
Thresholds of Endoglin Expression in Endothelial Cells Explains Vascular Etiology in Hereditary Hemorrhagic Telangiectasia Type 1

 , , ,
, , , 
Abstract
1. Introduction
2. Results
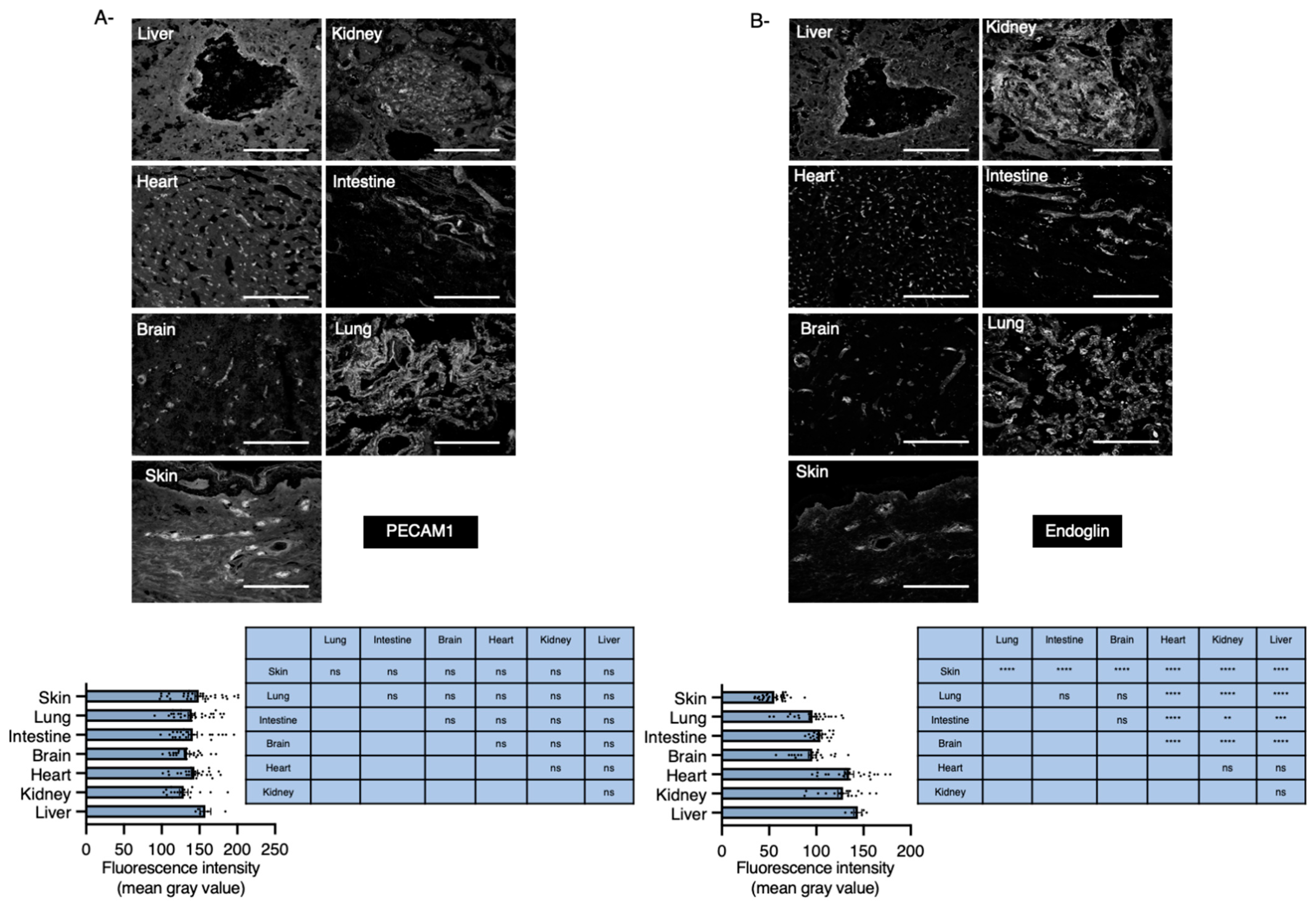
2.1. Endoglin mRNA Expression Levels in Various Organs
2.2. Organs with Basal Low Levels of Endoglin Associate with High Risk to Develop HHT
2.3. Endoglin Levels Become Almost Undetectable in Skin Capillaries and Arteries of Eng+/− Mice
2.4. Levels of Endoglin Are Linked to Tissue Developing HHT1 in Humans
2.5. Signaling Pathways in ECs Are Dependent on Endoglin Thresholds
3. Discussion
4. Materials and Methods
4.1. Human Material
4.2. Mice
4.3. Mouse Endothelial Cell Isolation and Culture
4.4. Adenoviral Infection
4.5. Immunofluorescence Staining
4.6. Image Quantification
4.7. RNA Isolation and Real Time PCR Analysis
4.8. Western Blot and Quantification
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ⍺-SMA | ⍺-smooth muscle actin |
| ALK1 | Activin recepto-ike kinase 1 |
| AVMs | Arteriovenous Malformations |
| BMP9/10 | Bone Morphogenetic Protein 9/10 |
| HHT | Hereditary Hemorrhagic Telangiectasia |
| HHT1 | Hereditary Hemorrhagic Telangiectasia type 1 |
| HHT2 | Hereditary Hemorrhagic Telangiectasia type 2 |
| MAPK | Mitogen Activated Protein Kinase |
| NF | Normalization factors |
| PI3Kinase | Phosphatidyl inositol 3-kinase |
| PMT | Photomultiplier tube |
| TGF-β | Transforming growth factor- β |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR1 | Vascular Endothelial Growth Factor Receptor 1 |
| VEGFR2 | Vascular Endothelial Growth Factor Receptor 2 |
| VSMCs | Vascular smooth muscle cells |
References
- Shovlin, C.L. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 2010, 24, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Galaris, G.; Thalgott, J.H.; Lebrin, F.P.G. Pericytes in Hereditary Hemorrhagic Telangiectasia. Adv. Exp. Med. Biol. 2019, 1147, 215–246. [Google Scholar] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J.; et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Qumsiyeh, M.; Benkhalifa, M.; Marchuk, D.A. Assignment of human transforming growth factor-beta type I and type III receptor genes (TGFBR1 and TGFBR3) to 9q33-q34 and 1p32-p33, respectively. Genomics 1995, 28, 356–357. [Google Scholar] [CrossRef]
- Kilian, A.; Clancy, M.S.; Olitsky, S.; Gossage, J.R.; Faughnan, M.E. Screening for pulmonary and brain vascular malformations is the North American standard of care for patients with hereditary hemorrhagic telangiectasia (HHT): A survey of HHT Centers of Excellence. Vasc. Med. 2021, 26, 53–55. [Google Scholar] [CrossRef]
- Bayrak-Toydemir, P.; McDonald, J.; Markewitz, B.; Lewin, S.; Miller, F.; Chou, L.S.; Gedge, F.; Tang, W.; Coon, H.; Mao, R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: Mutations and manifestations. Am. J. Med. Genet. A 2006, 140, 463–470. [Google Scholar] [CrossRef]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef]
