
Transcriptomic Analysis Reveals Key Genes Involved in Oil and Linoleic Acid Biosynthesis during Artemisia sphaerocephala Seed Development


Abstract
:1. Introduction
2. Results and Discussion
2.1. Morphological Characteristics and Oil Accumulation during A. sphaerocephala Seed Development
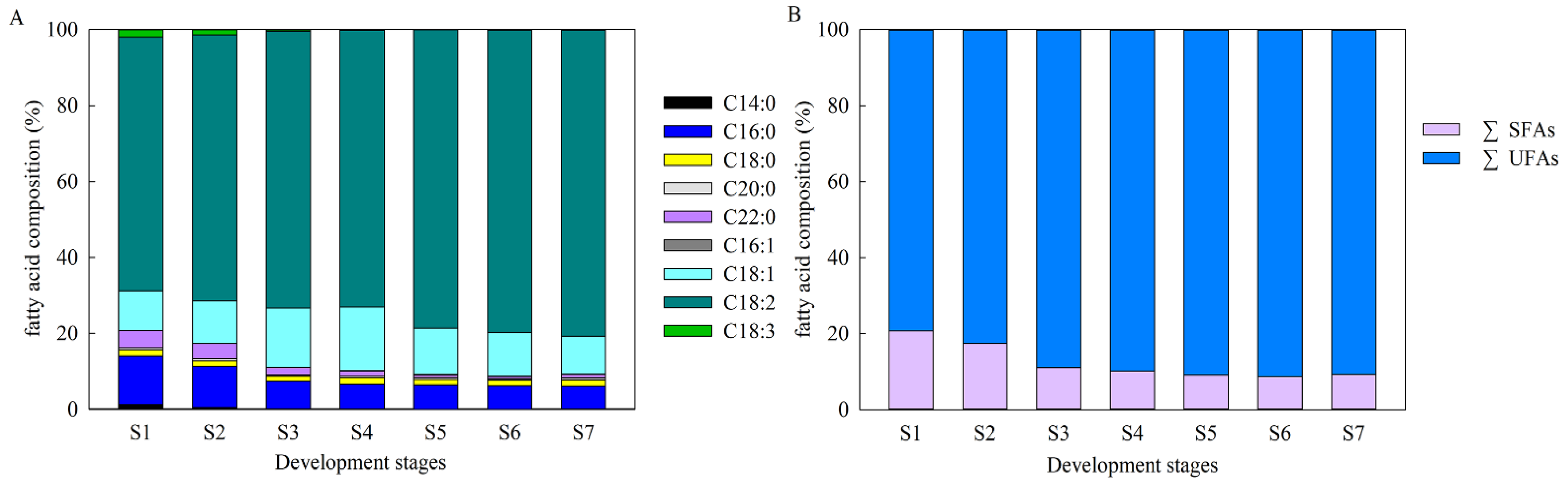
2.2. Characteristics of FA Compositions during A. sphaerocephala Seed Development
2.3. Transcriptomic Analysis and Functional Annotation
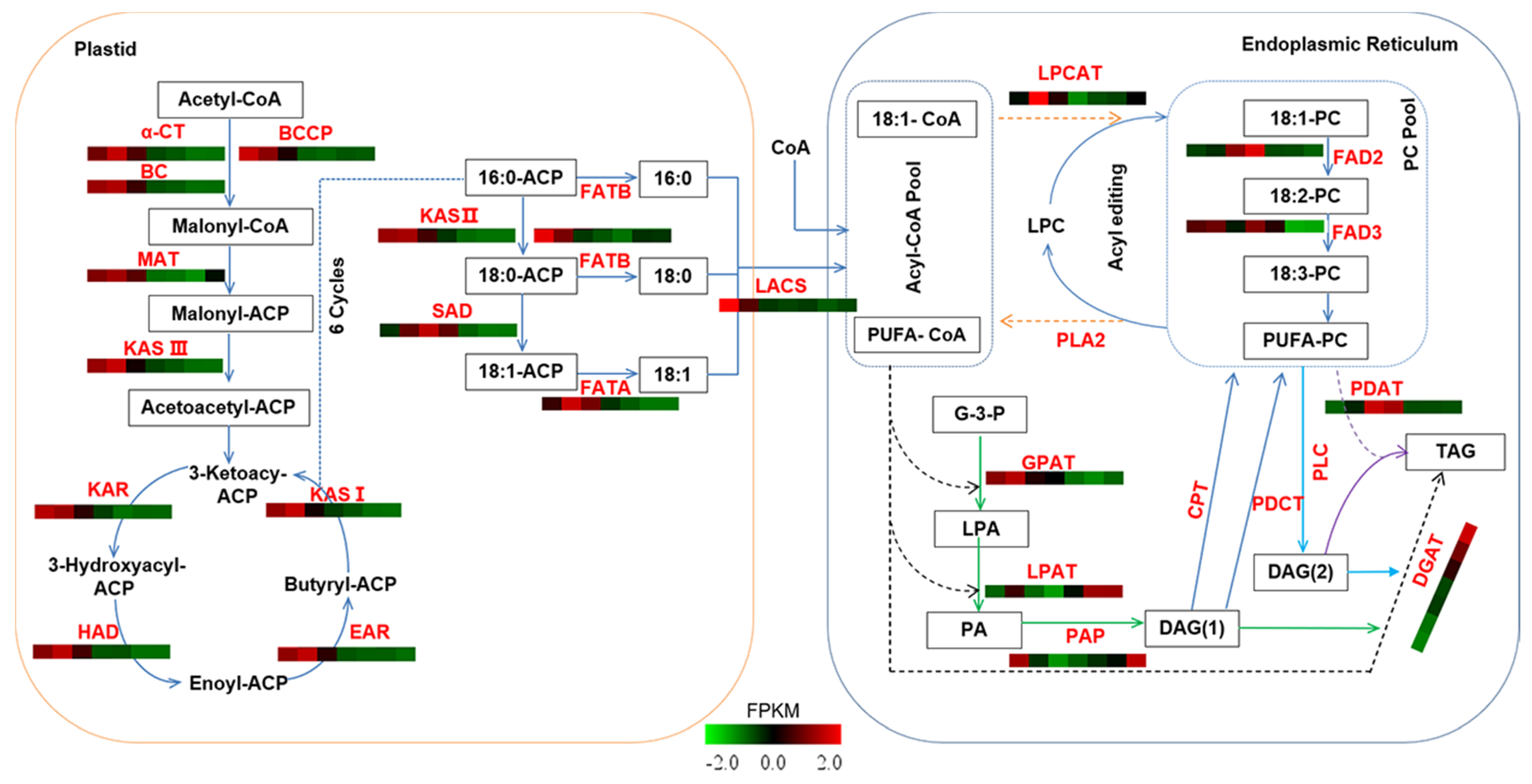
2.4. Identification of Candidate Genes Involved in Oil Biosynthesis
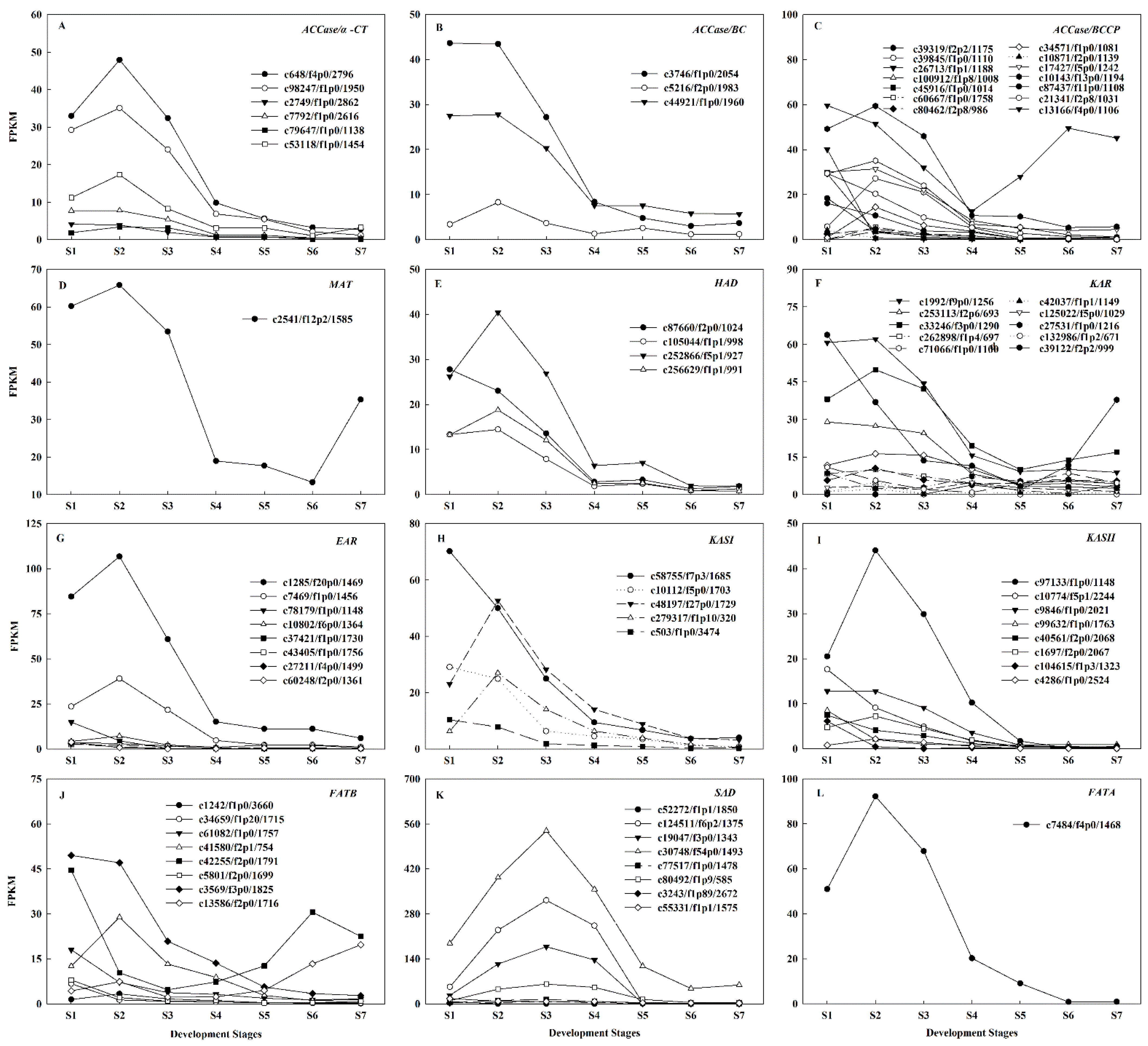
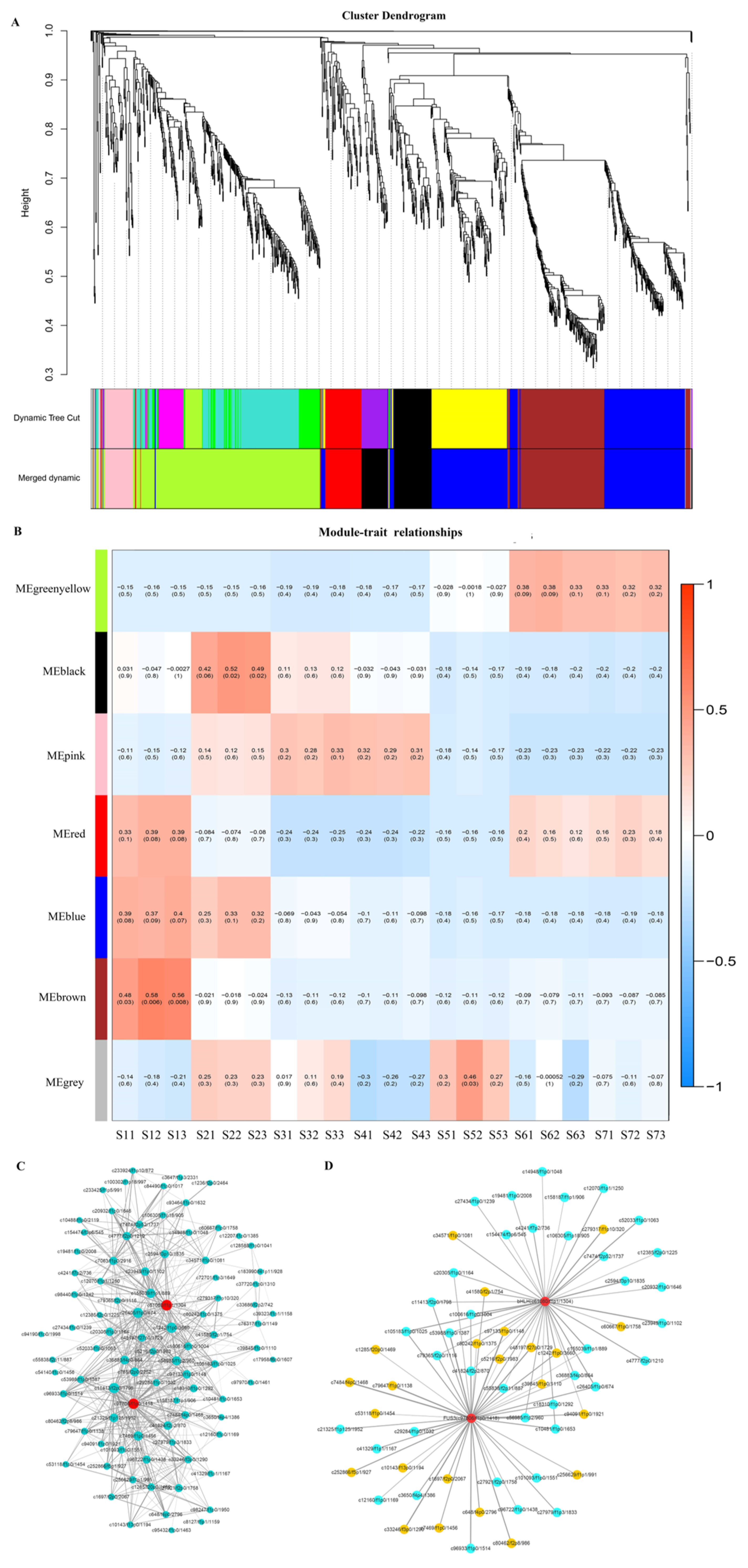
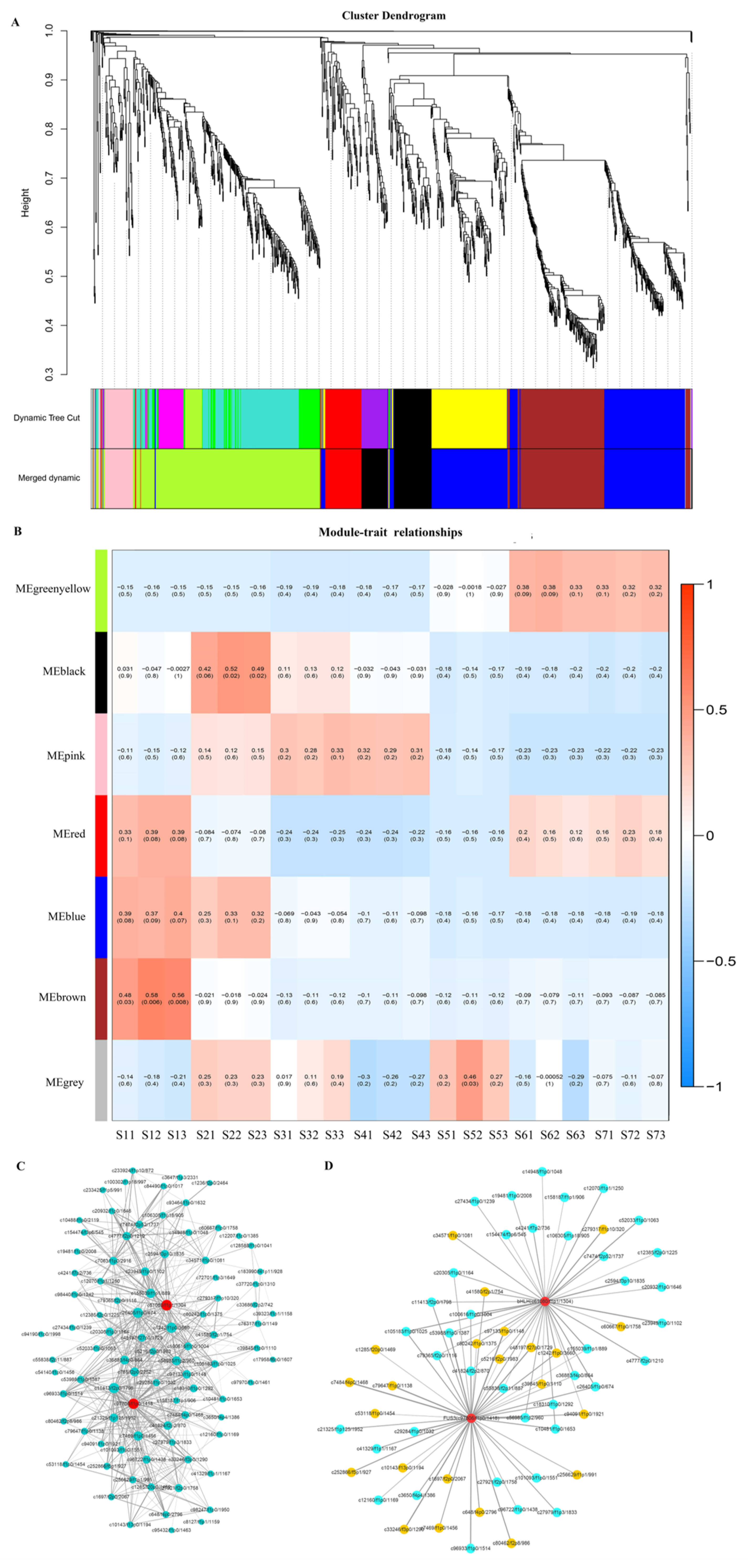
2.5. Regulatory Mechanisms of High Oil Accumulation during Early Seed Development
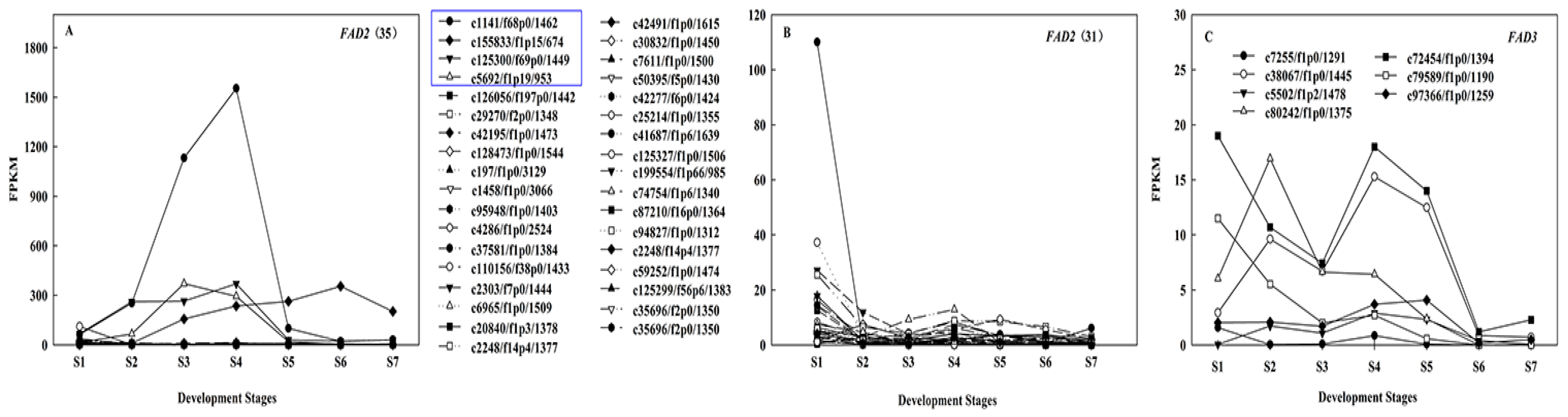
2.6. Regulation Mechanism of High C18:2 Content in Seed Oil
2.7. Regulation of Transcription Factors on Oil Accumulation
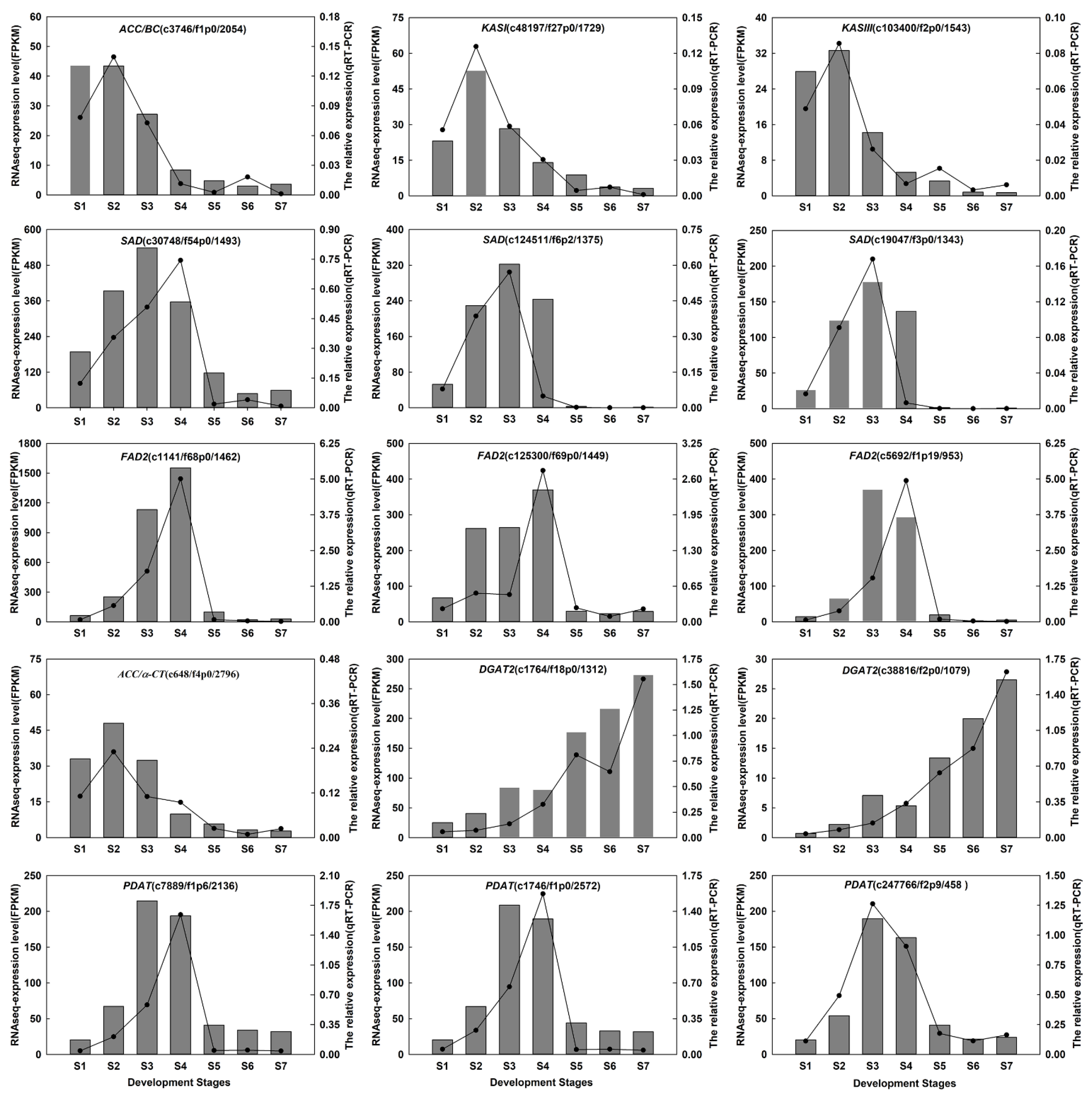
2.8. Validation of RNA Sequencing Results by RT-qPCR
3. Materials and Methods
3.1. Plant Materials
3.2. Oil Content Determination and Fatty Acid Analysis
3.3. RNA Extraction and Assessment
3.4. Illumina Transcriptome Library Preparation, Sequencing, and Data Analysis
3.5. PacBio Iso-Seq Library Construction, Sequencing, and Data Analysis
3.6. Transcriptome Sequence Correction and De-Redundancy
3.7. Functional Annotation and Enrichment Analysis
3.8. Quantification of Gene Expression Levels and Identification of DEGs
3.9. Quantitative Real Time PCR (RT-qPCR) Analysis
3.10. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACCase | acetyl-CoA carboxylase |
| BC | biotin carboxylase |
| BCCP | biotin carboxyl carrier protein |
| α/β-CT | α/β-carboxyltransferase |
| ACP | acyl carrier protein |
| MAT | malonyl-CoA ACP S-malonytransferase |
| KAS | 3-ketoacyl-ACP synthase |
| KAR | 3-ketoacyl-ACP reductase |
| HAD | 3-hydroxyacyl-ACP dehydratase |
| EAR | enoyl-ACP reductase |
| SAD | stearoyl-ACP desaturase |
| FAT | fatty acyl-ACP thioesterase |
