
Application of Novel Transcription Factor Machine Learning Model and Targeted Drug Combination Therapy Strategy in Triple Negative Breast Cancer
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
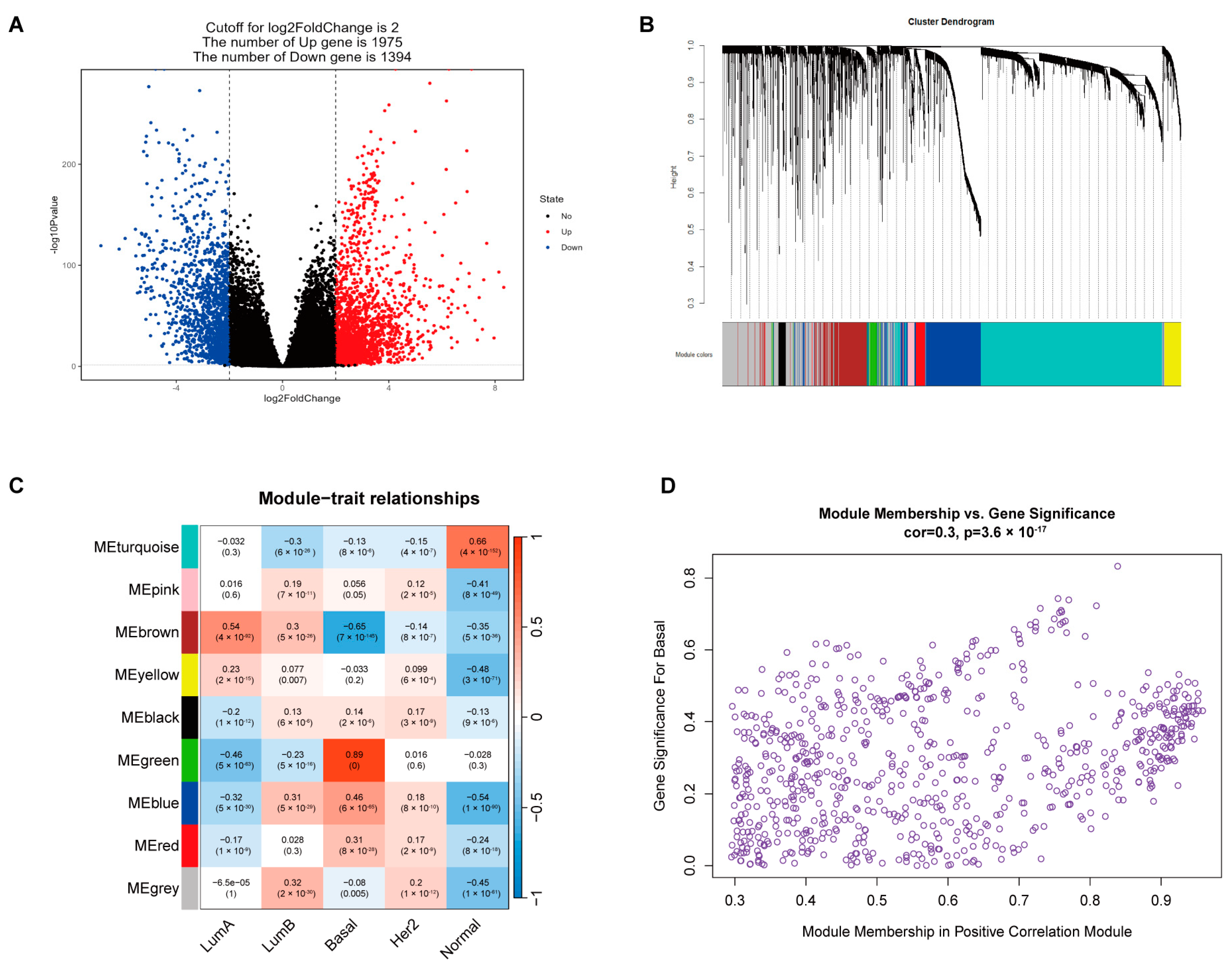
2.1. Identification of TF Gene Sets Highly Associated with TNBC
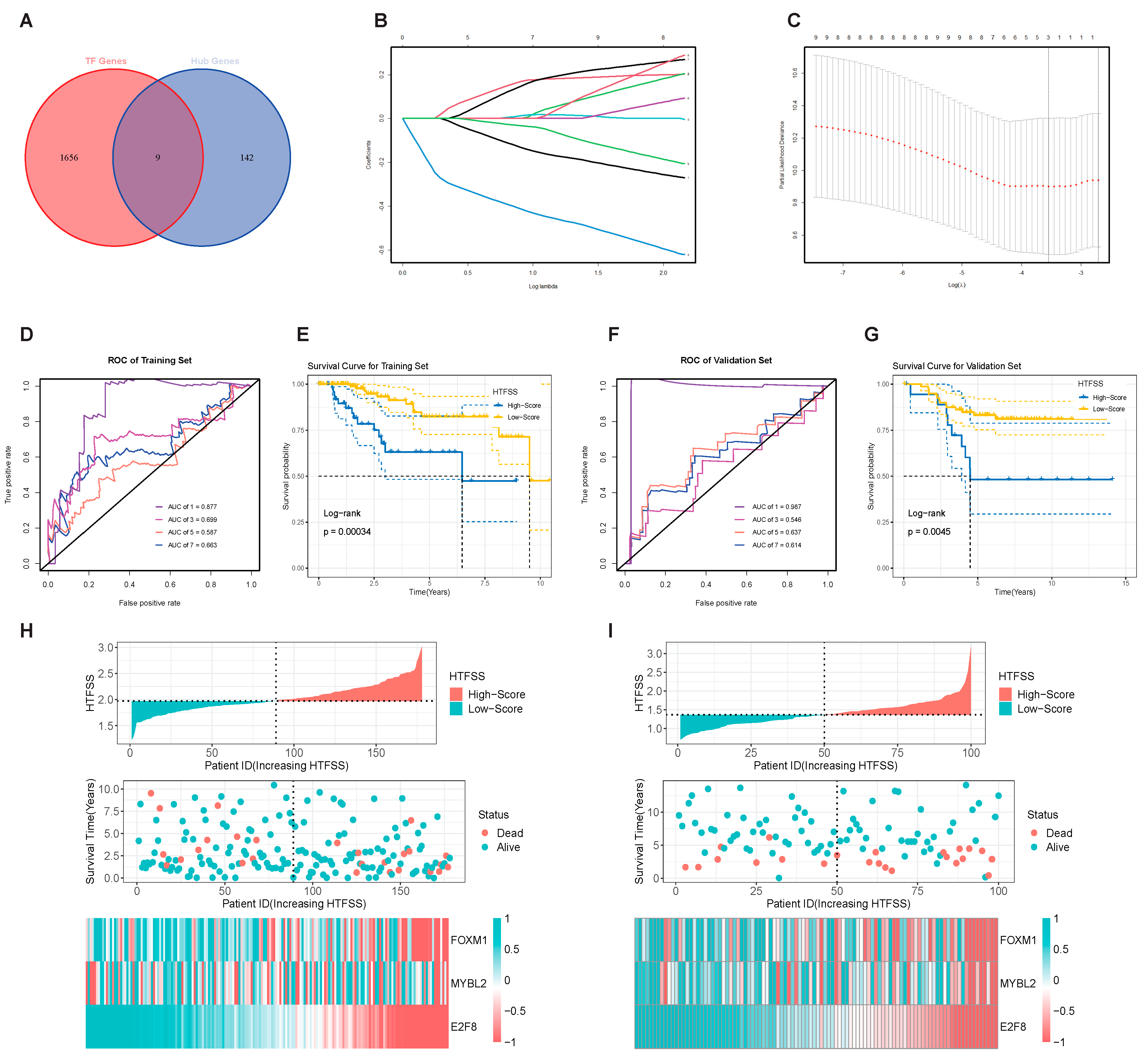
2.2. HTFSS Has Excellent Prognostic Prediction Performance
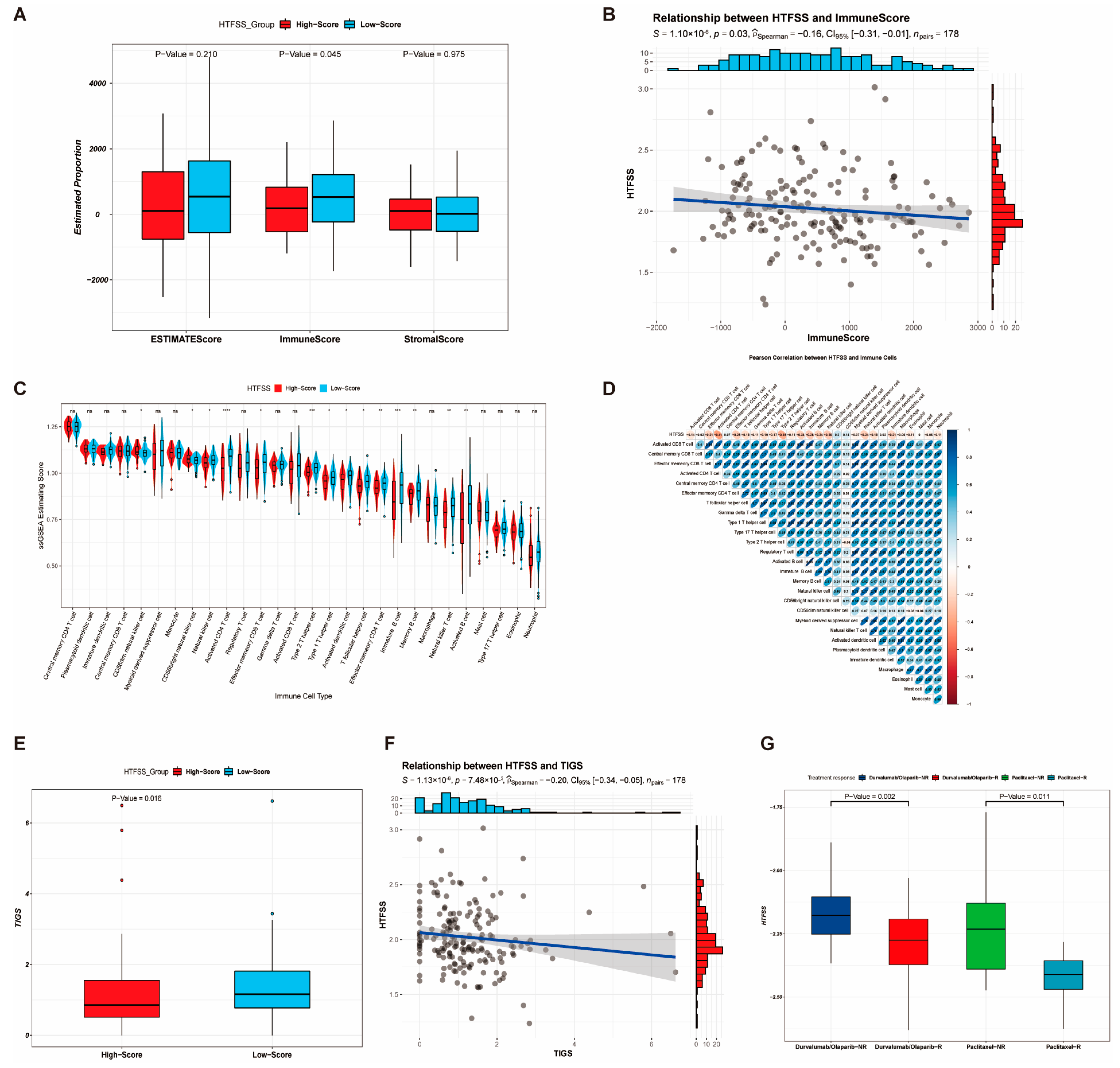
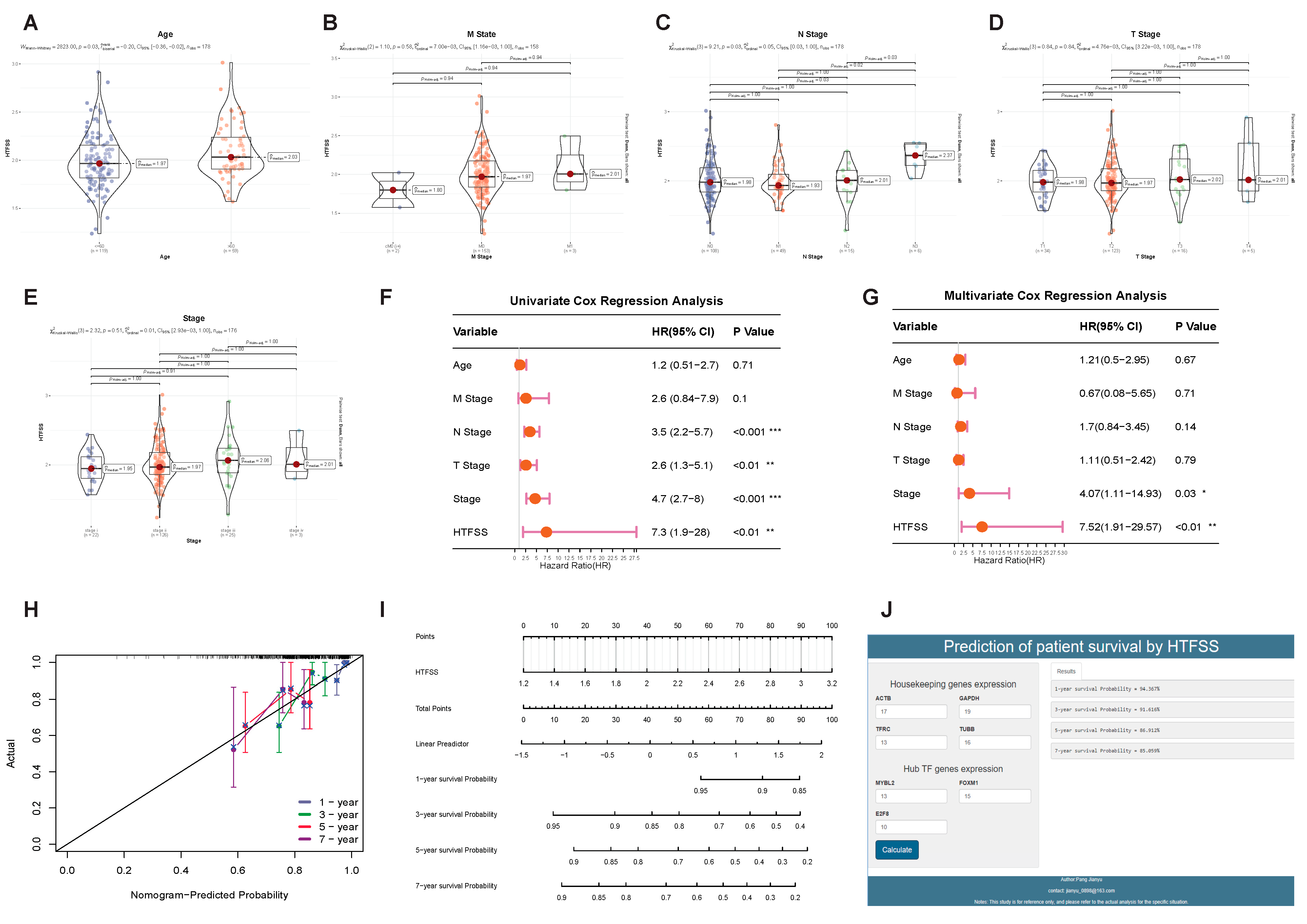
2.3. Application of HTFSS in Clinical Diagnosis and Prediction of Immunotherapy Response
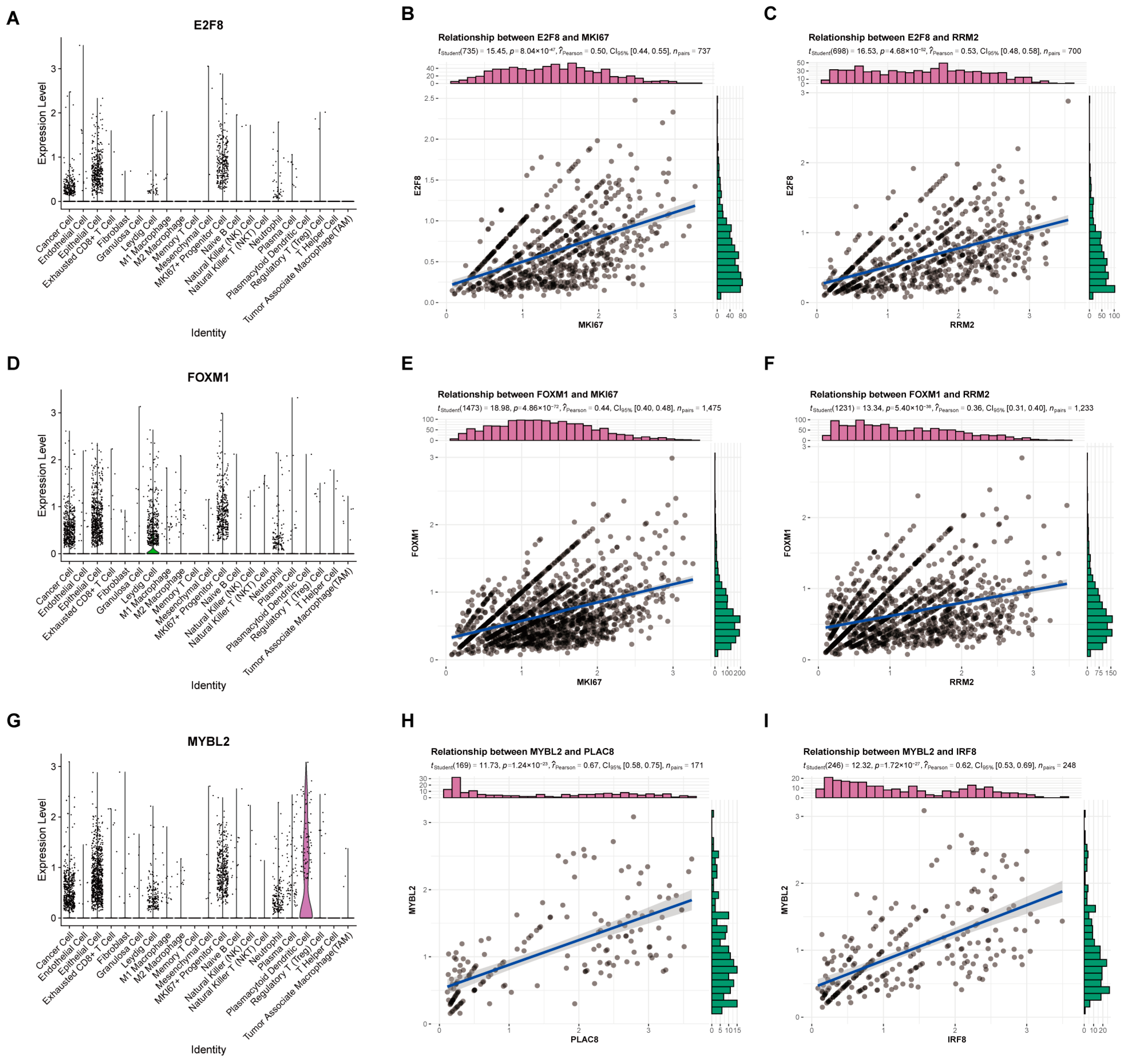
2.4. The Specific Relationship between HTFSS and TIME Was Analyzed by scRNA-Seq
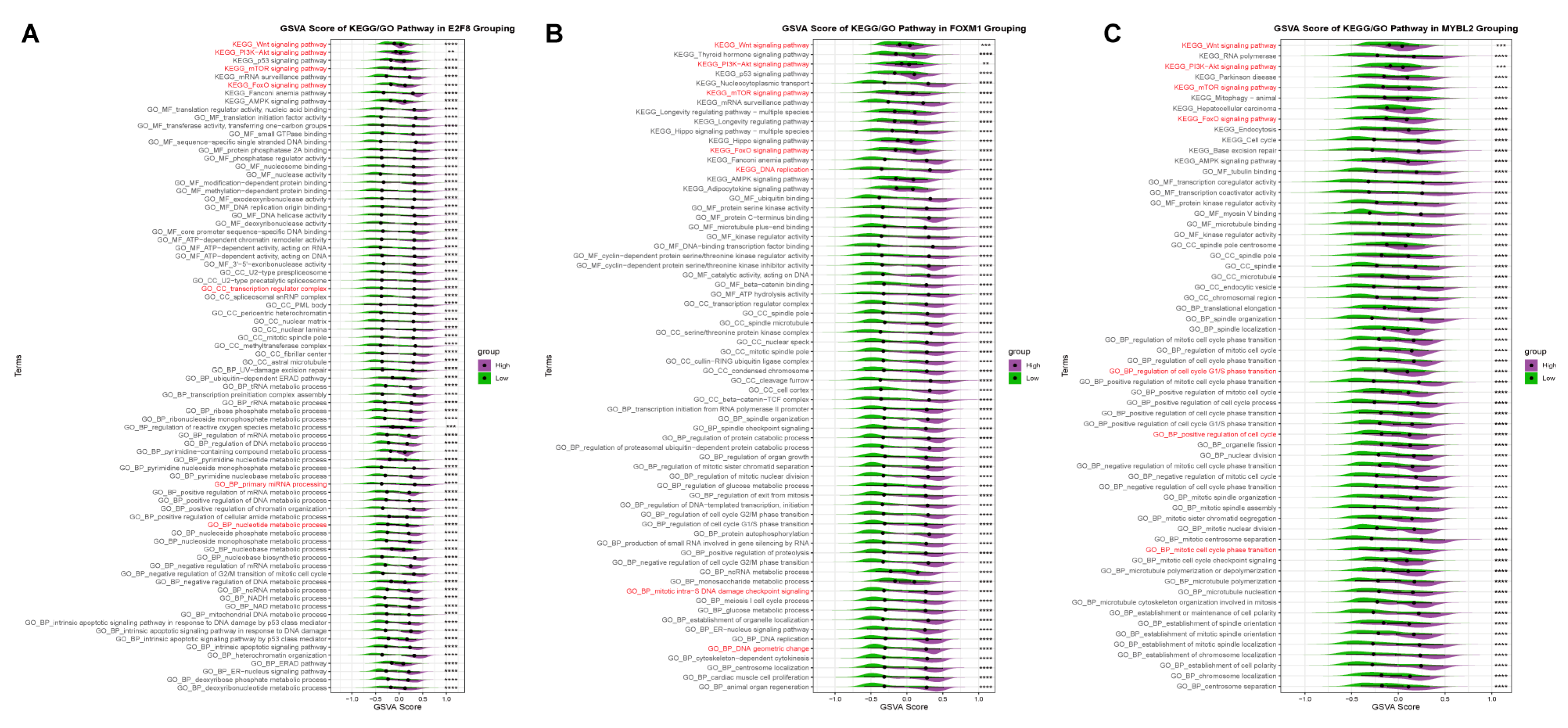
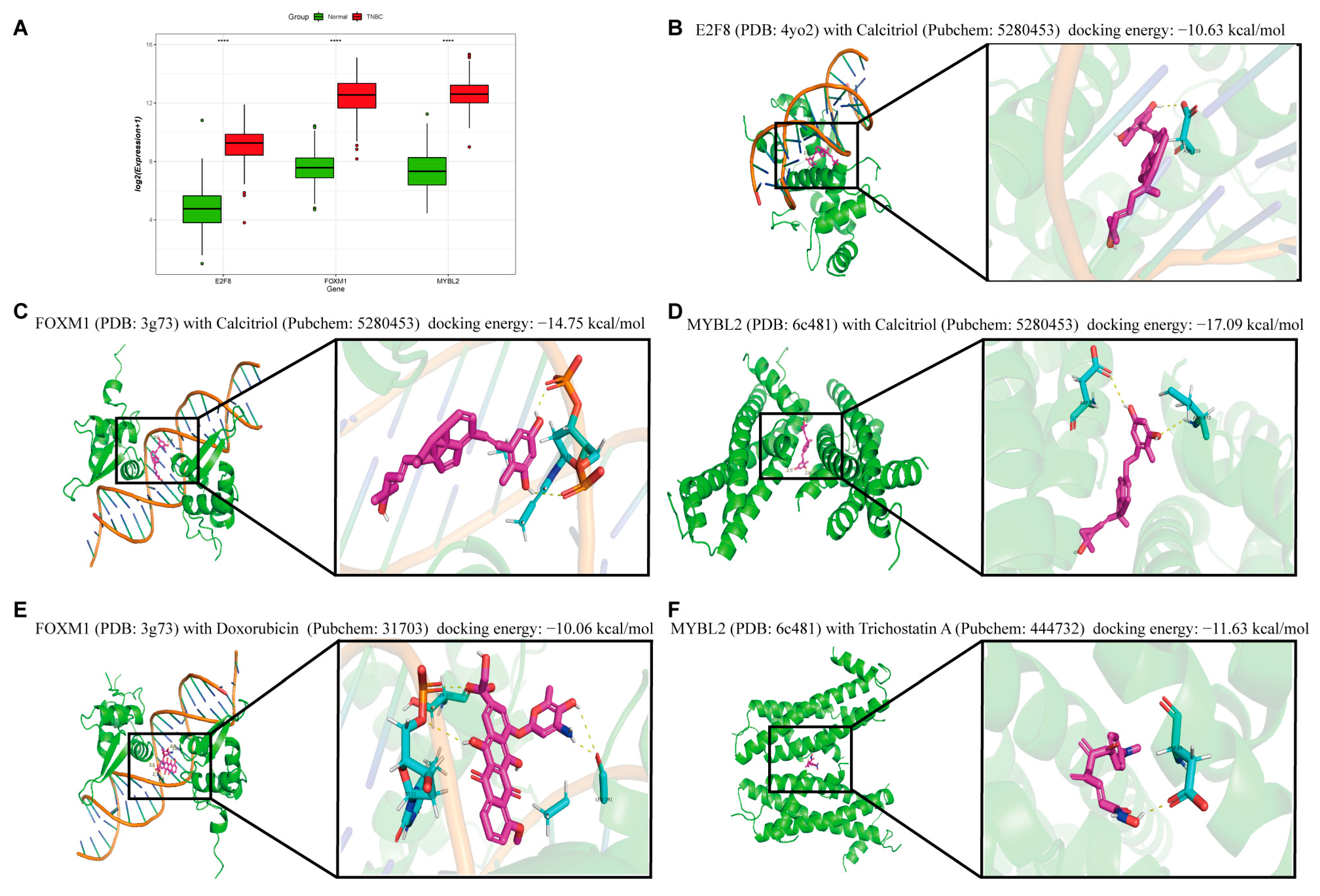
2.5. Virtual Docking of Hub TF Genes with Targeted Drugs
2.6. Research on the Prognostic Function of Hub TF Target Genes
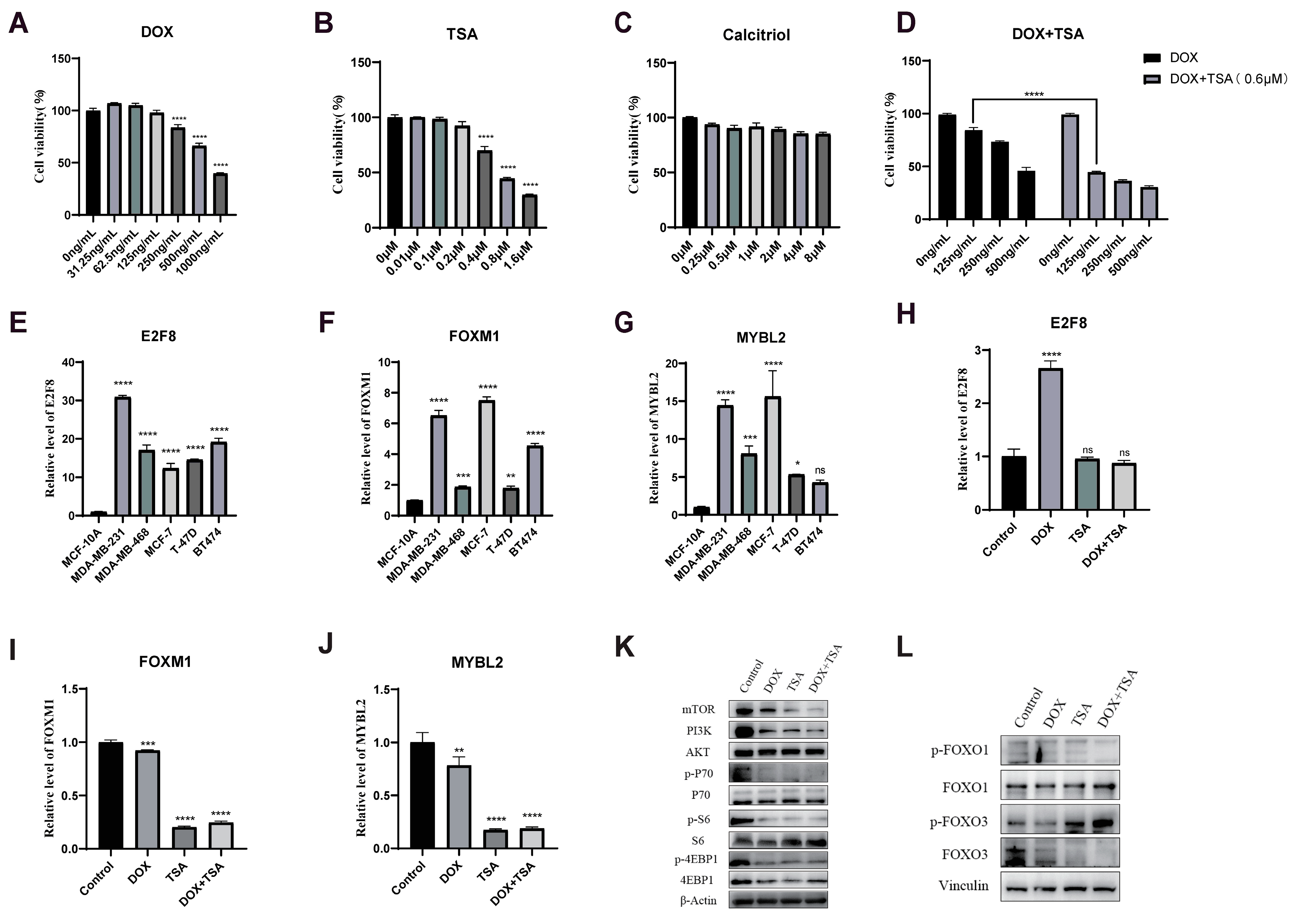
2.7. DOX Combined with TSA Significantly Inhibited the Proliferation of TNBC Cells
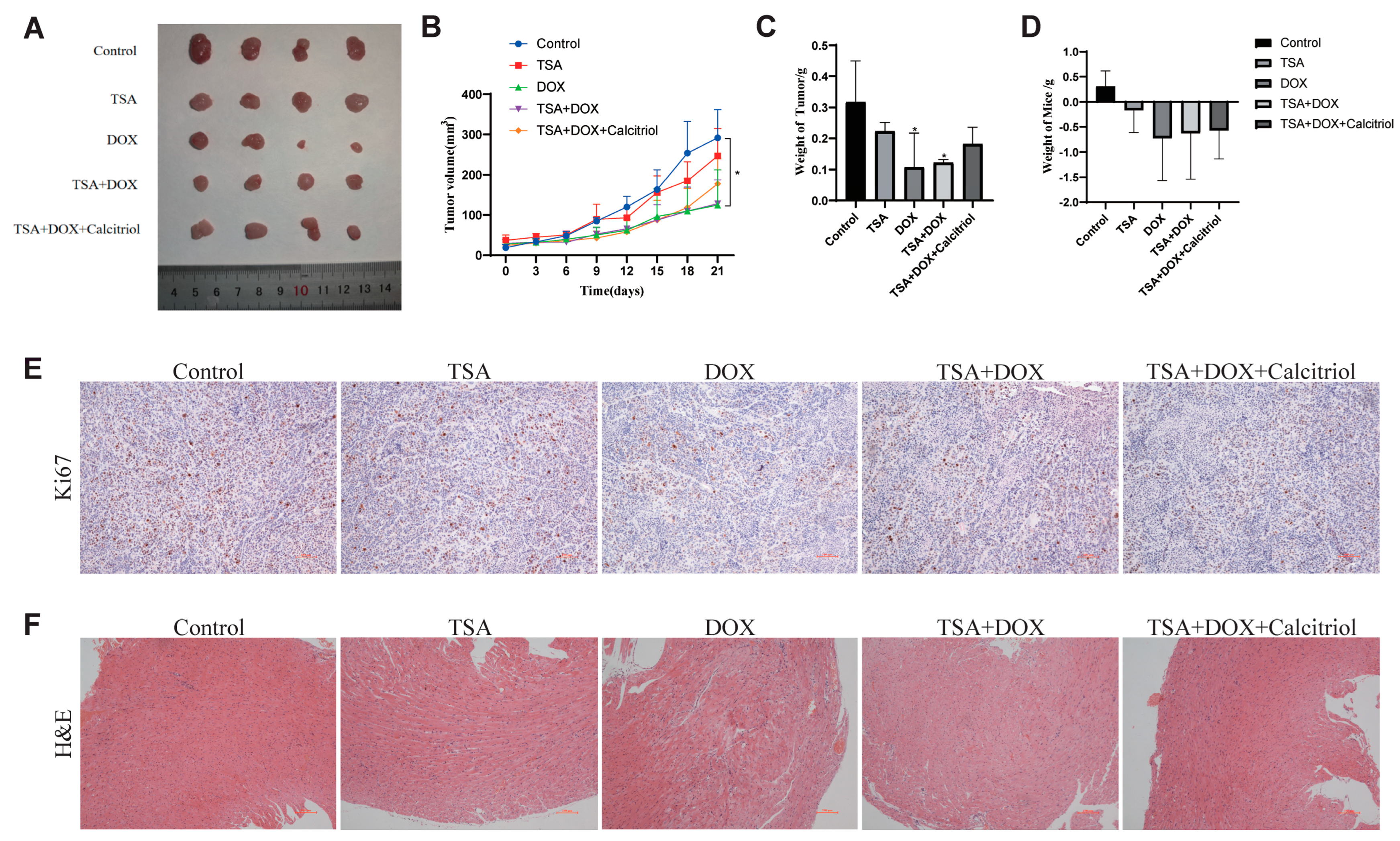
2.8. The Combination of DOX and TSA Significantly Inhibited the Progression of TNBC in Mice, and Calcitriol Could Improve the Cardiac Function of Mice
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Selection of TFs in Hub Gene Sets Highly Related to TNBC
4.3. Construction and Validation of the HTFSS
4.4. Explore the Clinical Application Value of HTFSS and the Predictive Value of Immunotherapy
4.5. scRNA-Seq Cohort Research
4.6. Targeted Drug Screening of Hub TF Genes