- Fernandez, L.A.; Sanz-Rodriguez, F.; Zarrabeitia, R.; Perez-Molino, A.; Hebbel, R.P.; Nguyen, J.; Bernabeu, C.; Botella, L.M. Blood outgrowth endothelial cells from Hereditary Haemorrhagic Telangiectasia patients reveal abnormalities compatible with vascular lesions. Cardiovasc. Res. 2005, 68, 235–248. [Google Scholar] [CrossRef]
- Brown, M.A.; Zhao, Q.; Baker, K.A.; Naik, C.; Chen, C.; Pukac, L.; Singh, M.; Tsareva, T.; Parice, Y.; Mahoney, A.; et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem. 2005, 280, 25111–25118. [Google Scholar] [CrossRef]
- David, L.; Mallet, C.; Mazerbourg, S.; Feige, J.J.; Bailly, S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007, 109, 1953–1961. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120 Pt 6, 964–972. [Google Scholar] [CrossRef]
- Crist, A.M.; Lee, A.R.; Patel, N.R.; Westhoff, D.E.; Meadows, S.M. Vascular deficiency of Smad4 causes arteriovenous malformations: A mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis 2018, 21, 363–380. [Google Scholar] [CrossRef]
- Larrivee, B.; Prahst, C.; Gordon, E.; del Toro, R.; Mathivet, T.; Duarte, A.; Simons, M.; Eichmann, A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 2012, 22, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Moya, I.M.; Umans, L.; Maas, E.; Pereira, P.N.; Beets, K.; Francis, A.; Sents, W.; Robertson, E.J.; Mummery, C.L.; Huylebroeck, D.; et al. Stalk cell phenotype depends on integration of Notch and Smad1/5 signaling cascades. Dev. Cell 2012, 22, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Jerkic, M.; Rivas-Elena, J.V.; Prieto, M.; Carron, R.; Sanz-Rodriguez, F.; Perez-Barriocanal, F.; Rodriguez-Barbero, A.; Bernabeu, C.; Lopez-Novoa, J.M. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004, 18, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Toporsian, M.; Gros, R.; Kabir, M.G.; Vera, S.; Govindaraju, K.; Eidelman, D.H.; Husain, M.; Letarte, M. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ. Res. 2005, 96, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, A.; Liso, A.; D’Ovidio, F.; Mestice, A.; Pasculli, G.; Gallitelli, M.; Rizzi, R.; Specchia, G.; Sabba, C. Vascular endothelial growth factor serum levels are elevated in patients with hereditary hemorrhagic telangiectasia. Acta Haematol. 2003, 110, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Sadick, H.; Naim, R.; Sadick, M.; Hormann, K.; Riedel, F. Plasma level and tissue expression of angiogenic factors in patients with hereditary hemorrhagic telangiectasia. Int. J. Mol. Med. 2005, 15, 591–596. [Google Scholar] [CrossRef]
- Sadick, H.; Riedel, F.; Naim, R.; Goessler, U.; Hormann, K.; Hafner, M.; Lux, A. Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression. Haematologica 2005, 90, 818–828. [Google Scholar]
- Botella, L.M.; Albinana, V.; Ojeda-Fernandez, L.; Recio-Poveda, L.; Bernabeu, C. Research on potential biomarkers in hereditary hemorrhagic telangiectasia. Front. Genet. 2015, 6, 115. [Google Scholar] [CrossRef]