| LACS | long chain acyl-CoA |
| GPAT | glycerol-3-phosphate acyltransferase |
| PDAT | phospholipid: diacylglycerol acyltransferase |
| LPAT | 1ysophosphatidic acid acyltransferase |
| DGAT | acyl-CoA: diacylglycerol acyltransferase |
| PDCT | phosphatidylcholine:diacylglycerol cholinephosphotransferase |
| CPT | diacylglycerol cholinephosphotransferas |
| LPCAT | lysophosphatidylcholine acyltransferase |
| PLA2 | phospholipase A2 |
| PAP | phosphate phosphatase |
| FAD2 | omega-6 desaturase |
| FAD3 | omega-3 desaturase |
| G-3-P | Glycerol-3-phosphate |
| LPA | Lyso-phosphatidic acid |
| PA | Phosphatidic acid |
| PC | phosphatidylcholine |
| LPC | lysophosphatidylcholine |
| DAG | 1,2-Diacylglycerol |
| TAG | Triacylglycerol |
| FA | fatty acid |
| C16:0 | palmitic acid |
| C18:0 | stearic acid |
| C18:1 | oleic acid |
| C18:2 | linoleic acid |
| C18:3 | linolenic acid |
| C20:0 | arachidic acid |
| C22:0 | behenic acid |
| CLA | conjugated linoleic acid |
| UFAs | unsaturated fatty acid |
| SFAs | Saturated fatty acid |
| WRI1 | WRINKLED1 |
| FUS3 | FUSCA3 |
| ABI3 | ABSCISIC ACID INSENSITIVE 3 |
| LEC1 | LEAFY COTYLEDON1 |
References
- Kaur, N.; Chugh, V.; Gupta, A.K. Essential fatty acids as functional components of foods-a review. J. Food Sci. Technol. 2014, 51, 2289–2303. [Google Scholar] [CrossRef] [Green Version]
- Den Hartigh, L.J. Conjugated linoleic acid effects on cancer, obesity, and atherosclerosis: A review of pre-clinical and human trials with current perspectives. Nutrients 2019, 11, 370. [Google Scholar] [CrossRef] [Green Version]
- Goettel, W.; Xia, E.; Upchurch, R.; Wang, M.L.; Chen, P.; An, Y.Q.C. Identification and characterization of transcript polymorphisms in soybean lines varying in oil composition and content. BMC Genomics 2014, 15, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, H.F.; Cui, Y.X.; Ding, Y.J.; Mei, J.Q.; Dong, H.L.; Zhang, W.X.; Wu, S.Q.; Liang, Y.; Zhang, C.Y.; Li, J.N.; et al. Time-series analyses of transcriptomes and proteomes reveal molecular networks underlying oil accumulation in Canola. Front. Plant Sci. 2017, 7, 2007. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.M.; Zhou, Y.; Zhang, J.P.; Ji, F.Y.; Jin, F.; Fan, W.; Pei, D. Transcriptome analysis of Walnut (Juglans regia L.) embryos reveals key developmental stages and genes involved in lipid biosynthesis and polyunsaturated fatty acid metabolism. J. Agric. Food Chem. 2021, 69, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, H.F.; Gu, J.Z.; Deng, L.; Ren, L.; Hong, Y.B.; Lu, Q.; Chen, X.P.; Liang, X.Q. Identification of the candidate proteins related to oleic acid accumulation during Peanut (Arachis hypogaea L.) seed development through comparative proteome analysis. Int. J. Mol. Sci. 2018, 19, 1235. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.P.; Wang, Y.M.; Huang, Y.; Cui, Y.P.; Hua, J.P. Gene network of oil accumulation reveals expression profiles in developing embryos and fatty acid composition in Upland cotton. J. Plant Physiol. 2018, 228, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.D.; Stymne, S.; Ohlrogge, J. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Li-Beisson, Y.; Shorrosh, B.; Beisson, F.; Andersson, M.X.; Arondel, V.; Bates, P.D.; Baud, S.; Bird, D.; Debono, A.; Durrett, T.P.; et al. Acyl-lipid metabolism. Arab. Book 2013, 11, e0161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuley, J.; Lightner, J.; Feldmann, K.; Yadav, N.; Lark, E.; Browse, J. Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid-synthesis. Plant Cell 1994, 6, 147–158. [Google Scholar] [CrossRef]
- Arondel, V.; Lemieux, B.; Hwang, I.; Gibson, S.; Goodman, H.M.; Somerville, C.R. Map-based cloning of a gene controlling omega-3-fatty-acid desaturation in Arabidopsis. Science 1992, 258, 1353–1355. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; An, J.Y.; Wang, L.B.; Fang, C.L.; Ha, D.L.; Fu, C.Y.; Qiu, L.; Yu, H.Y.; Zhao, H.Y.; Hou, X.Y.; et al. Transcriptomic analysis revealed the mechanism of oil dynamic accumulation during developing Siberian apricot (Prunus sibirica L.) seed kernels for the development of woody biodiesel. Biotechnol. Biofuels 2015, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Q.; Zhang, T.; Zhang, Q.X.; Chen, M.; Wang, Z.J.; Zheng, B.S.; Xia, G.H.; Yang, X.Y.; Huang, C.Y.; Huang, Y.J. The mechanism of high contents of oil and oleic acid revealed by transcriptomic and lipidomic analysis during embryogenesis in Carya cathayensis Sarg. BMC Genomics 2016, 17, 113. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Wang, K.L.; Zhou, C.F.; Xie, Y.H.; Yao, X.H.; Yin, H.F. Seed transcriptomics analysis in Camellia oleifera uncovers genes associated with oil content and fatty acid composition. Int. J. Mol. Sci. 2018, 19, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.S.; Wang, L.S.; Shu, Q.Y.; Wu, J.; Chen, L.G.; Shao, S.; Yin, D.D. Fatty acid composition of developing tree peony (Paeonia section Moutan DC.) seeds and transcriptome analysis during seed development. BMC Genomics 2015, 16, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.U.; Lee, K.R.; Shim, D.; Lee, J.H.; Chen, G.Q.; Hwang, S. Transcriptome analysis and identification of genes associated with omega-3 fatty acid biosynthesis in Perilla frutescens (L.) var. frutescens. BMC Genomics 2016, 17, 474. [Google Scholar] [CrossRef] [Green Version]
- Correa, S.M.; Fernie, A.R.; Nikoloski, Z.; Brotman, Y. Towards model-driven characterization and manipulation of plant lipid metabolism. Prog. Lipid Res. 2020, 80, 101051. [Google Scholar] [CrossRef]
- Troncoso-Ponce, M.A.; Kilaru, A.; Cao, X.; Durrett, T.P.; Fan, J.; Jensen, J.K.; Thrower, N.A.; Pauly, M.; Wilkerson, C.; Ohlrogge, J.B. Comparative deep transcriptional profiling of four developing oilseeds. Plant J. 2011, 68, 1014–1027. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Guschina, I.A.; O′Hara, P.; Slabas, A.R.; Quant, P.A.; Fawcett, T.; Harwood, J.L. Metabolic control analysis of developing oilseed rape (Brassica napus cv. Westar) embryos shows that lipid assembly exerts significant control over oil accumulation. New Phytol. 2012, 196, 414–426. [Google Scholar] [CrossRef] [Green Version]