4.7. Research on the Prognostic Function of Hub TF Target Genes
4.8. In Vitro Experimental Verification
4.8.1. Cell Culture
4.8.2. CCK-8
4.8.3. Real-Time q-PCR (RT q-PCR)
4.8.4. Western Blotting Analysis
4.9. In Vivo Tumor Model
4.9.1. Construction of Orthotopic Transplanted Tumor in Nude Mice
4.9.2. Immunohistochemical Staining
4.9.3. Hematoxylin and Eosin (H&E) Staining
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Greco, S.; Fabbri, N.; Spaggiari, R.; De Giorgi, A.; Fabbian, F.; Giovine, A. Update on Classic and Novel Approaches in Metastatic Triple-Negative Breast Cancer Treatment: A Comprehensive Review. Biomedicines 2023, 11, 1772. [Google Scholar] [CrossRef]
- Derakhshan, F.; Reis-Filho, J.S. Pathogenesis of Triple-Negative Breast Cancer. Annu. Rev. Pathol. 2022, 17, 181–204. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Herold, C.I.; Anders, C.K. New targets for triple-negative breast cancer. Oncology 2013, 27, 846–854. [Google Scholar]
- Ong, C.T.; Corces, V.G. Enhancer function: New insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.H.; Al-Hallak, N.; Philip, P.A.; Chen, H.; El-Rayes, B.; Azmi, A.S. Aberrant transcription factors in the cancers of the pancreas. Semin. Cancer Biol. 2022, 86, 28–45. [Google Scholar] [CrossRef]
- Asleh, K.; Riaz, N.; Nielsen, T.O. Heterogeneity of triple negative breast cancer: Current advances in subtyping and treatment implications. J. Exp. Clin. Cancer Res. 2022, 41, 265. [Google Scholar] [CrossRef] [PubMed]
