
Nicotinamide N-Methyltransferase in Acquisition of Stem Cell Properties and Therapy Resistance in Cancer

 , and
, and 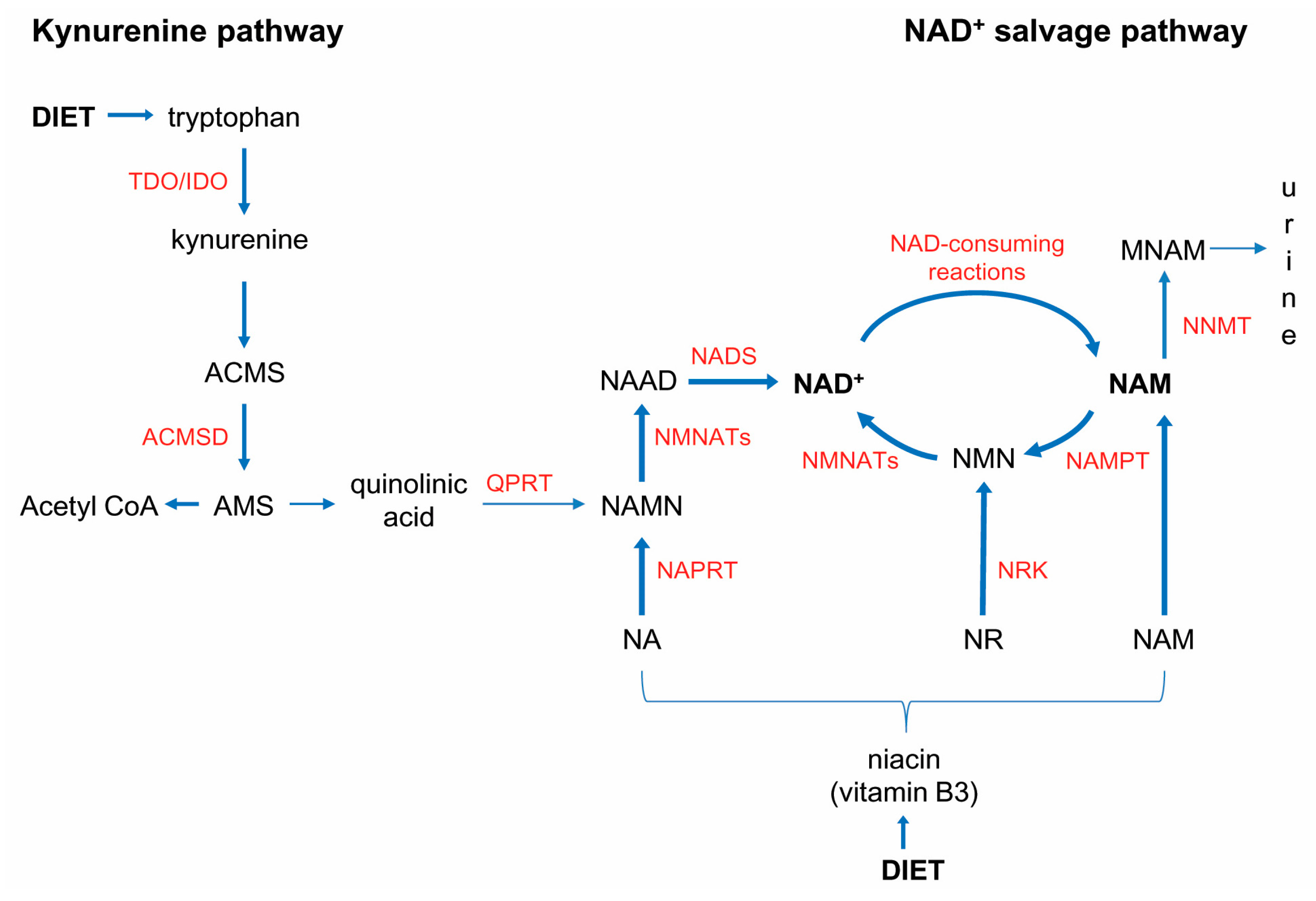{kind=link}
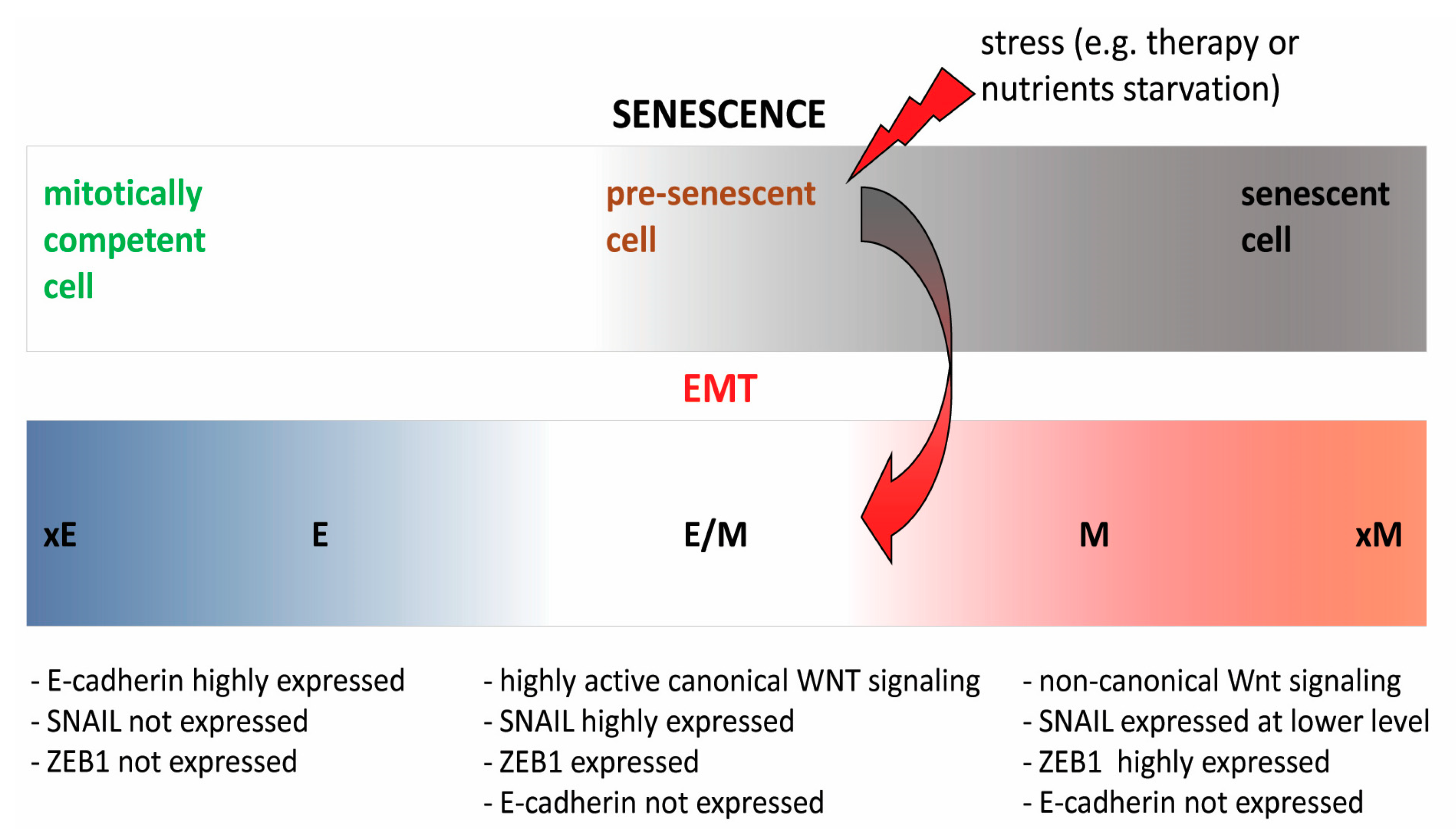{kind=link}
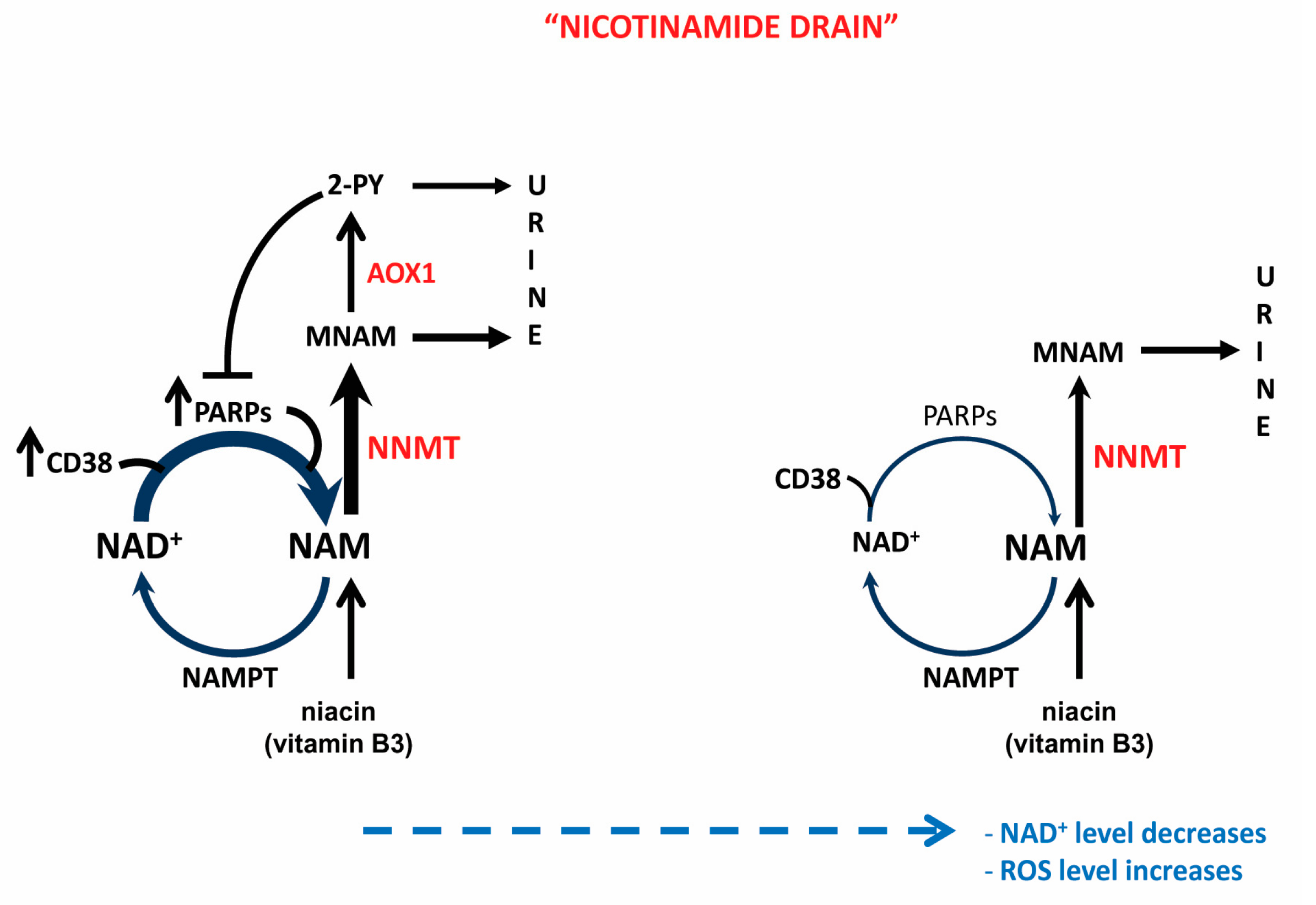{kind=link}
Abstract
1. Introduction
2. Cellular Senescence, Circumventing Senescence, and the Onset of Cancer Therapy Resistance
2.1. Dual Roles of Cellular Senescence in Cancer
2.2. Acquisition of Stem Cell Properties in Cancer
2.3. Interwoven Processes of Senescence and EMT—A Major Culprit in Therapy Resistance
3. The Role of NNMT in Acquisition of Stem Cell Properties and Therapy Resistance
3.1. NNMT
3.2. NNMT-Mediated “Nicotinamide Drain” and NAD+ Depletion
3.3. NNMT and Pluripotency
3.4. NNMT and Epigenomic Reprogramming in Cancer
3.5. The Role of NNMT in Acquisition of Metabolic Plasticity and Stem Cell Properties in Cancer
4. ROS and EMT
4.1. Aldehyde Oxidase (AOX1)
The Role of AOX1 in Cancer
5. Concluding Remarks and Open Questions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-PY | N1-methyl-2-pyridone-5-carboxamide |
| 4-PY | N1-methyl-4-pyridone-3-carboxamide |
| AADPR | Acetyl Adenosinediphosphate Ribose |
| ACMS | Alpha-Amino-Beta-Carboxy-Muconate Semialdehyde |
| ACMSD | Alpha-Amino-Beta-Carboxy-Muconate Semialdehyde Decarboxylase |
| ADP | Adenosine Diphosphate |
| ADPR | Adenosinediphosphate Ribose |
| Ahcy | Adenosylhomocysteinase |
| AMS | Aminomuconate Semialdehyde |
| AOX1 | Aldehyde Oxidase 1 |
| APC | Adenomatous Polyposis Coli Protein |
| BCL2 | B-cell Lymphoma 2 |
| Bhmt | Betaine-Homocysteine Methyltransferase |
| BRCA1 | Breast Cancer Type 1 Susceptibility Protein |
| cADPR | cyclic Adenosinediphosphate Ribose |
| CAF | Cancer Associated Fibroblasts |
| CD133 | Cluster of Differentiation 133 |
| CD38 | Cluster of Differentiation 38 |
| CD44 | Cluster of Differentiation 44 |
| CD44s | CD44 standard |
| CD44v | CD44 variable |
| CDH1 | E-Cadherin |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| CSC | Cancer Stem Cells |
| DRD2 | Dopamine Receptor 2 |
| EGLN1 | Egl Nine Homolog 1 |
| EMT | Epithelial to Mesenchymal Transition |
| EMT-TFs | EMT-Inducing Transcription Factors |
| ESRP1 | Epithelial Splicing Regulatory Protein 1 |
| EZH1 | Enhancer of Zeste 1 |
| EZH2 | Enhancer of Zeste 2 |
| GAD3 | Probable Aldehyde Oxidase gad-3 |
| GSH | Glutathione |
| GSK3β | Glycogen Synthase Kinase 3β |
| GSSG | Oxidized Glutathione |
| hESC | human Embryonic Stem Cells |
| HGSC | High Grade Ovarian Serous Carcinoma |
| HIF1α | Hypoxia Inducible Factor 1 Alpha |
| HNCC | Head and Neck Carcinoma Cells |
| HNF1 β | Hepatocyte Nuclear Factor 1β |
| IDO | Indoleamine 2,3-dioxygenase |
| IL-6 | Interleukin 6 |
| iPSC | induced Pluripotent Stem Cells |
| LCMT1 | Leucine Carboxyl Methyltransferase 1 |
| LCMT1 | Leucine Carboxyl Methyl Transferase 1 |
| mAOX1 | mouse Aldehyde Oxidase 1 |
| MAPK | Mitogen-Activated Protein Kinase |
| Mat1a | Methionine Adenosyltransferase I alpha |
| MET | Mesenchymal to Epithelial Transition |
| MNAM | 1-Methylnicotinamide |
| MOCOS | Molybdenium Cofactor Sulfurase |
| NA | Nicotinic acid |
| NAAD | Nicotinic Acid Adenine Dinucleotide |