- Woodfield, H.K.; Cazenave-Gassiot, A.; Haslam, R.P.; Guschina, I.A.; Wenk, M.R.; Harwood, J.L. Using lipidomics to reveal details of lipid accumulation in developing seeds from oilseed rape (Brassica napus L.). BBA Mol. Cell Biol. Lipids 2018, 1863, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.D.; Browse, J. The pathway of triacylglycerol synthesis through phosphatidylcholine in Arabidopsis produces a bottleneck for the accumulation of unusual fatty acids in transgenic seeds. Plant J. 2011, 68, 387–399. [Google Scholar] [CrossRef]
- Bates, P.D.; Browse, J. The significance of different diacylgycerol synthesis pathways on plant oil composition and bioengineering. Front. Plant Sci. 2012, 3, 147. [Google Scholar] [CrossRef] [Green Version]
- Baud, S.; Lepiniec, L. Physiological and developmental regulation of seed oil production. Prog. Lipid Res. 2010, 49, 235–249. [Google Scholar] [CrossRef]
- Zafar, S.; Li, Y.L.; Li, N.N.; Zhu, K.M.; Tan, X.L. Recent advances in enhancement of oil content in oilseed crops. J. Biotechnol. 2019, 301, 35–44. [Google Scholar] [CrossRef]
- Editorial Board of the Flora of China of Chinese Academy of Sciences. Flora of China; Science Press: Beijing, Chain, 1991; Volume 76, pp. 195–197. [Google Scholar]
- Han, X.X.; Zhang, L.J.; Miao, X.M.; Hu, X.W.; Nan, S.Z.; Fu, H. Transcriptome analysis reveals the molecular mechanisms of mucilage biosynthesis during Artemisia sphaerocephala seed development. Ind. Crop. Prod. 2020, 145, 111991. [Google Scholar] [CrossRef]
- Yang, X.J.; Baskin, J.M.; Baskin, C.C.; Huang, Z.Y. More than just a coating: Ecological importance.; taxonomic occurrence and phylogenetic relationships of seed coat mucilage. Perspect. Plant Ecol. Evol. Syst. 2012, 14, 434–442. [Google Scholar] [CrossRef]
- Kakar, M.U.; Kakar, I.U.; Mehboob, M.Z.; Zada, S.; Soomro, H.; Umair, M.; Iqbal, I.; Umer, M.; Shaheen, S.; Syed, S.F.; et al. A review on polysaccharides from Artemisia sphaerocephala Krasch seeds, their extraction, modification, structure and applications. Carbohyd. Polym. 2021, 252, 117113. [Google Scholar] [CrossRef] [PubMed]
