J. Endocrinol. Metab. 2021, 11, 42–48. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Song, Q.; Xu, X.; Cao, D. Insulin promotes progression of colon cancer by upregulation of ACAT1. Lipids Health Dis. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Arcidiakono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef] [PubMed]
- Webster, N.J.; Resnik, J.L.; Reichart, D.B.; Struss, B.; Haas, M.; Seely, B.L. Repression of the insulin receptor promoter by the tumor suppressor gene product p53: A possible mechanism for receptor overexpression in breast cancer. Cancer Res. 1996, 56, 2781–2788. [Google Scholar]
- Werner, H.; Maor, S. The insulin-like growth factor-1 receptor gene: A downstream target for oncogene and tumor suppressor action. Trends Endocrinol. Metab. 2006, 17, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Gallhager, E.J.; LeRoith, D. Hyperinsulinemia in cancer. Nat. Rev. Cancer 2020, 20, 629–644. [Google Scholar] [CrossRef]
- Madsen, R.R.; Vanhaese, B.; Semple, R.K. Cancer-associated PBK mutations in overgrowth disorder. Trends Mol. Med. 2018, 24, 856–870. [Google Scholar] [CrossRef]
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef]
- Vatseba, T. Study of insulin resistance in patients with cancer. Arch. Clin. Med. 2020, 26, 15–19. [Google Scholar] [CrossRef]
- Sędzikowska, A.; Szablewski, L. Insulin and insulin resistance in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 9987. [Google Scholar] [CrossRef]
- Griffith, C.M.; Eid, T.; Rose, G.M.; Patrylo, P.R. Evidence for altered insulin receptor signaling in Alzheimer’s disease. Neuropharmacology 2018, 136, 202–215. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, L.I.; Brown, J. Glucose inhibition of fatty acid oxidation by rat diaphragm. Am. J. Physiol. 1960, 199, 235–237. [Google Scholar] [CrossRef]
- Christe, M.; Rodgers, R.L. Cardiac glucose and fatty acid oxidation in the streptozotocin-induced diabetic spontaneously hypersensitive rat. Hypertension 1995, 25, 235–241. [Google Scholar] [CrossRef]
- Cline, G.W.; Petersen, K.F.; Krssak, M.; Shen, J.; Hundal, R.S.; Trajanoski, Z.; Inzucchi, S.; Dresner, A.; Rothman, D.L.; Shulman, G.I. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N. Engl. J. Med. 1999, 341, 240–246. [Google Scholar] [CrossRef]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, M.; Barzilai, N.; Liu, R.; Hu, M.; Chen, W.; Rossetti, L. Role of the glucosamine pathway in fat-induced insulin resistance. J. Clin. Investig. 1997, 99, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, Y.; Hart, G.W.; Wells, L.; Vosseller, K.; Yamamoto, K.; Munetomo, E.; Ohara-Imaizumi, M.; Nishiwaki, C.; Nagamatsu, S.; Hirano, H.; et al. Elevation of the post-translational modification of the glucose-stimulated insulin secretion in the pancreas of diabetic Goto-Kakizaki rats. Glycobiology 2007, 17, 127–140. [Google Scholar] [CrossRef]
- Akimoto, Y.; Kawakami, H.; Yamamoto, K.; Munetomo, E.; Hida, T.; Hirano, H. Elevated expression of O-GlcNAc-modified proteins and O-GlcNAc transferase in corneas of diabetic Goto-Kakizaki rats. Investig. Ophtalmol. Vis. Sci. 2003, 44, 3802–3809. [Google Scholar] [CrossRef]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Complete inhibition of glucose-induced desensitization of the glucose transport system by inhibitors of mRNA synthesis. Evidence for rapid turnover of glutamine:fructose-6-phosphate aminotransferase. J. Biol. Chem. 1991, 266, 10155–10161. [Google Scholar] [CrossRef]
- Copeland, R.J.; Bullen, J.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Roles of insulin resistance and glucose toxicity. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E17–E28. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.E.; Berkaw, M.N.; Buse, M.G. Identification of the major site of O-linked β-N-acetylglucosamine modification in the C terminus of insulin receptor substrate-1. Mol. Cell Proteom. 2006, 5, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.S.; Han, D.; Park, J.; Kwak, T.K.; Oh, M.A.; Lee, S.A.; Choi, S.; Park, Z.Y.; Kim, Y.; Lee, J. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic beta cells. Exp. Cell Res. 2008, 314, 2238–2248. [Google Scholar] [CrossRef] [PubMed]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 2008, 283, 16283–16292. [Google Scholar] [CrossRef]