- Tual-Chalot, S.; Garcia-Collado, M.; Redgrave, R.E.; Singh, E.; Davison, B.; Park, C.; Lin, H.; Luli, S.; Jin, Y.; Wang, Y.; et al. Loss of Endothelial Endoglin Promotes High-Output Heart Failure Through Peripheral Arteriovenous Shunting Driven by VEGF Signaling. Circ. Res. 2020, 126, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Muhl, L.; Burmakin, M.; Wang, Y.; Duchez, A.C.; Betsholtz, C.; Arthur, H.M.; Jakobsson, L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 2017, 19, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, C.; Cannavicci, A.; Faughnan, M.E.; Kutryk, M.J.B. Endoglin deficiency impairs VEGFR2 but not FGFR1 or TIE2 activation and alters VEGF-mediated cellular responses in human primary endothelial cells. Transl. Res. 2021, 5, 22. [Google Scholar]
- Ola, R.; Dubrac, A.; Han, J.; Zhang, F.; Fang, J.S.; Larrivee, B.; Lee, M.; Urarte, A.A.; Kraehling, J.R.; Genet, G.; et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat. Commun. 2016, 7, 13650. [Google Scholar] [CrossRef]
- Thalgott, J.H.; Dos-Santos-Luis, D.; Hosman, A.E.; Martin, S.; Lamande, N.; Bracquart, D.; Srun, S.; Galaris, G.; de Boer, H.C.; Tual-Chalot, S.; et al. Decreased Expression of Vascular Endothelial Growth Factor Receptor 1 Contributes to the Pathogenesis of Hereditary Hemorrhagic Telangiectasia Type 2. Circulation 2018, 138, 2698–2712. [Google Scholar] [CrossRef]
- Mahmoud, M.; Allinson, K.R.; Zhai, Z.; Oakenfull, R.; Ghandi, P.; Adams, R.H.; Fruttiger, M.; Arthur, H.M. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 2010, 106, 1425–1433. [Google Scholar] [CrossRef]
- Choi, E.J.; Walker, E.J.; Shen, F.; Oh, S.P.; Arthur, H.M.; Young, W.L.; Su, H. Minimal homozygous endothelial deletion of Eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc. Dis. 2012, 33, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Park, S.O.; Wankhede, M.; Lee, Y.J.; Choi, E.J.; Fliess, N.; Choe, S.W.; Oh, S.H.; Walter, G.; Raizada, M.K.; Sorg, B.S.; et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 2009, 119, 3487–3496. [Google Scholar] [CrossRef]
- Jerkic, M.; Peter, M.; Ardelean, D.; Fine, M.; Konerding, M.A.; Letarte, M. Dextran sulfate sodium leads to chronic colitis and pathological angiogenesis in Endoglin heterozygous mice. Inflamm. Bowel. Dis. 2010, 16, 1859–1870. [Google Scholar] [CrossRef]
- Garrido-Martin, E.M.; Nguyen, H.L.; Cunningham, T.A.; Choe, S.W.; Jiang, Z.; Arthur, H.M.; Lee, Y.J.; Oh, S.P. Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models--brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2232–2236. [Google Scholar] [CrossRef]
- Han, C.; Choe, S.W.; Kim, Y.H.; Acharya, A.P.; Keselowsky, B.G.; Sorg, B.S.; Lee, Y.J.; Oh, S.P. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis 2014, 17, 823–830. [Google Scholar] [CrossRef]