- Han, X.X.; Zhang, L.J.; Niu, D.C.; Nan, S.Z.; Miao, X.M.; Hu, X.W.; Li, C.S.; Fu, H. Transcriptome and co-expression network analysis reveal molecular mechanisms of mucilage formation during seed development in Artemisia sphaerocephala. Carbohyd. Polym. 2021, 251, 117044. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wang, J.L.; Wang, Z.Q.; Chen, L.P. Fatty acid and amino acid compositions of Artemisia sphaerocephala seed and its influence on mouse hyperlipidemia. Chem. Nat. Compd. 2011, 47, 675–678. [Google Scholar] [CrossRef]
- Zhang, L.J.; Hu, X.W.; Miao, X.M.; Chen, X.L.; Nan, S.Z.; Fu, H. Genome-scale transcriptome analysis of the desert shrub Artemisia sphaerocephala. PLoS ONE 2016, 11, e0154300. [Google Scholar] [CrossRef] [Green Version]
- Hymowitz, T.J.; Dudley, J.W.; Collins, F.I.; Brown, C.M. Estimations of protein and oil concentration in corn.; soybean.; and oat seed by near-infrared light reflectance1. Crop Sci. 1974, 14, 713. [Google Scholar] [CrossRef]
- Uzun, B.; Arslan, C.; Furat, S. Variation in fatty acid compositions.; oil content and oil yield in a germplasm collection of Sesame (Sesamum indicum L.). J. Am. Oil Chem. Soc. 2008, 85, 1135–1142. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Y.P.; Chen, J.Z.; Li, P.W.; Li, C.Z.; Niu, G.H.; Jiang, L.J. Transcriptome analysis revealed the dynamic oil accumulation in Symplocos paniculata fruit. BMC Genomics 2016, 17, 929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banas, W.; Sanchez Garcia, A.; Banas, A.; Stymne, S. Activities of acyl-CoA:diacylglycerol acyltransferase (DGAT) and phospholipid:diacylglycerol acyltransferase (PDAT) in microsomal preparations of developing sunflower and safflower seeds. Planta 2013, 237, 1627–1636. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.Z.; Lou, H.Q.; Chen, W.C.; Zhou, Y.; Zhang, Z.H.; Xiao, M.H.; Wang, Z.Q.; Yang, Y.; Yang, L.; Zhang, F.C.; et al. Comparative transcriptome analysis of the genes involved in lipid biosynthesis pathway and regulation of oil body formation in Torreya grandis kernels. Ind. Crop. Prod. 2020, 145, 112051. [Google Scholar] [CrossRef]