| NAD+ | Nicotinamide Adenine Dinucleotide |
| NADS | Nicotinamide Adenine Dinucleotide Synthetase |
| NAM | Nicotinamide |
| NAMN | Nicotinic Acid Mononucleotide |
| NAMPT | Nicotinamide Phosphoribosyltransferase |
| NAPRT | Nicotinic Acid Phosphoribosyltransferase |
| NMN | Nicotinamide Mononucleotide |
| NMNAT | Nicotinamide Mononucleotide Adenylyl-Transferase |
| NNMT | Nicotinamide N-Methyltransferase |
| NR | Nicotinamide Riboside |
| NRK | Nicotinamide Riboside Kinase |
| NRF1 | NF-E2-Related Factor 1 |
| NRF2 | NF-E2-Related Factor 2 |
| OXPHOS | Oxidative Phosphorylation |
| PARP | Poly [ADP-ribose] Polymerase |
| PHD2 | Prolyl Hydroxylase 2 |
| PI3K | Phosphatidylinositol 3-Kinase |
| PP2A | Protein Phosphatase 2 |
| QPRT | Quinolinate Phosphoribosyltransferase |
| ROS | Reactive Oxygen Species |
| SAM | S-Adenosyl Methionine |
| SASP | Senescence-Associated Secretory Phenotype |
| SIRT1 | NAD-Dependent Protein Deacetylase Sirtuin-1 |
| SNP | Single Nucleotide Polymorphism |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| SUV39H1 | Suppressor of Variegation 3-9 Homolog 1 |
| TDO | Tryptophan 2,3-dioxygenase |
| TGFβ | Transforming Growth Factor-β |
| TIS | Therapy-Induced Senescence |
| TME | Tumor Microenvironment |
| TP53 | Tumor Suppressor p53 |
| Trp | Tryptophan |
| ZEB1 | Zinc Finger E-box-Binding Homeobox 1 |
References
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Sarmento-Ribeiro, A.B.; Scorilas, A.; Gonçalves, A.C.; Efferth, T.; Trougakos, I.P. The emergence of drug resistance to targeted cancer therapies: Clinical evidence. Drug Resist. Updates 2019, 47, 100646. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.L.; Francescangeli, F.; La Torre, F.; Zeuner, A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front. Oncol. 2019, 9, 626. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Deschênes-Simard, X.; Parisotto, M.; Rowell, M.C.; Le Calvé, B.; Igelmann, S.; Moineau-Vallée, K.; Saint-Germain, E.; Kalegari, P.; Bourdeau, V.; Kottakis, F.; et al. Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 2019, 18, e12889. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Freinkman, E.; Comb, W.C.; Cantor, J.R.; Tam, W.L.; Thiru, P.; Kim, D.; Kanarek, N.; Pacold, M.E.; Chen, W.W.; et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 2014, 158, 1094–1109. [Google Scholar] [CrossRef] [PubMed]
- Guillot, B.; Lecomte, C.; Cousson, A.; Scherf, C.; Jelsch, C. High-resolution neutron structure of nicotinamide adenine dinucleotide. Acta Cryst. D Biol. Cryst. 2001, 57, 981–989. [Google Scholar] [CrossRef]
- Yang, Y.; Sauve, A.A. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [PubMed]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Strømland, Ø.; Niere, M.; Nikiforov, A.A.; VanLinden, M.R.; Heiland, I.; Ziegler, M. Keeping the balance in NAD metabolism. Biochem. Soc. Trans. 2019, 47, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef]
- Bütepage, M.; Eckei, L.; Verheugd, P.; Lüscher, B. Intracellular Mono-ADP-Ribosylation in Signaling and Disease. Cells 2015, 4, 569–595. [Google Scholar] [CrossRef] [PubMed]