- Chen, G.; Liu, P.; Thurmond, D.C.; Elmendorf, J.S. Glucosamine-induced insulin resistance is coupled to O-linked glycosylation of Munc 18c. FEBS Lett. 2003, 534, 54–60. [Google Scholar] [CrossRef]
- Wondmkun, Y.T. Obesity, insulin resistance, and type 2 diabetes: Associations and therapeutic implications. Diabet. Metab. Syndr. Obes. Targets Ther. 2020, 13, 3611–3616. [Google Scholar] [CrossRef]
- Eshima, H.; Tamura, Y.; Kakehi, S.; Kurebayashi, N.; Murayama, T.; Nakamura, K.; Kakigi, R.; Okada, T.; Sakurai, T.; Kawamori, R.; et al. Long-term, but not short-term high-fat diet induces fiber composition changes and impaired contractile force in mouse fast-twitch skeletal muscle. Physiol. Rep. 2017, 5, e13250. [Google Scholar] [CrossRef]
- Badin, M.; Louche, K.; Mairal, A.; Liebisch, G.; Schmitz, G.; Rustan, A.C.; Smith, S.R.; Langin, D.; Moro, C. Altered skeletal muscle lipase expression and activity contribute to insulin resistance in humans. Diabetes 2011, 60, 1734–1742. [Google Scholar] [CrossRef]
- Mann, J.P.; Savage, D.B. What lipodystrophies teach us about the metabolic syndrome. J. Clin. Investig. 2019, 129, 4009–4021. [Google Scholar] [CrossRef]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef]
- Krssak, M.; Falk Petersen, K.; Dresner, A.; DiPietro, L.; Vogel, S.M.; Rothman, D.L.; Roden, M.; Shulman, G.I. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1H NMR spectroscopy study. Diabetologia 1999, 42, 113–116. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef]
- Goudriaan, J.R.; Dahlmans, C.E.H.; Teusink, B.; Ouwers, M.; Fabbriao, M.; Maassen, J.A.; Romijn, J.A.; Havekes, L.M.; Voshol, P.J. CD36 deficiency increases insulin sensitivity in muscle, but induces insulin resistance in the liver in mice. J. Lipid Res. 2003, 44, P2270–P2277. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Gimeno, R.E.; Higoshimori, T.; Kim, H.-J.; Choi, H.; Punreddy, S.; Mozell, R.L.; Tan, C.; Stricker-Krongrad, A.; Hirsch, D.J.; et al. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J. Clin. Investig. 2004, 113, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Doege, H.; Grimm, D.; Falcon, A.; Tsang, B.; Storm, T.A.; Xu, H.; Ortegon, A.M.; Kazantzis, M.; Kay, M.A.; Stahl, A. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J. Biol. Chem. 2008, 283, 22186–22192. [Google Scholar] [CrossRef] [PubMed]
- Falcon, A.; Doege, H.; Fliitt, A.; Tsang, B.; Watson, N.; Kay, M.A.; Stahl, A. FATP2 is a hepatic fatty acid transporter and proximal very long-chain acyl-CoA synthase. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E384–E393. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Nyce, M.R.; Allen, L.E.; Morales, L.M.; Triester, S.; Serrano, J.; Colberg, J.; Lanza-Jacoby, S.; Caro, J.F. Protein kinase C is increased in the liver of humans and rats with non-insulin-dependent diabetes mellitus: An alteration not due to hyperglycemia. J. Clin. Investig. 1995, 95, 2938–2944. [Google Scholar] [CrossRef] [PubMed]
- Karasik, A.; Rothenberg, P.L.; Yamada, K.; White, M.F.; Kahn, C.R. Increased protein kinase C activity is linked to reduced insulin receptor autophosphorylation in liver of starved rats. J. Biol. Chem. 1990, 265, 10226–10231. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Madiraju, A.K.; Gassaway, B.M.; Marcel, M.; Nasiri, A.R.; Butrico, G.; Marcucci, M.J.; Zhang, D.; Abulizi, A.; Zhang, X.-M.; et al. Insulin receptor thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Investig. 2016, 126, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.-m.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C.; Browne, C.L.; Oakes, N.D.; Watkinson, A.; Chisholm, D.J.; Kraegen, E.W.; Biden, T.J. Alterations in the expression and cellular localization of proteins kinase C isoenzymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes 1997, 46, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Soos, T.J.; Li, X.; Wu, J.; Degennaro, M.; Sun, X.; Littman, D.R.; Birnbaum, M.J.; Polakiewicz, R.D. Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J. Biol. Chem. 2004, 279, 45304–45307. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Müller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Avignon, A.; Yamada, K.; Zhou, X.; Spencer, B.; Cardona, O.; Saba-Sidolique, S.; Galloway, M.l.; Standaert, M.L.; Farese, R.V. Chronic activation of protein kinase C in soleus muscles and other tissues of insulin-resistant type II diabetic Goto-Kakizaki (GK), obese/aged, and obese/Zucker rats: A mechanism for inhibiting glycogen synthesis. Diabetes 1996, 45, 1396–1404. [Google Scholar] [CrossRef]
- Chavez, J.A.; Summers, S.A. A ceramide—Centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef]
- Powell, D.J.; Hajduch, E.; Kular, G.; Hundal, H.S. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Aky by a PKCζ-dependent mechanism. Mol. Cell Biol. 2003, 23, 7794–7808. [Google Scholar] [CrossRef]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of insulin action by ceramide: Dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef]
- Westwick, J.K.; Bielawska, A.E.; Dbaibo, G.; Hannun, Y.A.; Brenner, D.A. Ceramide activates the stress-activated protein kinases. J. Biol. Chem. 1995, 270, 22689–22692. [Google Scholar] [CrossRef]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Yoshida, H.; Akazawa, R.; Negishi, M.; Mori, K. Distinct role of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 2002, 366 Pt 2, 585–594. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef]
- Marchetti, P.; Bugliani, M.; Lupi, R.; Marselli, L.; Masini, M.; Boggi, U.; Filipponi, F.; Weir, G.C.; Eizirik, D.L.; Cnop, M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007, 50, 2486–2494. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminsky, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2102. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Miyachi, H.; Maeda, S.; Egashira, K.; Kasuga, M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yu, Y.; Shi, Y.; Sun, W.; Xie, M.; Ge, N.; Mao, R.; Chang, A.; Chang, A.; Xu, G.; et al. Lysine 63-linked polyubiquination of TAK1 at lysine 158 is required for tumor necrosis factor-α- and interleukin-1/3-induced IKK/NF-κB and JNK/AP-1 activation. J. Biol. Chem. 2010, 285, 5347–5360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, Z.; Yin, J.; Quon, M.J.; Ye, J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382. [Google Scholar] [CrossRef]
- Jager, J.; Gremeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.E. Interleukin-1β-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef]
- Steppan, C.M.; Wang, J.; Whiteman, E.L.; Birnbaum, M.J.; Lazar, M.A. Activation of SOCS-3 by resistin. Mol. Cell Biol. 2005, 25, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Morgensen, T.H. Pathogen recognition and inflammation signaling in innate immune defects. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Mahadev, K.; Zilbering, A.; Zhu, L.; Goldstein, B.J. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphate 1b in vivo and enhances the early insulin action cascade. J. Biol. Chem. 2001, 276, 21938–21942. [Google Scholar] [CrossRef]
- Krieger-Bauer, H.I.; Kather, H. Human fat cells possess a plasma membrane-bound H2O2-generating system that is activated by insulin via a mechanism bypassing the receptor kinase. J. Clin. Investig. 1992, 89, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Chuang, L.M. The role of oxidative stress in the pathogenesis of type 2 diabetes: From molecular mechanism to clinical implication. Am. J. Transl. Res. 2010, 2, 316–331. [Google Scholar]
- Fridlyand, L.E.; Philipson, L.H. Reactive species and early manifestation of insulin resistance in type 2 diabetes. Diabetes Obes. Metab. 2006, 8, 136–145. [Google Scholar] [CrossRef]