- Liao, B.N.; Hao, Y.J.; Lu, J.X.; Bai, H.Y.; Guan, L.; Zhang, T. Transcriptomic analysis of Perilla frutescens seed to insight into the biosynthesis and metabolic of unsaturated fatty acids. BMC Genomics 2018, 19, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.K.; Cao, Y.Y.; Chen, C.; Gao, Z.Z.; Yu, F.Y.; Guy, R.D. Transcriptome analysis of metabolic pathways associated with oil accumulation in Developing seed kernels of Styrax tonkinensis, a woody biodiesel species. BMC Plant Biol. 2020, 20, 121. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Wu, L.M.; Li, Y.H.; Huang, H.L.; Qian, M.C.; Sun, W.; Zhu, H.; Xu, Y.F.; Fan, Y.H.; Mahmood, U.; et al. Deciphering the transcriptional regulatory networks that control size, color, and oil content in Brassica rapa seeds. Biotechnol. Biofuels 2020, 13, 90. [Google Scholar] [CrossRef]
- Gong, W.F.; Song, Q.L.; Ji, K.; Gong, S.F.; Wang, L.K.; Chen, L.; Zhang, J.; Yuan, D.Y. Full-length transcriptome from Camellia oleifera seed provides insight into the transcript variants involved in oil biosynthesis. J. Agric. Food Chem. 2020, 68, 14670–14683. [Google Scholar] [CrossRef]
- Ge, Y.; Dong, X.S.; Liu, Y.Z.; Yang, Y.; Zhan, R.L. Molecular and biochemical analyses of avocado (Persea americana) reveal differences in the oil accumulation pattern between the mesocarp and seed during the fruit developmental period. Sci. Hortic. 2021, 276, 109717. [Google Scholar] [CrossRef]
- Mao, Z.X.; Fu, H.; Nan, Z.B.; Wang, J.; Wan, C.G. Fatty acid content of common vetch (Vicia sativa L.) in different regions of Northwest China. Biochem. Syst. Ecol. 2012, 44, 347–351. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.T.; Ntagkas, N.; Fanourakis, D.; Tsaniklidis, G.; Zhao, J.T.; Cheng, R.F.; Yang, Q.C.; Li, T. The role of light intensity in mediating ascorbic acid content during postharvest tomato ripening: A transcriptomic analysis. Postharvest Biol. Technol. 