- Osborne, B.; Bentley, N.L.; Montgomery, M.K.; Turner, N. The role of mitochondrial sirtuins in health and disease. Free Radic. Biol. Med. 2016, 100, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef]
- Bockwoldt, M.; Houry, D.; Niere, M.; Gossmann, T.I.; Reinartz, I.; Schug, A.; Ziegler, M.; Heiland, I. Identification of evolutionary and kinetic drivers of NAD-dependent signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 15957–15966. [Google Scholar] [CrossRef]
- Burgos, E.S.; Schramm, V.L. Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry 2008, 47, 11086–11096. [Google Scholar] [CrossRef]
- Palzer, L.; Bader, J.J.; Angel, F.; Witzel, M.; Blaser, S.; McNeil, A.; Wandersee, M.K.; Leu, N.A.; Lengner, C.J.; Cho, C.E.; et al. Alpha-Amino-Beta-Carboxy-Muconate-Semialdehyde Decarboxylase Controls Dietary Niacin Requirements for NAD+ Synthesis. Cell Rep. 2018, 25, 1359–1370.e4. [Google Scholar] [CrossRef] [PubMed]
- Fukuwatari, T.; Ohta, M.; Kimtjra, N.; Sasaki, R.; Shibata, K. Conversion ratio of tryptophan to niacin in Japanese women fed a purified diet conforming to the Japanese Dietary Reference Intakes. J. Nutr. Sci. Vitaminol. 2004, 50, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD metabolome-a key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Kincaid, J.W.; Berger, N.A. NAD metabolism in aging and cancer. Exp. Biol. Med. 2020, 245, 1594–1614. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef]
- Menon, D.R.; Hammerlindl, H.; Torrano, J.; Schaider, H.; Fujita, M. Epigenetics and metabolism at the crossroads of stress-induced plasticity, stemness and therapeutic resistance in cancer. Theranostics 2020, 10, 6261–6277. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef]
- Martins-Neves, S.R.; Cleton-Jansen, A.M.; Gomes, C.M.F. Therapy-induced enrichment of cancer stem-like cells in solid human tumors: Where do we stand? Pharmacol. Res. 2018, 137, 193–204. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Bhang, H.E.; Ruddy, D.A.; Krishnamurthy Radhakrishna, V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P.; Werdell, J.; Santner, S.J.; Pauley, R.J.; Tait, L. Breast stroma plays a dominant regulatory role in breast epithelial growth and differentiation: Implications for tumor development and progression. Cancer Res. 2001, 61, 1320–1326. [Google Scholar]
- Varga, J.; De Oliveira, T.; Greten, F.R. The architect who never sleeps: Tumor-induced plasticity. FEBS Lett. 2014, 588, 2422–2427. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.; Sarma, A.; Nangia-Makker, P.; Shekhar, M.P. Breast cancer complexity: Implications of intratumoral heterogeneity in clinical management. Cancer Metastasis Rev. 2017, 36, 547–555. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef] [PubMed]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Mikuła-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Książek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell. Mol. Life Sci. 2020, 77, 213–229. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mancera, P.A.; Young, A.R.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Guillon, J.; Petit, C.; Toutain, B.; Guette, C.; Lelièvre, E.; Coqueret, O. Chemotherapy-induced senescence, an adaptive mechanism driving resistance and tumor heterogeneity. Cell Cycle 2019, 18, 2385–2397. [Google Scholar] [CrossRef]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Hay, E.D. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 2005, 233, 706–720. [Google Scholar] [CrossRef]
