
An Extensively Hydrolyzed Formula Supplemented with Two Human Milk Oligosaccharides Modifies the Fecal Microbiome and Metabolome in Infants with Cow’s Milk Protein Allergy
 ,
,  , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Results
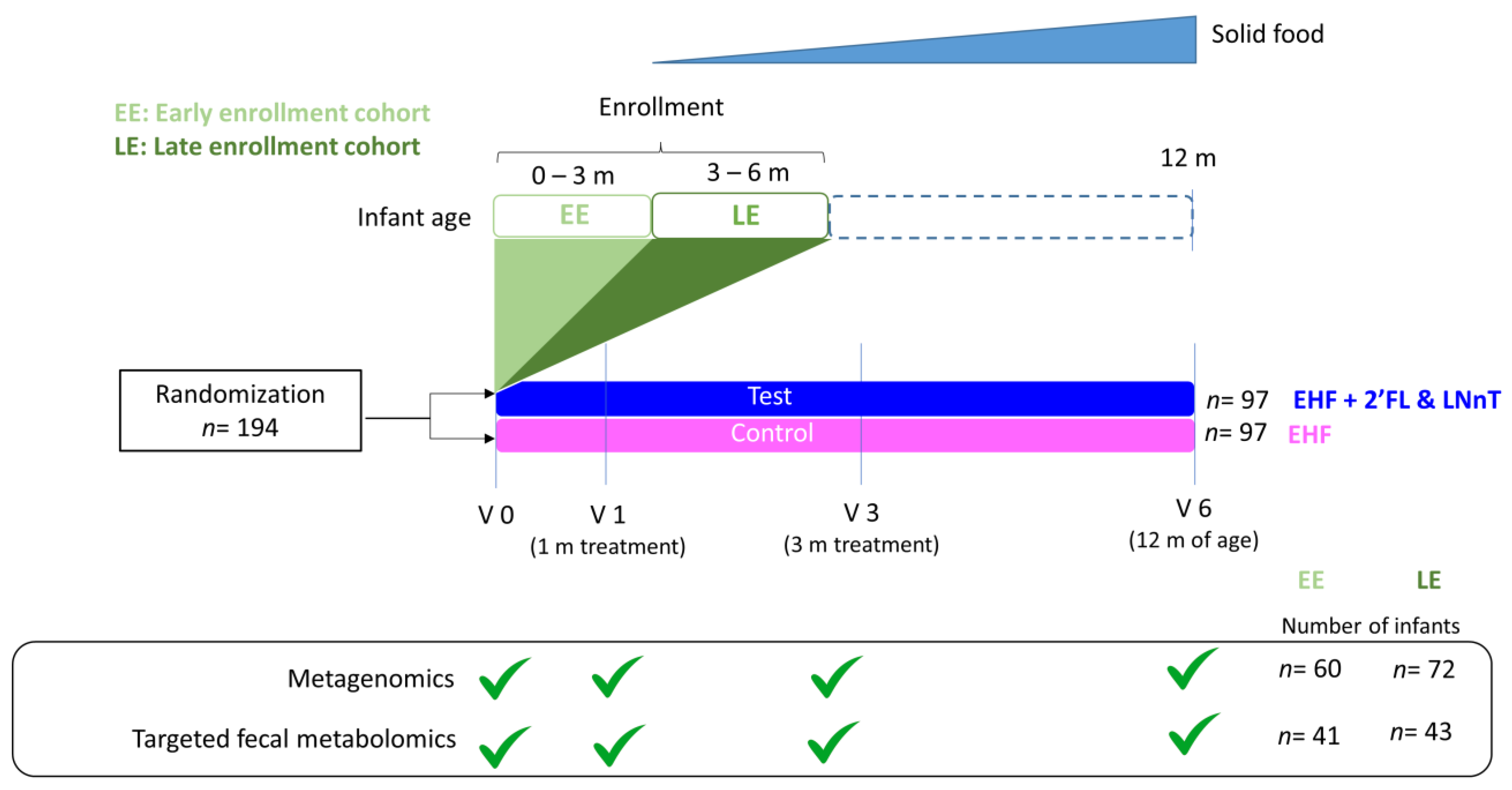
2.1. Cohort Description
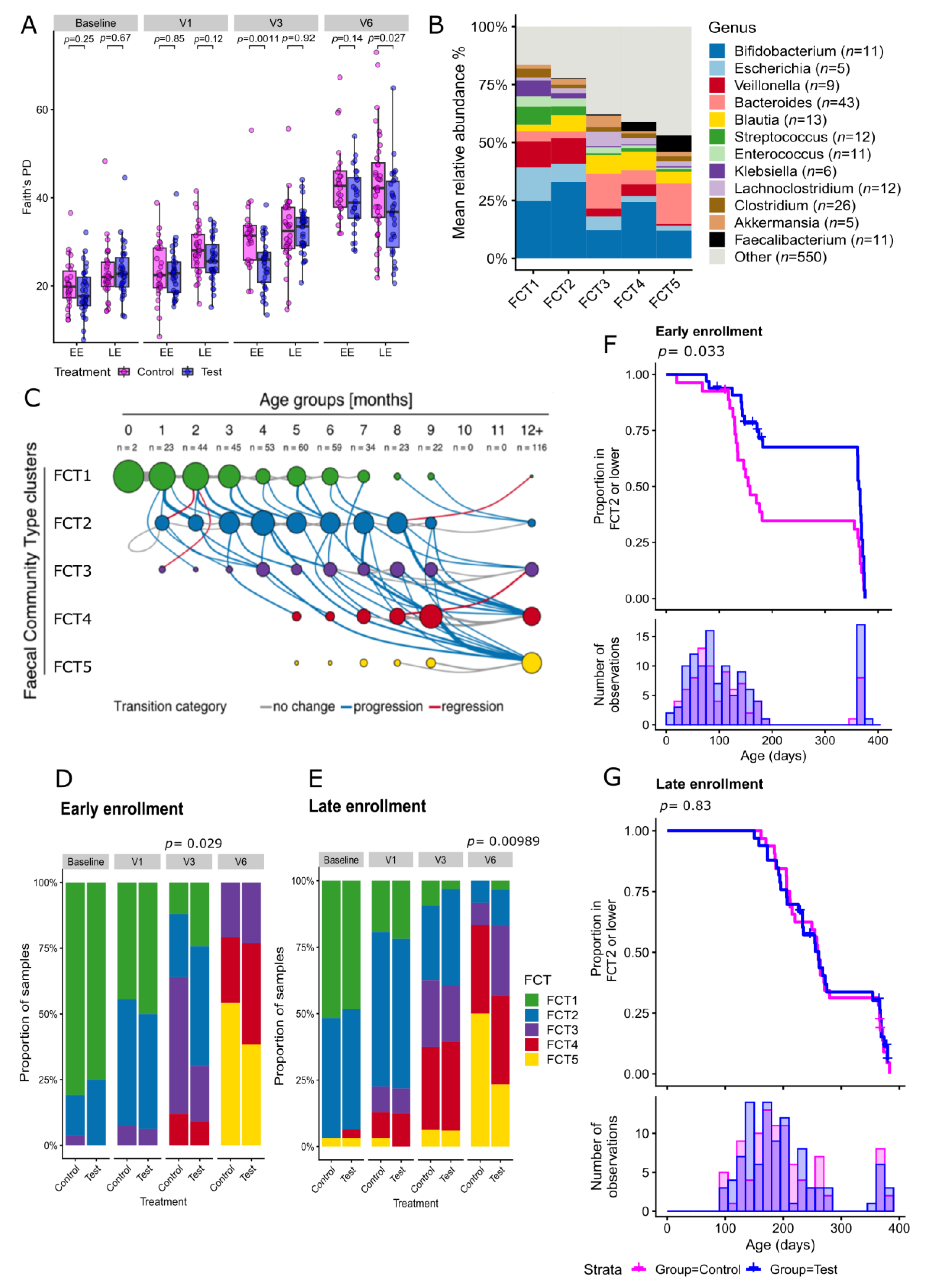
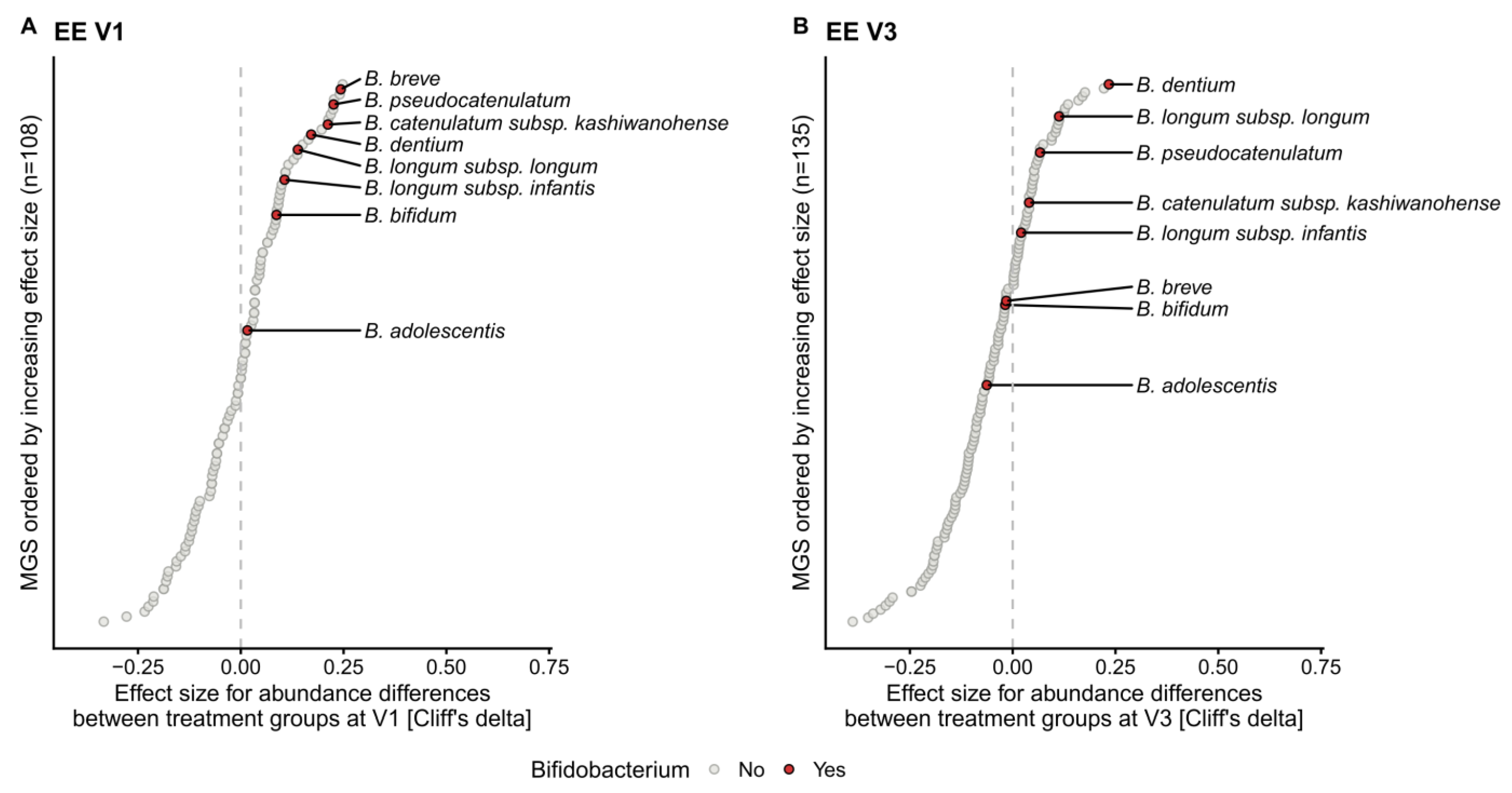
2.2. Human Milk Oligosaccharides Supplementation Shifts Microbiome Development by Favoring a Bifidobacteria-Enriched Microbiome
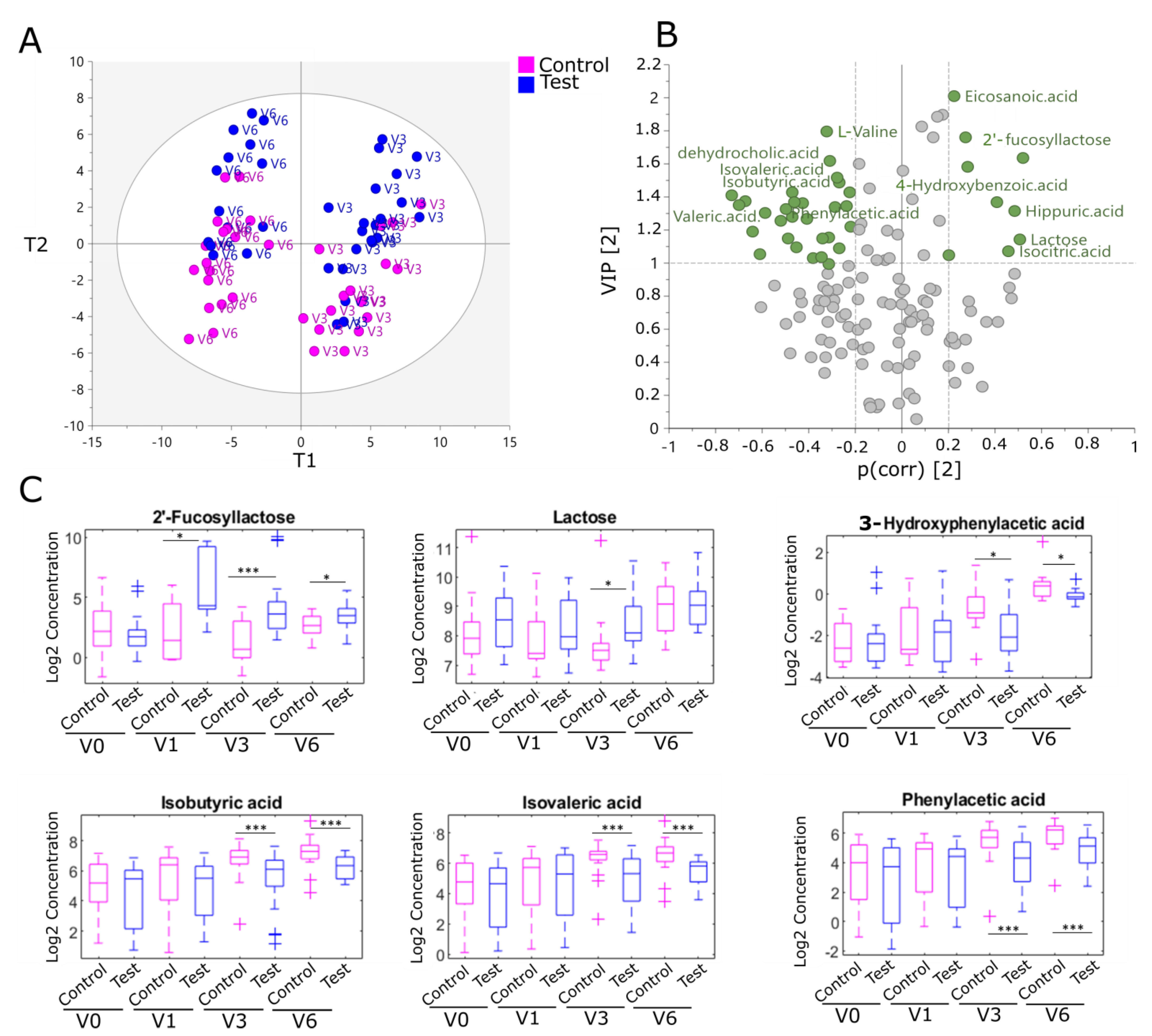
2.3. Human Milk Oligosaccharides Supplementation Impacts Fecal Metabolites from Colonic Amino Acid and Bile Acid Metabolism in the EE Cohort
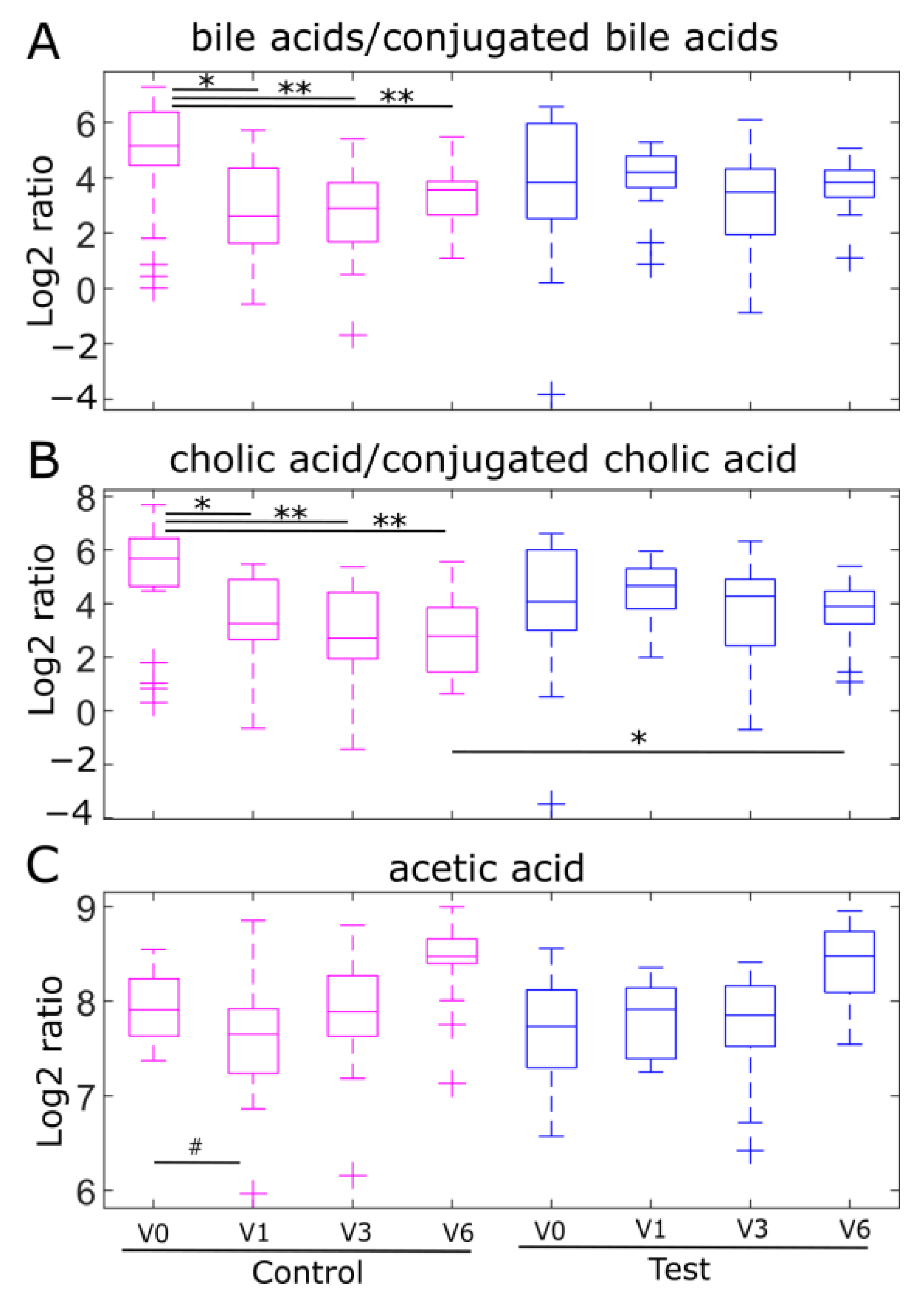
2.4. Fecal Ratios of Unconjugated/Conjugated Bile Acids and Acetic Acid Levels, Markers of Bifidobacterial Metabolism, Are Modulated by Human Milk Oligosaccharides in the EE Cohort
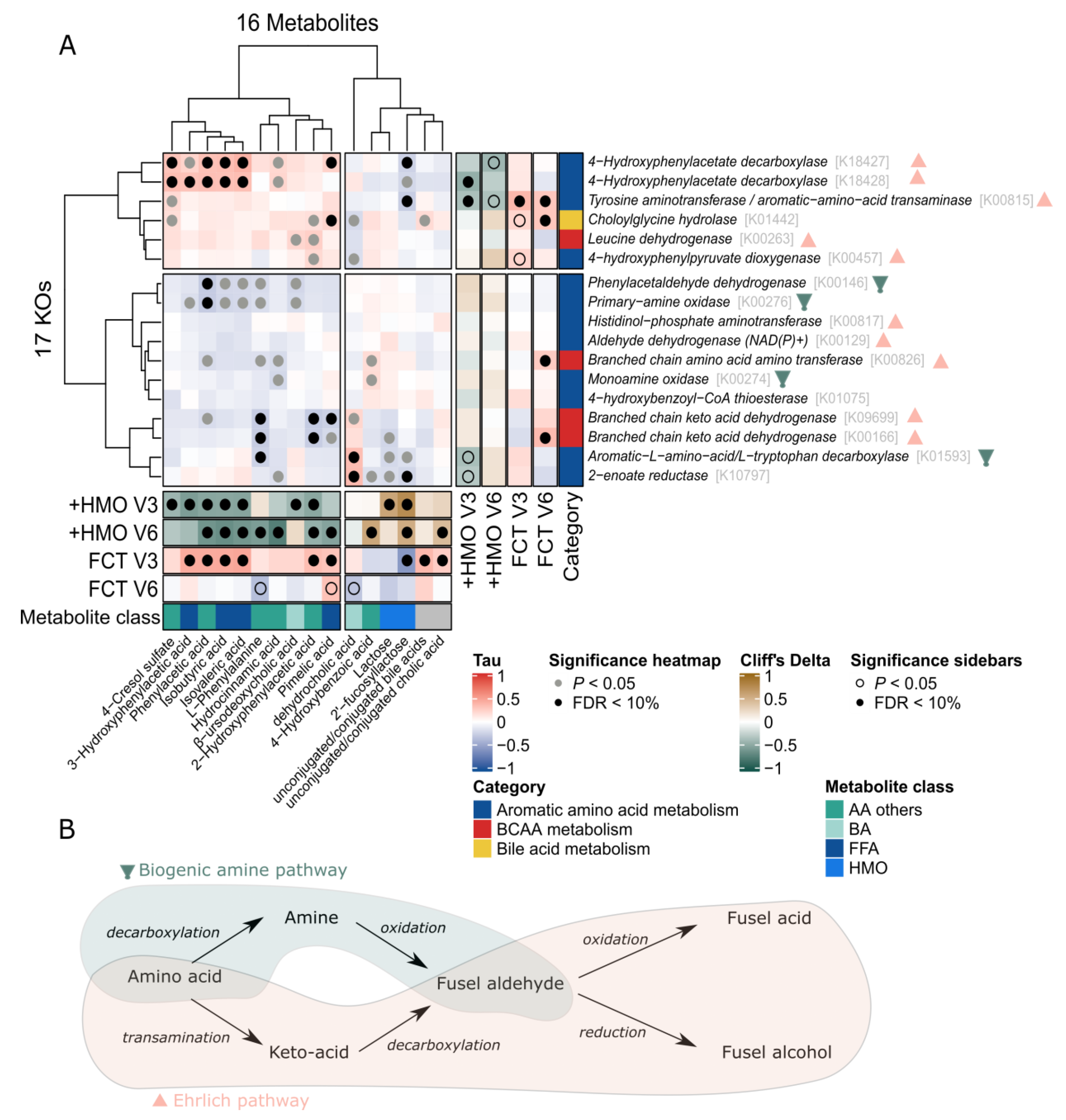
2.5. Omics Integration Describes Association between Bacterial Function and Fecal Metabolites Involved in Amino Acid and Bile Acids Metabolism
3. Discussion
4. Materials and Methods
4.1. Study Design and Participants
4.2. Interventions
4.3. Stool Collection
4.4. Fecal DNA Extraction and Ecological Measures
4.5. Fecal Community Type Clustering and Visualization
4.6. Statistical Analysis of Microbiome Data
4.7. Metabolomics of Fecal Samples and Statistical Analysis of Metabolomics Data
4.8. Metagenomics—Metabolomics Data Integration
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vatanen, T.; Plichta, D.R.; Somani, J.; Munch, P.C.; Arthur, T.D.; Hall, A.B.; Rudolf, S.; Oakeley, E.J.; Ke, X.; Young, R.A.; et al. Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nat. Microbiol. 2019, 4, 470–479. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2018, 562, 583–588. [Google Scholar] [CrossRef]
- WHO. Infant and Young Child Feeding; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Bode, L. Human Milk Oligosaccharides: Structure and Functions. Nestlé Nutr. Inst. Workshop Ser. 2020, 94, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, N.; Tytgat, H.L.P.; Binia, A.; Austin, S.; Singhal, A. Biology of human milk oligosaccharides: From basic science to clinical evidence. J. Hum. Nutr. Diet. 2022, 35, 280–299. [Google Scholar] [CrossRef]
- Plaza-Diaz, J.; Fontana, L.; Gil, A. Human Milk Oligosaccharides and Immune System Development. Nutrients 2018, 10, 1038. [Google Scholar] [CrossRef] [PubMed]