- Porta, F.M.; Sajjadi, E.; Venetis, K.; Frascarelli, C.; Cursano, G.; Guerini-Rocco, E.; Fusco, N.; Ivanova, M. Immune Biomarkers in Triple-Negative Breast Cancer: Improving the Predictivity of Current Testing Methods. J. Pers. Med. 2023, 13, 1176. [Google Scholar] [CrossRef] [PubMed]
- Mai, N.; Abuhadra, N.; Jhaveri, K. Molecularly Targeted Therapies for Triple Negative Breast Cancer: History, Advances, and Future Directions. Clin. Breast Cancer 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.; Sorolla, M.A.; Urdanibia, I.; Parisi, E.; Hidalgo, I.; Morales, S.; Salud, A.; Sorolla, A. Are Transcription Factors Plausible Oncotargets for Triple Negative Breast Cancers? Cancers 2022, 14, 1101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, M.; De Wilde, R.L.; Feng, R.; Su, M.; Torres-de la Roche, L.A.; Shi, W. A Machine Learning Model to Predict the Triple Negative Breast Cancer Immune Subtype. Front. Immunol. 2021, 12, 749459. [Google Scholar] [CrossRef] [PubMed]
- Manoochehri, M.; Borhani, N.; Gerhauser, C.; Assenov, Y.; Schonung, M.; Hielscher, T.; Christensen, B.C.; Lee, M.K.; Grone, H.J.; Lipka, D.B.; et al. DNA methylation biomarkers for noninvasive detection of triple-negative breast cancer using liquid biopsy. Int. J. Cancer 2023, 152, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Hu, J.; Zheng, Y.; Zhao, S.; Ma, J. Artificial intelligence: Opportunities and challenges in the clinical applications of triple-negative breast cancer. Br. J. Cancer 2023, 128, 2141–2149. [Google Scholar] [CrossRef]