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am. J. Pathol. 2010, 176, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Weinberg, R.A. Targeting the Epithelial-to-Mesenchymal Transition: The Case for Differentiation-Based Therapy. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 11–19. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Ocaña, O.H.; Córcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Lowe, S.W. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef]
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Marcarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602. [Google Scholar] [CrossRef]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Wang, W.; Hu, Y.; Yang, C.; Zhu, S.; Wang, X.; Zhang, Z.; Deng, H. Decreased NAD Activates STAT3 and Integrin Pathways to Drive Epithelial-Mesenchymal Transition. Mol. Cell. Proteom. 2018, 17, 2005–2017. [Google Scholar] [CrossRef]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline. Biochem. Biophys. Res. Commun. 2019, 513, 486–493. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.M.; Long, H. Nicotinamide N-methyltransferase as a potential marker for cancer. Neoplasma 2018, 65, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Pissios, P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Tomida, M.; Ohtake, H.; Yokota, T.; Kobayashi, Y.; Kurosumi, M. Stat3 up-regulates expression of nicotinamide N-methyltransferase in human cancer cells. J. Cancer Res. Clin. Oncol. 2008, 134, 551–559. [Google Scholar] [CrossRef]
- Xu, J.; Capezzone, M.; Xu, X.; Hershman, J.M. Activation of nicotinamide N-methyltransferase gene promoter by hepatocyte nuclear factor-1beta in human papillary thyroid cancer cells. Mol. Endocrinol. 2005, 19, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Kanakkanthara, A.; Kurmi, K.; Ekstrom, T.L.; Hou, X.; Purfeerst, E.R.; Heinzen, E.P.; Correia, C.; Huntoon, C.J.; O’Brien, D.; Wahner Hendrickson, A.E.; et al. BRCA1 Deficiency Upregulates NNMT, Which Reprograms Metabolism and Sensitizes Ovarian Cancer Cells to Mitochondrial Metabolic Targeting Agents. Cancer Res. 2019, 79, 5920–5929. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Hataya, N.; Katsuoka, F.; Yamamoto, M. NF-E2-Related Factor 1 (Nrf1) Serves as a Novel Regulator of Hepatic Lipid Metabolism through Regulation of the Lipin1 and PGC-1β Genes. Mol. Cell. Biol. 2012, 32, 2760–2770. [Google Scholar] [CrossRef]
- Roberti, A.; Fernández, A.F.; Fraga, M.F. Nicotinamide N-methyltransferase: At the crossroads between cellular metabolism and epigenetic regulation. Mol. Metab. 2021, 45, 101165. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liu, H.; Wang, Y.; Zhou, Y.; Yu, H.; Li, G.; Ruan, Z.; Li, F.; Wang, X.; Zhang, J. Nicotinamide N-methyltransferase enhances resistance to 5-fluorouracil in colorectal cancer cells through inhibition of the ASK1-p38 MAPK pathway. Oncotarget 2016, 7, 45837–45848. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, K.; Kanji, S.; Gordon, N.; Thirumoorthy, K.; Jacob, J.R.; Litzenberg, K.T.; Patel, D.; Chakravarti, A. NNMT Silencing Activates Tumor Suppressor PP2A, Inactivates Oncogenic STKs, and Inhibits Tumor Forming Ability. Clin. Cancer Res. 2017, 23, 2325–2334. [Google Scholar] [CrossRef]