- Choi, E.J.; Chen, W.; Jun, K.; Arthur, H.M.; Young, W.L.; Su, H. Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS ONE 2014, 9, e88511. [Google Scholar] [CrossRef]
- Snellings, D.A.; Gallione, C.J.; Clark, D.S.; Vozoris, N.T.; Faughnan, M.E.; Marchuk, D.A. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am. J. Hum. Genet. 2019, 105, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Benzinou, M.; Clermont, F.F.; Letteboer, T.G.; Kim, J.H.; Espejel, S.; Harradine, K.A.; Arbelaez, J.; Luu, M.T.; Roy, R.; Quigley, D.; et al. Mouse and human strategies identify PTPN14 as a modifier of angiogenesis and hereditary haemorrhagic telangiectasia. Nat. Commun. 2012, 3, 616. [Google Scholar] [CrossRef]
- Kawasaki, K.; Freimuth, J.; Meyer, D.S.; Lee, M.M.; Tochimoto-Okamoto, A.; Benzinou, M.; Clermont, F.F.; Wu, G.; Roy, R.; Letteboer, T.G.; et al. Genetic variants of Adam17 differentially regulate TGFbeta signaling to modify vascular pathology in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 7723–7728. [Google Scholar] [CrossRef] [PubMed]
- Letteboer, T.G.; Benzinou, M.; Merrick, C.B.; Quigley, D.A.; Zhau, K.; Kim, I.J.; To, M.D.; Jablons, D.M.; van Amstel, J.K.; Westermann, C.J.; et al. Genetic variation in the functional ENG allele inherited from the non-affected parent associates with presence of pulmonary arteriovenous malformation in hereditary hemorrhagic telangiectasia 1 (HHT1) and may influence expression of PTPN14. Front. Genet. 2015, 6, 67. [Google Scholar] [CrossRef]
- Bourdeau, A.; Faughnan, M.E.; McDonald, M.L.; Paterson, A.D.; Wanless, I.R.; Letarte, M. Potential role of modifier genes influencing transforming growth factor-beta1 levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am. J. Pathol. 2001, 158, 2011–2020. [Google Scholar] [CrossRef]
- Sanchez-Elsner, T.; Botella, L.M.; Velasco, B.; Langa, C.; Bernabeu, C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J. Biol. Chem. 2002, 277, 43799–43808. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef]
- Waters, J.C. Accuracy and precision in quantitative fluorescence microscopy. J. Cell Biol. 2009, 185, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Shihan, M.H.; Novo, S.G.; Le Marchand, S.J.; Wang, Y.; Duncan, M.K. A simple method for quantitating confocal fluorescent images. Biochem. Biophys. Rep. 2021, 25, 100916. [Google Scholar] [PubMed]
- Bourdeau, A.; Dumont, D.J.; Letarte, M. A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Investig. 1999, 104, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Torsney, E.; Charlton, R.; Diamond, A.G.; Burn, J.; Soames, J.V.; Arthur, H.M. Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation 2003, 107, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Srun, S.; Raymond, K.; Martin, S.; van den Brink, S.; Freitas, C.; Breant, C.; Mathivet, T.; Larrivee, B.; Thomas, J.L.; et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat. Med. 2010, 16, 420–428. [Google Scholar] [CrossRef]