- Popovic, L.S.; Matovina-Brko, G.; Popovic, M.; Punie, K.; Cvetanovic, A.; Lambertini, M. Targeting triple-negative breast cancer: A clinical perspective. Oncol. Res. 2023, 31, 221–238. [Google Scholar] [CrossRef]
- Abdelwahed, M.; Yurtsever, N.; Savant, D.; Karam, P.; Gimenez, C.; Das, K.; Sheikh-Fayyaz, S.; Khutti, S. Utility of TRPS-1 immunohistochemistry in diagnosis of metastatic breast carcinoma in cytology specimens. J. Am. Soc. Cytopathol. 2022, 11, 345–351. [Google Scholar] [CrossRef]
- Moreno, R.; Casares, L.; Higgins, M.; Ali, K.X.; Honda, T.; Wiel, C.; Sayin, V.I.; Dinkova-Kostova, A.T.; de la Vega, L. Biotinylation of an acetylenic tricyclic bis(cyanoenone) lowers its potency as an NRF2 activator while creating a novel activity against BACH1. Free Radic. Biol. Med. 2022, 191, 203–211. [Google Scholar] [CrossRef]
- Christensen, J.; Cloos, P.; Toftegaard, U.; Klinkenberg, D.; Bracken, A.P.; Trinh, E.; Heeran, M.; Di Stefano, L.; Helin, K. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res. 2005, 33, 5458–5470. [Google Scholar] [CrossRef]
- Le Boiteux, E.; Guichet, P.O.; Masliantsev, K.; Montibus, B.; Vaurs-Barriere, C.; Gonthier-Gueret, C.; Chautard, E.; Verrelle, P.; Karayan-Tapon, L.; Fogli, A.; et al. The Long Non-Coding RNA HOXA-AS2 Promotes Proliferation of Glioma Stem Cells and Modulates Their Inflammation Pathway Mainly through Post-Transcriptional Regulation. Int. J. Mol. Sci. 2022, 23, 4743. [Google Scholar] [CrossRef]