- Jung, J.; Kim, L.J.; Wang, X.; Wu, Q.; Sanvoranart, T.; Hubert, C.G.; Prager, B.C.; Wallace, L.C.; Jin, X.; Mack, S.C.; et al. Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight 2017, 2, e90019. [Google Scholar] [CrossRef]
- Slominska, E.M.; Kowalik, K.; Smolenski, R.T.; Szolkiewicz, M.; Rutkowski, P.; Rutkowski, B.; Swierczynski, J. Accumulation of poly(ADP-ribose) polymerase inhibitors in children with chronic renal failure. Pediatr. Nephrol. 2006, 21, 800–806. [Google Scholar] [CrossRef]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic Oxidative Bioenergetics Transitions into Pluripotency-Dependent Glycolysis to Facilitate Nuclear Reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Thompson, C.B. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef]
- Kim, J.; Hong, S.J.; Lim, E.K.; Yu, Y.S.; Kim, S.W.; Roh, J.H.; Do, I.G.; Joh, J.W.; Kim, D.S. Expression of nicotinamide N-methyltransferase in hepatocellular carcinoma is associated with poor prognosis. J. Exp. Clin. Cancer Res. 2009, 28, 20. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Park, C.W.; Yoon, G.; Hong, S.M.; Choi, K.Y. NNMT depletion contributes to liver cancer cell survival by enhancing autophagy under nutrient starvation. Oncogenesis 2018, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; You, S.; Zhang, S.; Hu, Q.; Wang, F.; Chi, X.; Zhao, W.; Xie, C.; Zhang, C.; Yu, Y.; et al. Elevated N-methyltransferase expression induced by hepatic stellate cells contributes to the metastasis of hepatocellular carcinoma via regulation of the CD44v3 isoform. Mol. Oncol. 2019, 13, 1993–2009. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Zhai, B.; Pissios, P. Nicotinamide N-Methyltransferase Interacts with Enzymes of the Methionine Cycle and Regulates Methyl Donor Metabolism. Biochemistry 2018, 57, 5775–5779. [Google Scholar] [CrossRef]
- Eckert, M.A.; Coscia, F.; Chryplewicz, A.; Chang, J.W.; Hernandez, K.M.; Pan, S.; Tienda, S.M.; Nahotko, D.A.; Li, G.; Blaženović, I.; et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 2019, 569, 723–728. [Google Scholar] [CrossRef]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef]
- Hong, S.; Moreno-Navarrete, J.M.; Wei, X.; Kikukawa, Y.; Tzameli, I.; Prasad, D.; Lee, Y.; Asara, J.M.; Fernandez-Real, J.M.; Maratos-Flier, E.; et al. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat. Med. 2015, 21, 887–894. [Google Scholar] [CrossRef]