- Tual-Chalot, S.; Oh, S.P.; Arthur, H.M. Mouse models of hereditary hemorrhagic telangiectasia: Recent advances and future challenges. Front. Genet. 2015, 6, 25. [Google Scholar] [CrossRef]
- Wang, J.; Niu, N.; Xu, S.; Jin, Z.G. A simple protocol for isolating mouse lung endothelial cells. Sci. Rep. 2019, 9, 1458. [Google Scholar] [CrossRef]
- Marcu, R.; Choi, Y.J.; Xue, J.; Fortin, C.L.; Wang, Y.; Nagao, R.J.; Xu, J.; MacDonald, J.W.; Bammler, T.K.; Murry, C.E.; et al. Human Organ-Specific Endothelial Cell Heterogeneity. Science 2018, 4, 20–35. [Google Scholar] [CrossRef]
- Ricard, N.; Bailly, S.; Guignabert, C.; Simons, M. The quiescent endothelium: Signalling pathways regulating organ-specific endothelial normalcy. Nat. Rev. Cardiol. 2021, 24, 1–16. [Google Scholar]
- Prost, S.; Sheahan, S.; Rannie, D.; Harrison, D.J. Adenovirus-mediated Cre deletion of floxed sequences in primary mouse cells is an efficient alternative for studies of gene deletion. Nucleic Acids Res. 2001, 29, E80. [Google Scholar] [CrossRef][Green Version]
- Paquet, M.E.; Pece-Barbara, N.; Vera, S.; Cymerman, U.; Karabegovic, A.; Shovlin, C.; Letarte, M. Analysis of several endoglin mutants reveals no endogenous mature or secreted protein capable of interfering with normal endoglin function. Hum. Mol. Genet. 2001, 10, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Guttmacher, A.E.; Marchuk, D.A.; White, R.I., Jr. Hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 1995, 333, 918–924. [Google Scholar] [CrossRef]
- Plauchu, H.; de Chadarevian, J.P.; Bideau, A.; Robert, J.M. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am. J. Med. Genet. 1989, 32, 291–297. [Google Scholar] [CrossRef]
- Kjeldsen, A.D.; Moller, T.R.; Brusgaard, K.; Vase, P.; Andersen, P.E. Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J. Intern. Med. 2005, 258, 349–355. [Google Scholar] [CrossRef]
- Letteboer, T.G.; Mager, J.J.; Snijder, R.J.; Koeleman, B.P.; Lindhout, D.; Ploos van Amstel, J.K.; Westermann, C.J. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J. Med. Genet. 2006, 43, 371–377. [Google Scholar] [CrossRef]
- Bossler, A.D.; Richards, J.; George, C.; Godmilow, L.; Ganguly, A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): Correlation of genotype with phenotype. Hum. Mutat. 2006, 27, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Lesca, G.; Olivieri, C.; Burnichon, N.; Pagella, F.; Carette, M.F.; Gilbert-Dussardier, B.; Goizet, C.; Roume, J.; Rabilloud, M.; Saurin, J.C.; et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: Data from the French-Italian HHT network. Genet. Med. 2007, 9, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Singh, E.; Redgrave, R.E.; Phillips, H.M.; Arthur, H.M. Arterial endoglin does not protect against arteriovenous malformations. Angiogenesis 2020, 23, 559–566. [Google Scholar] [CrossRef]