- Han, Z.; Mo, R.; Cai, S.; Feng, Y.; Tang, Z.; Ye, J.; Liu, R.; Cai, Z.; Zhu, X.; Deng, Y.; et al. Differential Expression of E2F Transcription Factors and Their Functional and Prognostic Roles in Human Prostate Cancer. Front. Cell Dev. Biol. 2022, 10, 831329. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Raychaudhuri, P.; Costa, R.H. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell. Biol. 2007, 27, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Noben-Trauth, K.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Sonoda, G.; Testa, J.R.; Klempnauer, K.H. Mybl2 (Bmyb) maps to mouse chromosome 2 and human chromosome 20q 13.1. Genomics 1996, 35, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.K.; Hoffmann, S.; Ahlskog, J.K.; O’Hanlon, K.; Quaas, M.; Larsen, B.D.; Rolland, B.; Rosner, H.I.; Walter, D.; Kousholt, A.N.; et al. Cyclin F suppresses B-Myb activity to promote cell cycle checkpoint control. Nat. Commun. 2015, 6, 5800. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef]
- Brawer, M.K. Recent Progress in the Treatment of Advanced Prostate Cancer With Intermittent Dose-Intense Calcitriol (DN-101). Rev. Urol. 2007, 9, 1–8. [Google Scholar]
- Rodriguez, M.; Munoz-Castaneda, J.R.; Almaden, Y. Therapeutic use of calcitriol. Curr. Vasc. Pharmacol. 2014, 12, 294–299. [Google Scholar] [CrossRef]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef]
- Datta, S.; Sinha, D. Low dose epigallocatechin-3-gallate revives doxorubicin responsiveness by a redox-sensitive pathway in A549 lung adenocarcinoma cells. J. Biochem. Mol. Toxicol. 2022, 36, e22999. [Google Scholar] [CrossRef]