- Evans, J.L.; Maddeux, B.A.; Goldfine, I.D. The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef]
- West, I.C. Radicals and oxidative stress in diabetes. Diabet. Med. 2000, 17, 171–180. [Google Scholar] [CrossRef]
- Rosen, P.; Nawroth, P.P.; King, G.; Moller, W.; Tritschler, H.J.; Packer, I. The role of oxidative stress in the onset and progression of diabetes and complications: A summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab. Res. Rev. 2001, 17, 189–212. [Google Scholar] [CrossRef]
- Dokhen, D.B.; Saengsirisuwan, V.; Kim, J.S.; Teachy, M.K.; Henriksen, E.J. Oxidative stress-induced insulin resistance in rat skeletal muscle: Role of glycogen synthase kinase-3. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E615–E621. [Google Scholar] [CrossRef]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Z.X.; Choi, C.S.; Tian, I.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.J. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef]
- Petersen, K.F.; Shulman, G.I. Etiology of insulin resistance. Am. J. Med. 2006, 119, S10–S16. [Google Scholar] [CrossRef]
- St-Pierre, J.; Lin, J.; Krauss, S.; Tarr, P.T.; Yang, R.; Newgard, C.B.; Spigelman, B.M. Bioenergetic analysis of peroxisome proliferator-activated receptor γ coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J. Biol. Chem. 2003, 278, 26597–26603. [Google Scholar] [CrossRef]
- Yuan, D.; Xiao, D.; Gao, Q.; Zeng, L. PGC-1α activation: A therapeutic target for type 2 diabetes. Eat. Weight Disord. 2019, 24, 385–395. [Google Scholar] [CrossRef]
- Wu, X.; Chen, K.; Williams, K.J. The role of pathway-selective insulin resistance and responsiveness, in diabetic dyslipoproteinemia. Curr. Opin. Lipidol. 2012, 23, 334–344. [Google Scholar] [CrossRef]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thorens, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTOR complement raptor, rictor, or mLST8 reveals that mTOR2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef]
- Kim, K.; Qiang, L.; Hayden, M.S.; Sparling, D.P.; Purcell, N.H.; Pajvani, U.B. mTORC1-independent raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat. Commun. 2016, 7, 10255. [Google Scholar] [CrossRef]
- Kim, K.; Ryu, D.; Dongiovanni, P.; Ozcan, L.; Nayak, S.; Ueberheide, B.; Valenti, L.; Auwerx, J.; Pajvani, U.B. Degradation of PHLPP2 by KCTD17, via a glucagon-dependent pathway promotes hepatic steatosis. Gastroenterology 2017, 153, 1568–1580. [Google Scholar] [CrossRef]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weissmann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Löffer, M.G.; et al. The role of peroxisome proliferator-activated receptor γ coactivator-1 β in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef]
- Bindesboll, C.; Fan, Q.; Norgaard, R.C.; MacPherson, L.; Ruan, H.B.; Wu, J.; Pedersen, T.; Steffensen, K.R.; Yang, X.; Mathews, J.; et al. Liver X receptor regulates hepatic nuclear O-GlcNAc signaling and carbohydrate responsive element-binding protein activity. J. Lipid Res. 2015, 56, 771–785. [Google Scholar] [CrossRef]
- Postic, C.; Dentin, R.; Denechaud, P.D.; Girard, J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu. Rev. Nutr. 2007, 27, 179–192. [Google Scholar] [CrossRef]
- Erion, D.M.; Popov, V.; Hsiao, J.J.; Vatner, D.; Mitchell, K.; Yonemitsu, S.; Nagai, Y.; Kahn, M.; Gillum, M.P.; Dong, J.; et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology 2013, 154, 36–44. [Google Scholar] [CrossRef]
- Uyeda, K.; Repa, J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef]
- Magkos, F.; Yannakoulia, M.; Chan, J.L.; Mantzoros, C.S. Management of the metabolic syndrome and type 2 diabetes through life style modification. Annu. Rev. Nutr. 2009, 29, 223–256. [Google Scholar] [CrossRef]
- Lloyd-Jons, D.M.; Liu, K.; Colangelo, L.A.; Yan, L.L.; Klein, L.; Loria, C.M.; Lewis, C.E.; Savage, P. Consistently stable or decreased body mass index in young adulhood and longitudal changes in metabolic syndrome components: The Coronary Artery Risk Development in Young Adults Study. Circulation 2007, 115, 1004–1011. [Google Scholar] [CrossRef]