- Hao, Q.; Zhu, Y.; Su, H.; Shen, F.; Yang, G.Y.; Kim, H.; Young, W.L. VEGF Induces More Severe Cerebrovascular Dysplasia in Endoglin than in Alk1 Mice. Transl. Stroke Res. 2010, 1, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wu, Y.Q.; Huey, M.; Arthur, H.M.; Marchuk, D.A.; Hashimoto, T.; Young, W.L.; Yang, G.Y. Vascular endothelial growth factor induces abnormal microvasculature in the endoglin heterozygous mouse brain. J. Cereb. Blood Flow Metab. 2004, 24, 237–244. [Google Scholar] [CrossRef]
- Chi, J.T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; van de Rijn, M.; Botstein, D.; et al. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef]
- Allinson, K.R.; Carvalho, R.L.; van den Brink, S.; Mummery, C.L.; Arthur, H.M. Generation of a floxed allele of the mouse Endoglin gene. Genesis 2007, 45, 391–395. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Product Size | Annealing Temperature |
|---|---|---|---|
| Flk1 | 5′-GGCGGTGGTGACAGTATCTT-3′ 5′-GAGGCGATGAATGGTGATCT-3′ | 152bp | 60 °C |
| Tie1 | 5′-CATCGAGACTTTGCAGGTGA-3′ 5′-GTTTCCATAGGGGGCGTATT-3′ | 132bp | 60 °C |
| Tie2 | 5′-AAGCATGCCCATCTGGTTAC-3′ 5′-GCCTGCCTTCTTTCTCACAC-3′ | 138bp | 60 °C |
| Pecam1 | 5′-GCACCCATCACTTACCACCT-3′ 5′-GCTCGTCCCCTCTTTCACA-3′ | 279bp | 60 °C |
| Icam2 | 5′-CATCCTCAAGGGAAGTGGAA-3′ 5′-ACTTGAGCTGGAGGCTGGTA-3′ | 137bp | 60 °C |
| m Endoglin | 5′-CTTCCAAGGACAGCCAAGAG-3′ 5′-GTGGTTGCCATTCAAGTGTG-3′ | 221bp | 60 °C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galaris, G.; Montagne, K.; Thalgott, J.H.; Goujon, G.J.P.E.; van den Driesche, S.; Martin, S.; Mager, H.-J.J.; Mummery, C.L.; Rabelink, T.J.; Lebrin, F. Thresholds of Endoglin Expression in Endothelial Cells Explains Vascular Etiology in Hereditary Hemorrhagic Telangiectasia Type 1. Int. J. Mol. Sci. 2021, 22, 8948. https://doi.org/10.3390/ijms22168948
Galaris G, Montagne K, Thalgott JH, Goujon GJPE, van den Driesche S, Martin S, Mager H-JJ, Mummery CL, Rabelink TJ, Lebrin F. Thresholds of Endoglin Expression in Endothelial Cells Explains Vascular Etiology in Hereditary Hemorrhagic Telangiectasia Type 1. International Journal of Molecular Sciences. 2021; 22(16):8948. https://doi.org/10.3390/ijms22168948
Chicago/Turabian StyleGalaris, Georgios, Kévin Montagne, Jérémy H. Thalgott, Geoffroy J. P. E. Goujon, Sander van den Driesche, Sabrina Martin, Hans-Jurgen J. Mager, Christine L. Mummery, Ton J. Rabelink, and Franck Lebrin. 2021. "Thresholds of Endoglin Expression in Endothelial Cells Explains Vascular Etiology in Hereditary Hemorrhagic Telangiectasia Type 1" International Journal of Molecular Sciences 22, no. 16: 8948. https://doi.org/10.3390/ijms22168948
APA StyleGalaris, G., Montagne, K., Thalgott, J. H., Goujon, G. J. P. E., van den Driesche, S., Martin, S., Mager, H.-J. J., Mummery, C. L., Rabelink, T. J., & Lebrin, F. (2021). Thresholds of Endoglin Expression in Endothelial Cells Explains Vascular Etiology in Hereditary Hemorrhagic Telangiectasia Type 1. International Journal of Molecular Sciences, 22(16), 8948. https://doi.org/10.3390/ijms22168948