2021, 180, 111622. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.W.; Zhang, L.J.; Nan, S.Z.; Miao, X.M.; Yang, P.F.; Duan, G.Q.; Fu, H. Selection and validation of reference genes for quantitative real-time PCR in Artemisia sphaerocephala based on transcriptome sequence data. Gene 2018, 657, 39–49. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detabase | Pathway Level | Number (Percentage (%)) | ||
|---|---|---|---|---|
| KEGG | 21,525 (25.55%) | |||
| metabolism | 9823 (45.64%) | |||
| Lipid metabolism | 856 (8.71%) | |||
| Glycerolipid metabolism | 149 | |||
| Glycerophospholipid metabolism | 168 | |||
| Sphingolipid metabolism | 48 | |||
| Steroid biosynthesis | 78 | |||
| Ether lipid metabolism | 39 | |||
| Synthesis and degradation of ketone bodies | 25 | |||
| Fatty acid biosynthesis | 137 | |||
| Biosynthesis of unsaturated fatty acids | 293 | |||
| Arachidonic acid metabolism | 52 | |||
| alpha-Linolenic acid metabolism | 46 | |||
| Linoleic acid metabolism | 1 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, S.; Zhang, L.; Hu, X.; Miao, X.; Han, X.; Fu, H. Transcriptomic Analysis Reveals Key Genes Involved in Oil and Linoleic Acid Biosynthesis during Artemisia sphaerocephala Seed Development. Int. J. Mol. Sci. 2021, 22, 8369. https://doi.org/10.3390/ijms22168369
Nan S, Zhang L, Hu X, Miao X, Han X, Fu H. Transcriptomic Analysis Reveals Key Genes Involved in Oil and Linoleic Acid Biosynthesis during Artemisia sphaerocephala Seed Development. International Journal of Molecular Sciences. 2021; 22(16):8369. https://doi.org/10.3390/ijms22168369
Chicago/Turabian StyleNan, Shuzhen, Lijing Zhang, Xiaowei Hu, Xiumei Miao, Xiaoxu Han, and Hua Fu. 2021. "Transcriptomic Analysis Reveals Key Genes Involved in Oil and Linoleic Acid Biosynthesis during Artemisia sphaerocephala Seed Development" International Journal of Molecular Sciences 22, no. 16: 8369. https://doi.org/10.3390/ijms22168369