- Xu, W.; Pang, C.; Song, C.; Qian, J.; Feola, S.; Cerullo, V.; Fan, L.; Yu, H.; Lehto, V.P. Black Porous Silicon as a Photothermal Agent and Immunoadjuvant for Efficient Antitumor Immunotherapy. Acta Biomater. 2022, 152, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Kavoosi, F. Effects of trichostatin A on the intrinsic and extrinsic apoptotic pathway, cell viability, and apoptosis induction in hepatocellular carcinoma cell lines. Gastroenterol. Hepatol. Bed Bench 2021, 14, 323–333. [Google Scholar]
- Donia, T.; Khedr, S.; Salim, E.I.; Hessien, M. Trichostatin A sensitizes hepatoma cells to Taxol more than 5-Aza-dC and dexamethasone. Drug Metab. Pers. Ther. 2021. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, T.; Yang, L.; Liu, C.; Wang, C.; Liu, H.; Wang, Y.A.; Su, Z. General route to multifunctional uniform yolk/mesoporous silica shell nanocapsules: A platform for simultaneous cancer-targeted imaging and magnetically guided drug delivery. Chemistry 2012, 18, 12512–12521. [Google Scholar] [CrossRef] [PubMed]
- Javanbakht, S.; Shaabani, A. Encapsulation of graphene quantum dot-crosslinked chitosan by carboxymethylcellulose hydrogel beads as a pH-responsive bio-nanocomposite for the oral delivery agent. Int. J. Biol. Macromol. 2019, 123, 389–397. [Google Scholar] [CrossRef]
- Hafez, A.; Nassef, E.; Fahmy, M.; Elsabagh, M.; Bakr, A.; Hegazi, E. Impact of dietary nano-zinc oxide on immune response and antioxidant defense of broiler chickens. Environ. Sci. Pollut. Res. Int. 2020, 27, 19108–19114. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas. Available online: https://portal.gdc.cancer.gov/ (accessed on 5 April 2022).