- Nieman, D.C.; Wentz, L.M. The compelling link between physical activity and the body’s defense system. J. Sport Health Sci. 2019, 8, 201–217. [Google Scholar] [CrossRef]
- Wolosowicz, M.; Prokopiuk, S.; Kaminski, T.W. Recent advances in the treatment of insulin resistance targeting molecular and metabolic pathways: Fighting a losing battle? Medicina 2022, 58, 472. [Google Scholar] [CrossRef]
- Moore, M.M.; Bailey, A.M.; Flannery, A.H.; Baum, R.A. Treatment of diabetic ketoacidosis with intravenous U-500 insulin in patient with Rabson-Mendenhall syndrome: A case report. J. Pharm. Pract. 2017, 30, 468–475. [Google Scholar] [CrossRef]
- Semple, R.K.; Williams, R.M.; Dunger, D.B. What is the best management strategy for patients with severe insulin resistance? Clin. Endocrinol. 2010, 73, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Nishimura, R.; Utsunomiya, A.; Kagawa, R.; Funata, H.; Fujimoto, M.; Hanaki, K.; Kanzaki, S. Leprechaunism (Donohue syndrome): A case bearing novel compound heterozygous mutations in the insulin receptor gene. Endocr. J. 2013, 60, 107–112. [Google Scholar] [CrossRef]
- Caldwell, S.H. Efficacy and safety of troglitazone for lipodystrophy syndrome. Ann. Intern. Med. 2001, 134, 1008. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Thomas, N.J.; Lynam, A.L.; Hill, A.V.; Weedon, M.N.; Shields, B.M.; Oram, R.A.; McDonald, T.J.; Hattersley, A.T.; Jones, A.G. Type 1 diabetes defined by severe insulin deficiency occurs after 30 years of age and is commonly treated as type 2 diabetes. Diabetologia 2019, 62, 1167–1172. [Google Scholar] [CrossRef]
- Zhang, A.M.Y.; Magrill, J.; de Winter, T.J.J.; Hu, X.; Skovsø, S.; Schaeffer, D.E.; Kopp, J.L.; Johnson, J.D. Endogenous hyperinsulinemia contributes to pancreatic cancer development. Cell Metab. 2019, 30, 403–404. [Google Scholar] [CrossRef]
- Zhong, W.; Mao, Y. Daily insulin dose and cancer risk among patients with type 1 diabetes. JAMA Oncol. 2022, 8, 1356. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Siddiqui, F.A.; Gupta, K.; Chattopadhyay, S.; Gopinath, P.; Kumar, B.; Manvati, S.; Chaman, N.; Bamezai, R.N.K. Insulin enhances metabolic capacities of cancer cells by dual regulation of glycolytic enzyme pyruvate kinase M2. Mol. Cancer 2013, 12, 72. [Google Scholar] [CrossRef]
- Frasca, F.; Pandini, P.; Scalia, P.; Sciacca, L.; Mineo, R.; Constantino, A.; Goldfine, I.D.; Belfore, A.; Vigneri, R. Insulin receptor isoforms A, a newly recognized high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef]
- Vigneri, P.; Frasca, F.; Sciacca, L.; Pandini, G.; Vigneri, R. Diabetes and cancer. Endocr.-Relat. Cancer 2009, 16, 1103–1123. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Stambolic, V. Obesity and insulin resistance in breast cancer—Chemoprevention strategies with a focus on metformin. Breast 2011, 20, S31–S35. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, M.E.; Fantus, I.G.; Ezzat, S.; McKeown-Eyssen, G.; Page, D.; Goodwin, P.J. Insulin and related factors in premenopausal breast cancer risk. Breast Cancer Res. Treat. 1998, 47, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Li, J.; Ho, G.Y.F.; Xue, X.; Anderson, G.I.; et al. Insulin, insulin-like growth factor-1 and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2009, 101, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Kaaks, R.; Toniolo, P.; Akmedkhanov, A.; Lukanova, A.; Biessy, C.; Dechaud, H.; Rinaldi, S.; Zeleniuch-Jacquotte, A.; Shore, P.E.; Riboli, E. Serum C-peptide, insulin-like growth factor (IGF)-I, IGF-binding protein, and colorectal cancer risk in women. J. Natl. Cancer Inst. 2000, 92, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Schoen, R.E.; Tangen, C.M.; Kuller, L.H.; Burke, G.L.; Cushman, M.; Tracy, R.P.; Dobs, A.; Savage, P.J. Increased blood glucose and insulin, body size, and incident colorectal cancer. J. Natl. Cancer Inst. 1999, 91, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Koohestani, N.; Tran, T.T.; Lee, W.; Wolever, T.M.; Bruce, W.R. Insulin resistance and promotion of aberrant crypt foci in the colons of rats on a high-fat diet. Nutr. Cancer 1997, 29, 69–76. [Google Scholar] [CrossRef]