- Triantis, V.; Bode, L.; van Neerven, R.J.J. Immunological Effects of Human Milk Oligosaccharides. Front. Pediatr. 2018, 6, 190. [Google Scholar] [CrossRef] [PubMed]
- Frese, S.A.; Hutton, A.A.; Contreras, L.N.; Shaw, C.A.; Palumbo, M.C.; Casaburi, G.; Xu, G.; Davis, J.C.C.; Lebrilla, C.B.; Henrick, B.M.; et al. Persistence of Supplemented Bifidobacterium longum subsp. infantis EVC001 in Breastfed Infants. mSphere 2017, 2, 1–15. [Google Scholar] [CrossRef]
- Fukuda, S.; Toh, H.; Taylor, T.D.; Ohno, H.; Hattori, M. Acetate-producing bifidobacteria protect the host from enteropathogenic infection via carbohydrate transporters. Gut Microbes 2012, 3, 449–454. [Google Scholar] [CrossRef]
- Laursen, M.F.; Sakanaka, M.; von Burg, N.; Morbe, U.; Andersen, D.; Moll, J.M.; Pekmez, C.T.; Rivollier, A.; Michaelsen, K.F.; Molgaard, C.; et al. Bifidobacterium species associated with breastfeeding produce aromatic lactic acids in the infant gut. Nat. Microbiol. 2021, 6, 1367–1382. [Google Scholar] [CrossRef]
- Henrick, B.M.; Rodriguez, L.; Lakshmikanth, T.; Pou, C.; Henckel, E.; Arzoomand, A.; Olin, A.; Wang, J.; Mikes, J.; Tan, Z.; et al. Bifidobacteria-mediated immune system imprinting early in life. Cell 2021, 184, 3884–3898.e11. [Google Scholar] [CrossRef]
- Newburg, D.S.; Ruiz-Palacios, G.M.; Morrow, A.L. Human milk glycans protect infants against enteric pathogens. Annu. Rev. Nutr. 2005, 25, 37–58. [Google Scholar] [CrossRef]
- Puccio, G.; Alliet, P.; Cajozzo, C.; Janssens, E.; Corsello, G.; Sprenger, N.; Wernimont, S.; Egli, D.; Gosoniu, L.; Steenhout, P. Effects of Infant Formula With Human Milk Oligosaccharides on Growth and Morbidity: A Randomized Multicenter Trial. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 624–631. [Google Scholar] [CrossRef]
- Berger, B.; Porta, N.; Foata, F.; Grathwohl, D.; Delley, M.; Moine, D.; Charpagne, A.; Siegwald, L.; Descombes, P.; Alliet, P.; et al. Linking Human Milk Oligosaccharides, Infant Fecal Community Types, and Later Risk To Require Antibiotics. mBio 2020, 11, e03196-19. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.K.; Martin, F.P.; Donnicola, D.; Julita, M.; Berger, B.; Sprenger, N. Human Milk Oligosaccharide-Stimulated Bifidobacterium Species Contribute to Prevent Later Respiratory Tract Infections. Microorganisms 2021, 9, 1939. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.P.; Tytgat, H.L.P.; Krogh Pedersen, H.; Moine, D.; Eklund, A.C.; Berger, B.; Sprenger, N. Host-microbial co-metabolites modulated by human milk oligosaccharides relate to reduced risk of respiratory tract infections. Front. Nutr. 2022, 9, 935711. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.C.; McCartney, A.L. Longitudinal investigation of the faecal microbiota of healthy full-term infants using fluorescence in situ hybridization and denaturing gradient gel electrophoresis. Microbiology 2010, 156 Pt 11, 3317–3328. [Google Scholar] [CrossRef]
- Thompson, A.L.; Monteagudo-Mera, A.; Cadenas, M.B.; Lampl, M.L.; Azcarate-Peril, M.A. Milk- and solid-feeding practices and daycare attendance are associated with differences in bacterial diversity, predominant communities, and metabolic and immune function of the infant gut microbiome. Front. Cell. Infect. Microbiol. 2015, 5, 3. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Eberl, G. Imprinting of the immune system by the microbiota early in life. Mucosal Immunol. 2020, 13, 183–189. [Google Scholar] [CrossRef]
- Stephen-Victor, E.; Crestani, E.; Chatila, T.A. Dietary and Microbial Determinants in Food Allergy. Immunity 2020, 53, 277–289. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Dulauroy, S.; Marques, R.; Cousu, C.; Al Bounny, S.; Dejardin, F.; Sparwasser, T.; Berard, M.; Cerf-Bensussan, N.; Eberl, G. A Weaning Reaction to Microbiota Is Required for Resistance to Immunopathologies in the Adult. Immunity 2019, 50, 1276–1288.e5. [Google Scholar] [CrossRef]
- Tsukuda, N.; Yahagi, K.; Hara, T.; Watanabe, Y.; Matsumoto, H.; Mori, H.; Higashi, K.; Tsuji, H.; Matsumoto, S.; Kurokawa, K.; et al. Key bacterial taxa and metabolic pathways affecting gut short-chain fatty acid profiles in early life. ISME J. 2021, 15, 2574–2590. [Google Scholar] [CrossRef] [PubMed]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.; Coyte, K.Z.; Bainter, W.; Geha, R.S.; Martin, C.R.; Rakoff-Nahoum, S. Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature 2021, 591, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, M.R.; Blanton, L.V.; DiGiulio, D.B.; Relman, D.A.; Lebrilla, C.B.; Mills, D.A.; Gordon, J.I. A microbial perspective of human developmental biology. Nature 2016, 535, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, D.; Erlewyn-Lajeunesse, M.; Kurukulaaratchy, R.J.; Potter, S.; Roberts, G.; Matthews, S.; Arshad, S.H. Prevalence and longitudinal trends of food allergy during childhood and adolescence: Results of the Isle of Wight Birth Cohort study. Clin. Exp. Allergy 2018, 48, 394–402. [Google Scholar] [CrossRef]