- Trammell, S.A.; Brenner, C. NNMT: A Bad Actor in Fat Makes Good in Liver. Cell Metab. 2015, 22, 200–201. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Liu, Y.; Liu, X. Nicotinamide N-methyltransferase enhances the progression of prostate cancer by stabilizing sirtuin 1. Oncol. Lett. 2018, 15, 9195–9201. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, J.; Wu, W.; Xie, S.; Yu, H.; Li, G.; Zhu, T.; Li, F.; Lu, J.; Wang, G.Y.; et al. Nicotinamide N-methyltransferase enhances chemoresistance in breast cancer through SIRT1 protein stabilization. Breast Cancer Res. 2019, 21, 64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Are poly(ADP-ribosyl)ation by PARP-1 and deacetylation by Sir2 linked? Bioessays 2003, 25, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, A.; Liabeuf, S.; Bodeau, S.; Louvet, L.; Mary, A.; Boullier, A.; Lemaire-Hurtel, A.S.; Jonet, A.; Sonnet, P.; Kamel, S.; et al. N-methyl-2-pyridone-5-carboxamide (2PY)-Major Metabolite of Nicotinamide: An Update on an Old Uremic Toxin. Toxins 2016, 8, 339. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Zhu, P.X.; Yang, X.; Han, Z.P.; Jiang, J.H.; Zong, C.; Zhang, X.G.; Liu, W.T.; Zhao, Q.D.; Fan, T.T.; et al. Overexpression of SIRT1 promotes metastasis through epithelial-mesenchymal transition in hepatocellular carcinoma. BMC Cancer 2014, 14, 978. [Google Scholar] [CrossRef]
- Kanska, J.; Aspuria, P.P.; Taylor-Harding, B.; Spurka, L.; Funari, V.; Orsulic, S.; Karlan, B.Y.; Wiedemeyer, W.R. Glucose deprivation elicits phenotypic plasticity via ZEB1-mediated expression of NNMT. Oncotarget 2017, 8, 26200–26220. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional RepressorsZEB1andZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial–mesenchymal transition through modulation of the miR-130b–ZEB1 axis. Oncogene 2012, 32, 3286–3295. [Google Scholar] [CrossRef]
- Yaginuma, Y.; Westphal, H. Abnormal structure and expression of the p53 gene in human ovarian carcinoma cell lines. Cancer Res. 1992, 52, 4196–4199. [Google Scholar] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Mori, K.; Shibanuma, M.; Nose, K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004, 64, 7464–7472. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Venables, J.P. Unbalanced alternative splicing and its significance in cancer. Bioessays 2006, 28, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Reinke, L.M.; Xu, Y.; Cheng, C. Snail represses the splicing regulator epithelial splicing regulatory protein 1 to promote epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 36435–36442. [Google Scholar] [CrossRef] [PubMed]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef]
- Miyazaki, H.; Takahashi, R.U.; Prieto-Vila, M.; Kawamura, Y.; Kondo, S.; Shirota, T.; Ochiya, T. CD44 exerts a functional role during EMT induction in cisplatin-resistant head and neck cancer cells. Oncotarget 2018, 9, 10029–10041. [Google Scholar] [CrossRef] [PubMed]
- Barnett, P.; Arnold, R.S.; Mezencev, R.; Chung, L.W.K.; Zayzafoon, M.; Odero-Marah, V. Snail-mediated regulation of reactive oxygen species in ARCaP human prostate cancer cells. Biochem. Biophys. Res. Commun. 2011, 404, 34–39. [Google Scholar] [CrossRef]
- Maeda, K.; Ohno, T.; Igarashi, S.; Yoshimura, T.; Yamashiro, K.; Sakai, M. Aldehyde oxidase 1 gene is regulated by Nrf2 pathway. Gene 2012, 505, 374–378. [Google Scholar] [CrossRef]
- Shintani, Y.; Maruoka, S.; Gon, Y.; Koyama, D.; Yoshida, A.; Kozu, Y.; Kuroda, K.; Takeshita, I.; Tsuboi, E.; Soda, K.; et al. Nuclear factor erythroid 2-related factor 2 (Nrf2) regulates airway epithelial barrier integrity. Allergol. Int. 2015, 64, S54–S63. [Google Scholar] [CrossRef]