- Gene Expression Omnibus Database. Available online: https://www.ncbi.nlm.nih.gov/geo (accessed on 10 May 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Zhang, H.M.; Chen, H.; Liu, W.; Liu, H.; Gong, J.; Wang, H.; Guo, A.Y. AnimalTFDB: A comprehensive animal transcription factor database. Nucleic Acids Res. 2012, 40, D144–D149. [Google Scholar] [CrossRef]
- AnimalTFDB. Available online: http://bioinfo.life.hust.edu.cn/AnimalTFDB/#!/ (accessed on 22 May 2022).
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Mao, Q.; Xia, W.; Dong, G.; Chen, S.; Wang, A.; Jin, G.; Jiang, F.; Xu, L. A nomogram to predict the survival of stage IIIA-N2 non-small cell lung cancer after surgery. J. Thorac. Cardiovasc. Surg. 2018, 155, 1784–1792. [Google Scholar] [CrossRef]
- Foroutan, M.; Bhuva, D.D.; Lyu, R.; Horan, K.; Cursons, J.; Davis, M.J. Single sample scoring of molecular phenotypes. BMC Bioinform. 2018, 19, 404. [Google Scholar] [CrossRef] [PubMed]
- Franzen, O.; Gan, L.M.; Bjorkegren, J.L.M. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [PubMed]
- PanglaoDB Database. Available online: https://panglaodb.se/ (accessed on 25 July 2022).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- The Comparative Toxicogenomics Database. Available online: http://ctdbase.org/ (accessed on 14 October 2022).
- PubChem Database. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 14 October 2022).