- Nowak-Wegrzyn, A.; Czerkies, L.; Reyes, K.; Collins, B.; Heine, R.G. Confirmed Hypoallergenicity of a Novel Whey-Based Extensively Hydrolyzed Infant Formula Containing Two Human Milk Oligosaccharides. Nutrients 2019, 11, 1447. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.S.; Quinn, P.J.; Campbell, D.E.; Peake, J.; Smart, J.; Robinson, M.; O’Sullivan, M.; Vogt, J.K.; Pedersen, H.K.; Liu, X.; et al. Effects of an Amino Acid-Based Formula Supplemented with Two Human Milk Oligosaccharides on Growth, Tolerability, Safety, and Gut Microbiome in Infants with Cow’s Milk Protein Allergy. Nutrients 2022, 14, 2297. [Google Scholar] [CrossRef]
- Vandenplas, Y.; Zolnowska, M.; Berni Canani, R.; Ludman, S.; Tengelyi, Z.; Moreno-Alvarez, A.; Goh, A.E.N.; Gosoniu, M.L.; Kirwan, B.A.; Tadi, M.; et al. Effects of an Extensively Hydrolyzed Formula Supplemented with Two Human Milk Oligosaccharides on Growth, Tolerability, Safety and Infection Risk in Infants with Cow’s Milk Protein Allergy: A Randomized, Multi-Center Trial. Nutrients 2022, 14, 530. [Google Scholar] [CrossRef]
- Grill, J.P.; Manginot-Durr, C.; Schneider, F.; Ballongue, J. Bifidobacteria and probiotic effects: Action of Bifidobacterium species on conjugated bile salts. Curr. Microbiol. 1995, 31, 23–27. [Google Scholar] [CrossRef]
- Moriki, D.; Francino, M.P.; Koumpagioti, D.; Boutopoulou, B.; Rufian-Henares, J.A.; Priftis, K.N.; Douros, K. The Role of the Gut Microbiome in Cow’s Milk Allergy: A Clinical Approach. Nutrients 2022, 14, 4537. [Google Scholar] [CrossRef]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- van den Elsen, L.W.J.; Garssen, J.; Burcelin, R.; Verhasselt, V. Shaping the Gut Microbiota by Breastfeeding: The Gateway to Allergy Prevention? Front. Pediatr. 2019, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Curtis, N. Factors Influencing the Intestinal Microbiome During the First Year of Life. Pediatr. Infect. Dis. J. 2018, 37, e315–e335. [Google Scholar] [CrossRef]
- Zimmermann, P.; Messina, N.; Mohn, W.W.; Finlay, B.B.; Curtis, N. Association between the intestinal microbiota and allergic sensitization, eczema, and asthma: A systematic review. J. Allergy Clin. Immunol. 2019, 143, 467–485. [Google Scholar] [CrossRef] [PubMed]