- Watkins, L.F.; Lewis, L.R.; Lewine, A.E. Characterization of the synergistic effect of insulin and transferin and the regulation of their receptors on a human colon carcinoma cell line. Int. J. Cancer 1990, 45, 372–375. [Google Scholar] [CrossRef]
- Moschos, S.J.; Mantzoros, C.S. The role of the IGF system in cancer: From basic to clinical studies and clinical applications. Oncology 2002, 63, 317–332. [Google Scholar] [CrossRef]
- Pisani, P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch. Physiol. Biochem. 2008, 114, 63–70. [Google Scholar] [CrossRef]
- Chan, M.T.; Lim, G.E.; Skovsø, S.; Yang, Y.H.C.; Albrecht, T.; Alejandro, E.U.; Hoesli, C.A.; Piret, J.M.; Warnock, G.L.; Johnson, J.D. Effects of insulin on human pancreatic cancer progression modeled in vitro. BMC Cancer 2014, 14, 814. [Google Scholar] [CrossRef]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular partaits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.; Johnson, A.R.; Makowski, L. Obesity, metabolism and the microenvironment links to cancer. J. Carcinog. 2013, 12, 19. [Google Scholar] [PubMed]
- Yang, Z.Y.; Di, M.Y.; Yuan, J.Q.; Shen, W.X.; Zheng, D.Y.; Chen, J.Z.; Mao, C.; Tang, J.L. The prognostic value of phosphorylated Akt in breast cancer: A systematic review. Sci. Rep. 2015, 5, 7758. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Site of Defects | Cause | Syndrome | Features |
|---|---|---|---|
| Insulin gene | Mutation ValA3→Leu | Insulin Wakayama | Decreased binding of insulin to the INSR. |
| Mutation PheB24→Ser | Insulin Los Angeles | Significantly reduced bioactivity of insulin. | |
| Mutation PheB25→Leu | Insulin Chicago | Decreased binding of insulin to the INSR. | |
| Insulin receptor | Autosomal recessive mutations | Donohue syndrome | Affected patients seldom live beyond infancy. Most affected patients survive fewer than 2 years. |
| Rabson–Mendenhall syndrome | Hyperinsulinemia, fasting hyperglycemia, failed reaction on insulin. Most affected patients survive up to 15 years. | ||
| Autosomal dominant or recessive mutations | Type A insulin resistance syndrome (TAIRS) | Syndrome has a relatively good prognosis. Affected patients can live beyond middle age. Hypoglycemia is observed. | |
| Autosomal dominant mutations | Type C insulin resistance | It is a variant of Type A insulin resistance with less severe insulin resistance. It is so-called HAIR-AN (hyperandrogenic, insulin resistance, acanthosis nigricans). | |
| An autoimmune disorder caused by polyclonal autoantibodies acting against the insulin receptor. | Type B insulin resistance (TBIRS) | Depending on levels of autoantibodies, hypoglycemia or hyperglycemia may be observed. | |
| Depending on the type of lipodystrophy, these syndromes may be due to autosomal dominant, autosomal recessive or X-linked mutations. | Lipodystrophies | The lipodystrophy syndromes are a heterogenous group of disorders. Depending on the disorder, different pathologies, such as severe insulin resistance, severe hypertriglyceridemia, pancreatitis, complete or partial loss of adipose tissue and depletion of lipid storage capacity, are observed. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szablewski, L. Changes in Cells Associated with Insulin Resistance. Int. J. Mol. Sci. 2024, 25, 2397. https://doi.org/10.3390/ijms25042397
Szablewski L. Changes in Cells Associated with Insulin Resistance. International Journal of Molecular Sciences. 2024; 25(4):2397. https://doi.org/10.3390/ijms25042397
Chicago/Turabian StyleSzablewski, Leszek. 2024. "Changes in Cells Associated with Insulin Resistance" International Journal of Molecular Sciences 25, no. 4: 2397. https://doi.org/10.3390/ijms25042397
APA StyleSzablewski, L. (2024). Changes in Cells Associated with Insulin Resistance. International Journal of Molecular Sciences, 25(4), 2397. https://doi.org/10.3390/ijms25042397