- Uniport Database. Available online: https://www.uniprot.org (accessed on 16 October 2022).
- GTRD Database. Available online: https://gtrd.biouml.org/#! (accessed on 7 January 2023).
- Cistrome Database. Available online: http://cistrome.org/db/#/ (accessed on 7 January 2023).
- hTFtarget Database. Available online: http://bioinfo.life.hust.edu.cn/hTFtarget#!/ (accessed on 7 January 2023).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, J.; Li, H.; Zhang, X.; Luo, Z.; Chen, Y.; Zhao, H.; Lv, H.; Zheng, H.; Fu, Z.; Tang, W.; et al. Application of Novel Transcription Factor Machine Learning Model and Targeted Drug Combination Therapy Strategy in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2023, 24, 13497. https://doi.org/10.3390/ijms241713497
Pang J, Li H, Zhang X, Luo Z, Chen Y, Zhao H, Lv H, Zheng H, Fu Z, Tang W, et al. Application of Novel Transcription Factor Machine Learning Model and Targeted Drug Combination Therapy Strategy in Triple Negative Breast Cancer. International Journal of Molecular Sciences. 2023; 24(17):13497. https://doi.org/10.3390/ijms241713497
Chicago/Turabian StylePang, Jianyu, Huimin Li, Xiaoling Zhang, Zhengwei Luo, Yongzhi Chen, Haijie Zhao, Handong Lv, Hongan Zheng, Zhiqian Fu, Wenru Tang, and et al. 2023. "Application of Novel Transcription Factor Machine Learning Model and Targeted Drug Combination Therapy Strategy in Triple Negative Breast Cancer" International Journal of Molecular Sciences 24, no. 17: 13497. https://doi.org/10.3390/ijms241713497