- Alcazar, C.G.; Paes, V.M.; Shao, Y.; Oesser, C.; Miltz, A.; Lawley, T.D.; Brocklehurst, P.; Rodger, A.; Field, N. The association between early-life gut microbiota and childhood respiratory diseases: A systematic review. Lancet Microbe 2022, 3, e867–e880. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Schaal, M.; Jahn, D.; Schmidt-Hohagen, K. Metabolism the Difficile Way: The Key to the Success of the Pathogen Clostridioides difficile. Front. Microbiol. 2019, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Kok, C.R.; Brabec, B.; Chichlowski, M.; Harris, C.L.; Moore, N.; Wampler, J.L.; Vanderhoof, J.; Rose, D.; Hutkins, R. Stool microbiome, pH and short/branched chain fatty acids in infants receiving extensively hydrolyzed formula, amino acid formula, or human milk through two months of age. BMC Microbiol. 2020, 20, 337. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, H.; Van Treuren, W.; Hou, B.H.; Higginbottom, S.K.; Dodd, D. Clostridium sporogenes uses reductive Stickland metabolism in the gut to generate ATP and produce circulating metabolites. Nat. Microbiol. 2022, 7, 695–706. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Nyangale, E.P.; Mottram, D.S.; Gibson, G.R. Gut microbial activity, implications for health and disease: The potential role of metabolite analysis. J. Proteome Res. 2012, 11, 5573–5585. [Google Scholar] [CrossRef]
- Diether, N.E.; Willing, B.P. Microbial Fermentation of Dietary Protein: An Important Factor in Diet(-)Microbe(-)Host Interaction. Microorganisms 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.K.; Muir, J.G.; Gibson, P.R. Review article: Insights into colonic protein fermentation, its modulation and potential health implications. Aliment. Pharmacol. Ther. 2016, 43, 181–196. [Google Scholar] [CrossRef]
- Popkov, V.A.; Zharikova, A.A.; Demchenko, E.A.; Andrianova, N.V.; Zorov, D.B.; Plotnikov, E.Y. Gut Microbiota as a Source of Uremic Toxins. Int. J. Mol. Sci. 2022, 23, 483. [Google Scholar] [CrossRef]
- Evenepoel, P.; Meijers, B.K.; Bammens, B.R.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. Suppl. 2009, 76, S12–S19. [Google Scholar] [CrossRef]
- Zheng, Y.; Bek, M.K.; Prince, N.Z.; Peralta Marzal, L.N.; Garssen, J.; Perez Pardo, P.; Kraneveld, A.D. The Role of Bacterial-Derived Aromatic Amino Acids Metabolites Relevant in Autism Spectrum Disorders: A Comprehensive Review. Front. Neurosci. 2021, 15, 738220. [Google Scholar] [CrossRef]