- Obach, R.S. Potent inhibition of human liver aldehyde oxidase by raloxifene. Drug Metab. Dispos. 2004, 32, 89–97. [Google Scholar] [CrossRef]
- Takeuchi, K.; Yokouchi, C.; Goto, H.; Umehara, K.; Yamada, H.; Ishii, Y. Alleviation of fatty liver in a rat model by enhancing N1-methylnicotinamide bioavailability through aldehyde oxidase inhibition. Biochem. Biophys. Res. Commun. 2018, 507, 203–210. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, S.T.; Turner, A.R.; Young, T.; Smith, S.; Liu, W.; Lindberg, J.; Egevad, L.; Gronberg, H.; Isaacs, W.B.; et al. Identification of new differentially methylated genes that have potential functional consequences in prostate cancer. PLoS ONE 2012, 7, e48455. [Google Scholar] [CrossRef] [PubMed]
- Varisli, L. Identification of new genes downregulated in prostate cancer and investigation of their effects on prognosis. Genet. Test Mol. Biomark. 2013, 17, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Middha, M.; Bicak, M.; Sjoberg, D.D.; Vertosick, E.; Dahlin, A.; Häggström, C.; Hallmans, G.; Rönn, A.C.; Stattin, P.; et al. Genome-wide Scan Identifies Role for AOX1 in Prostate Cancer Survival. Eur. Urol. 2018, 74, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Crnogorac-Jurcevic, T.; Gangeswaran, R.; Bhakta, V.; Capurso, G.; Lattimore, S.; Akada, M.; Sunamura, M.; Prime, W.; Campbell, F.; Brentnall, T.A.; et al. Proteomic analysis of chronic pancreatitis and pancreatic adenocarcinoma. Gastroenterology 2005, 129, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Shoulson, R.; Chatterjee, A.; Ronghe, A.; Bhat, N.K.; Dim, D.C.; Bhat, H.K. Resveratrol inhibits estrogen-induced breast carcinogenesis through induction of NRF2-mediated protective pathways. Carcinogenesis 2014, 35, 1872–1880. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Y.; Zhang, Z.; Zou, J.; Li, J.; Wei, R.; Guo, Q.; Zhu, X.; Chu, C.; Fu, X.; et al. Meta-analysis based gene expression profiling reveals functional genes in ovarian cancer. Biosci. Rep. 2020, 40, BSR20202911. [Google Scholar] [CrossRef] [PubMed]
- Oster, B.; Thorsen, K.; Lamy, P.; Wojdacz, T.K.; Hansen, L.L.; Birkenkamp-Demtröder, K.; Sørensen, K.D.; Laurberg, S.; Orntoft, T.F.; Andersen, C.L. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int. J. Cancer 2011, 129, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Vantaku, V.; Putluri, V.; Bader, D.A.; Maity, S.; Ma, J.; Arnold, J.M.; Rajapakshe, K.; Donepudi, S.R.; von Rundstedt, F.C.; Devarakonda, V.; et al. Epigenetic loss of AOX1 expression via EZH2 leads to metabolic deregulations and promotes bladder cancer progression. Oncogene 2020, 39, 6265–6285. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, W.; Zhu, Z.; Li, X. Aldehyde oxidase 1 promoted the occurrence and development of colorectal cancer by up-regulation of expression of CD133. Int. Immunopharmacol. 2020, 85, 106618. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Damuni, Z. Okadaic acid and microcystin-LR directly inhibit the methylation of protein phosphatase 2A by its specific methyltransferase. Biochem. Biophys. Res. Commun. 1994, 202, 1023–1030. [Google Scholar] [CrossRef]