- Murphy, G.M.; Signer, E. Bile acid metabolism in infants and children. Gut 1974, 15, 151–163. [Google Scholar] [CrossRef]
- Ramirez-Perez, O.; Cruz-Ramon, V.; Chinchilla-Lopez, P.; Mendez-Sanchez, N. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann. Hepatol. 2017, 16 (Suppl. 1), S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Hammons, J.L.; Jordan, W.E.; Stewart, R.L.; Taulbee, J.D.; Berg, R.W. Age and diet effects on fecal bile acids in infants. J. Pediatr. Gastroenterol. Nutr. 1988, 7, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Khodakivskyi, P.V.; Lauber, C.L.; Yevtodiyenko, A.; Bazhin, A.A.; Bruce, S.; Ringel-Kulka, T.; Ringel, Y.; Betrisey, B.; Torres, J.; Hu, J.; et al. Noninvasive imaging and quantification of bile salt hydrolase activity: From bacteria to humans. Sci. Adv. 2021, 7, eaaz9857. [Google Scholar] [CrossRef]
- Sillner, N.; Walker, A.; Lucio, M.; Maier, T.V.; Bazanella, M.; Rychlik, M.; Haller, D.; Schmitt-Kopplin, P. Longitudinal Profiles of Dietary and Microbial Metabolites in Formula- and Breastfed Infants. Front. Mol. Biosci. 2021, 8, 660456. [Google Scholar] [CrossRef]
- Joyce, S.A.; Shanahan, F.; Hill, C.; Gahan, C.G. Bacterial bile salt hydrolase in host metabolism: Potential for influencing gastrointestinal microbe-host crosstalk. Gut Microbes 2014, 5, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Mullish, B.H.; McDonald, J.A.K.; Pechlivanis, A.; Allegretti, J.R.; Kao, D.; Barker, G.F.; Kapila, D.; Petrof, E.O.; Joyce, S.A.; Gahan, C.G.M.; et al. Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut 2019, 68, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.R.; Naik, S.R.; Vakil, B.V. Probiotics, prebiotics and synbiotics—A review. J. Food Sci. Technol. 2015, 52, 7577–7587. [Google Scholar] [CrossRef]
- Arifuzzaman, M.; Won, T.H.; Li, T.T.; Yano, H.; Digumarthi, S.; Heras, A.F.; Zhang, W.; Parkhurst, C.N.; Kashyap, S.; Jin, W.B.; et al. Inulin fibre promotes microbiota-derived bile acids and type 2 inflammation. Nature 2022, 611, 578–584. [Google Scholar] [CrossRef]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef]
- Crestani, E.; Harb, H.; Charbonnier, L.M.; Leirer, J.; Motsinger-Reif, A.; Rachid, R.; Phipatanakul, W.; Kaddurah-Daouk, R.; Chatila, T.A. Untargeted metabolomic profiling identifies disease-specific signatures in food allergy and asthma. J. Allergy Clin. Immunol. 2020, 145, 897–906. [Google Scholar] [CrossRef]
- Hornef, M.W.; Torow, N. ‘Layered immunity’ and the ‘neonatal window of opportunity’—Timed succession of non-redundant phases to establish mucosal host-microbial homeostasis after birth. Immunology 2020, 159, 15–25. [Google Scholar] [CrossRef]
- Hauser, J.; Pisa, E.; Arias Vasquez, A.; Tomasi, F.; Traversa, A.; Chiodi, V.; Martin, F.P.; Sprenger, N.; Lukjancenko, O.; Zollinger, A.; et al. Sialylated human milk oligosaccharides program cognitive development through a non-genomic transmission mode. Mol. Psychiatry 2021, 26, 2854–2871. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Faith, D.P.; Baker, A.M. Phylogenetic diversity (PD) and biodiversity conservation: Some bioinformatics challenges. Evol. Bioinform. Online 2007, 2, 121–128. [Google Scholar] [CrossRef]
- Hamady, M.; Lozupone, C.; Knight, R. Fast UniFrac: Facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010, 4, 17–27. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef]
- Xie, G.; Zhong, W.; Li, H.; Li, Q.; Qiu, Y.; Zheng, X.; Chen, H.; Zhao, X.; Zhang, S.; Zhou, Z.; et al. Alteration of bile acid metabolism in the rat induced by chronic ethanol consumption. FASEB J. 2013, 27, 3583–3593. [Google Scholar] [CrossRef]
- Xie, G.; Wang, L.; Chen, T.; Zhou, K.; Zhang, Z.; Li, J.; Sun, B.; Guo, Y.; Wang, X.; Wang, Y.; et al. A Metabolite Array Technology for Precision Medicine. Anal. Chem. 2021, 93, 5709–5717. [Google Scholar] [CrossRef]
- Bao, Y.; Chen, C.; Newburg, D.S. Quantification of neutral human milk oligosaccharides by graphitic carbon high-performance liquid chromatography with tandem mass spectrometry. Anal. Biochem. 2013, 433, 28–35. [Google Scholar] [CrossRef]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.; Barritt, A.S.t. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PLS Model at V3 and V6 | WMW Test V3 | WMW Test V6 | ||||
|---|---|---|---|---|---|---|
| Metabolites | Correlation with Treatment | VIP Score | FC Test/Control | p Value | FC Test/Control | p Value |
| 2′-Fucosyllactose | 0.52 | 1.63 | 19.07 | 0.0001 | 2.44 | 0.0200 |
| Lactose | 0.50 | 1.14 | 1.80 | 0.0033 | 0.98 | 0.5600 |
| Hydroxybenzoic acid | 0.41 | 1.37 | 1.01 | 0.8100 | 2.34 | 0.0140 |
| 2-Hydroxyphenylacetic acid | −0.58 | 1.30 | 0.31 | 0.0100 | 0.60 | 0.054 |