- Deris-Abdolahpour, F.; Abdolalipouran-Sadegh, L.; Dastmalchi, S.; Hamzeh-Mivehroud, M.; Zarei, O.; Dehgan, G.; Rashidi, M.R. Effects of Phenothiazines on Aldehyde Oxidase Activity Towards Aldehydes and N-Heterocycles: An In Vitro and In Silico Study. Eur. J. Drug Metab. Pharm. 2018, 44, 275–286. [Google Scholar] [CrossRef]
- Rao, R.K.; Clayton, L.W. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem. Biophys. Res. Commun. 2002, 293, 610–616. [Google Scholar] [CrossRef]
- Tegowski, M.; Fan, C.; Baldwin, A.S. Thioridazine inhibits self-renewal in breast cancer cells via DRD2-dependent STAT3 inhibition, but induces a G1 arrest independent of DRD2. J. Biol. Chem. 2018, 293, 15977–15990. [Google Scholar] [CrossRef]
- Nakata, B.; Albright, K.D.; Barton, R.M.; Howell, S.B.; Los, G. Synergistic interaction between cisplatin and tamoxifen delays the emergence of cisplatin resistance in head and neck cancer cell lines. Cancer Chemother. Pharmacol. 1995, 35, 511–518. [Google Scholar] [CrossRef]
- Chen, S.; Austin-Muttitt, K.; Zhang, L.H.; Mullins, J.G.L.; Lau, A.J. In Vitro and In Silico Analyses of the Inhibition of Human Aldehyde Oxidase by Bazedoxifene, Lasofoxifene, and Structural Analogues. J. Pharmacol. Exp. Ther. 2019, 371, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Vidugiriene, J.; Leippe, D.; Sobol, M.; Vidugiris, G.; Zhou, W.; Meisenheimer, P.; Gautam, P.; Wennerberg, K.; Cali, J.J. Bioluminescent cell-based NAD(P)/NAD(P)H assays for rapid dinucleotide measurement and inhibitor screening. Assay Drug Dev. Technol. 2014, 12, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Terao, M.; Garattini, E.; Romão, M.J.; Leimkühler, S. Evolution, expression, and substrate specificities of aldehyde oxidase enzymes in eukaryotes. J. Biol. Chem. 2020, 295, 5377–5389. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novak Kujundžić, R.; Prpić, M.; Đaković, N.; Dabelić, N.; Tomljanović, M.; Mojzeš, A.; Fröbe, A.; Trošelj, K.G. Nicotinamide N-Methyltransferase in Acquisition of Stem Cell Properties and Therapy Resistance in Cancer. Int. J. Mol. Sci. 2021, 22, 5681. https://doi.org/10.3390/ijms22115681
Novak Kujundžić R, Prpić M, Đaković N, Dabelić N, Tomljanović M, Mojzeš A, Fröbe A, Trošelj KG. Nicotinamide N-Methyltransferase in Acquisition of Stem Cell Properties and Therapy Resistance in Cancer. International Journal of Molecular Sciences. 2021; 22(11):5681. https://doi.org/10.3390/ijms22115681
Chicago/Turabian StyleNovak Kujundžić, Renata, Marin Prpić, Nikola Đaković, Nina Dabelić, Marko Tomljanović, Anamarija Mojzeš, Ana Fröbe, and Koraljka Gall Trošelj. 2021. "Nicotinamide N-Methyltransferase in Acquisition of Stem Cell Properties and Therapy Resistance in Cancer" International Journal of Molecular Sciences 22, no. 11: 5681. https://doi.org/10.3390/ijms22115681
APA StyleNovak Kujundžić, R., Prpić, M., Đaković, N., Dabelić, N., Tomljanović, M., Mojzeš, A., Fröbe, A., & Trošelj, K. G. (2021). Nicotinamide N-Methyltransferase in Acquisition of Stem Cell Properties and Therapy Resistance in Cancer. International Journal of Molecular Sciences, 22(11), 5681. https://doi.org/10.3390/ijms22115681