| 3-Hydroxyphenylacetic acid | −0.28 | 1.52 | 0.24 | 0.0040 | 0.77 | 0.011 |
| Phenylacetic acid | −0.70 | 1.35 | 0.24 | 0.0089 | 0.34 | 0.0065 |
| Hydrocinnamic acid | −0.46 | 1.37 | 0.21 | 0.1400 | 0.25 | 0.0007 |
| 4-Cresol sulfate | −0.31 | 1.00 | 0.28 | 0.0320 | 0.40 | 0.14 |
| L-Phenylalanine | −0.34 | 1.14 | 1.31 | 0.3700 | 0.41 | 0.0083 |
| Isobutyric acid | −0.67 | 1.37 | 0.44 | 0.0098 | 0.36 | 0.0035 |
| Isovaleric acid | −0.73 | 1.41 | 0.28 | 0.0080 | 0.44 | 0.00730 |
| Pimelic acid | −0.22 | 1.43 | 0.58 | 0.1230 | 0.57 | 0.0210 |
| Dehydrocholic acid | −0.30 | 1.62 | 0.51 | 0.0490 | 0.69 | 0.1800 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulangé, C.L.; Pedersen, H.K.; Martin, F.-P.; Siegwald, L.; Pallejà Caro, A.; Eklund, A.C.; Jia, W.; Zhang, H.; Berger, B.; Sprenger, N.; et al. An Extensively Hydrolyzed Formula Supplemented with Two Human Milk Oligosaccharides Modifies the Fecal Microbiome and Metabolome in Infants with Cow’s Milk Protein Allergy. Int. J. Mol. Sci. 2023, 24, 11422. https://doi.org/10.3390/ijms241411422
Boulangé CL, Pedersen HK, Martin F-P, Siegwald L, Pallejà Caro A, Eklund AC, Jia W, Zhang H, Berger B, Sprenger N, et al. An Extensively Hydrolyzed Formula Supplemented with Two Human Milk Oligosaccharides Modifies the Fecal Microbiome and Metabolome in Infants with Cow’s Milk Protein Allergy. International Journal of Molecular Sciences. 2023; 24(14):11422. https://doi.org/10.3390/ijms241411422
Chicago/Turabian StyleBoulangé, Claire L., Helle K. Pedersen, Francois-Pierre Martin, Léa Siegwald, Albert Pallejà Caro, Aron C. Eklund, Wei Jia, Huizhen Zhang, Bernard Berger, Norbert Sprenger, and et al. 2023. "An Extensively Hydrolyzed Formula Supplemented with Two Human Milk Oligosaccharides Modifies the Fecal Microbiome and Metabolome in Infants with Cow’s Milk Protein Allergy" International Journal of Molecular Sciences 24, no. 14: 11422. https://doi.org/10.3390/ijms241411422
APA StyleBoulangé, C. L., Pedersen, H. K., Martin, F.-P., Siegwald, L., Pallejà Caro, A., Eklund, A. C., Jia, W., Zhang, H., Berger, B., Sprenger, N., Heine, R. G., & CINNAMON Study Investigator Group. (2023). An Extensively Hydrolyzed Formula Supplemented with Two Human Milk Oligosaccharides Modifies the Fecal Microbiome and Metabolome in Infants with Cow’s Milk Protein Allergy. International Journal of Molecular Sciences, 24(14), 11422. https://doi.org/10.3390/ijms241411422