
PredNTS: Improved and Robust Prediction of Nitrotyrosine Sites by Integrating Multiple Sequence Features

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Sequence Preference Analysis
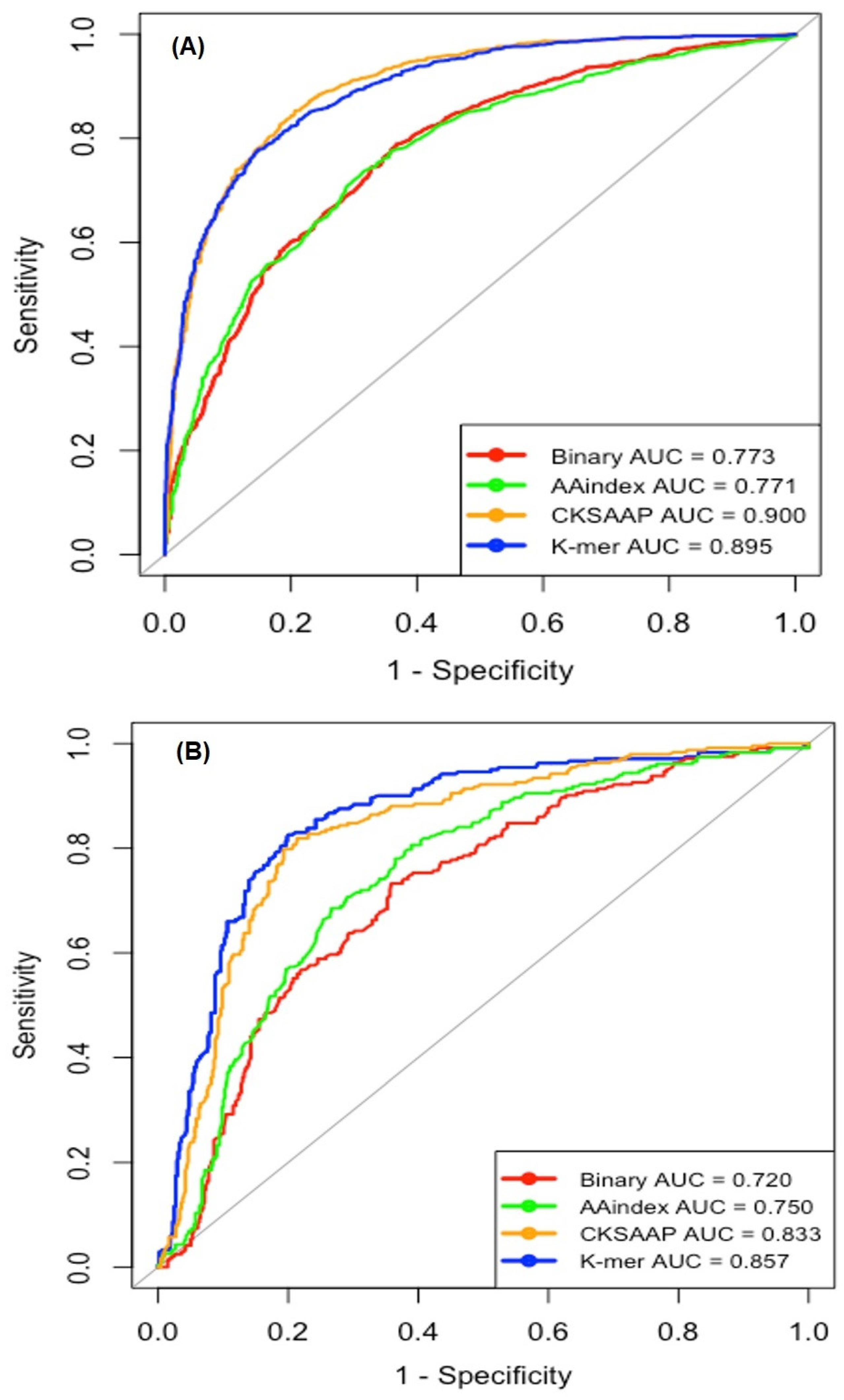
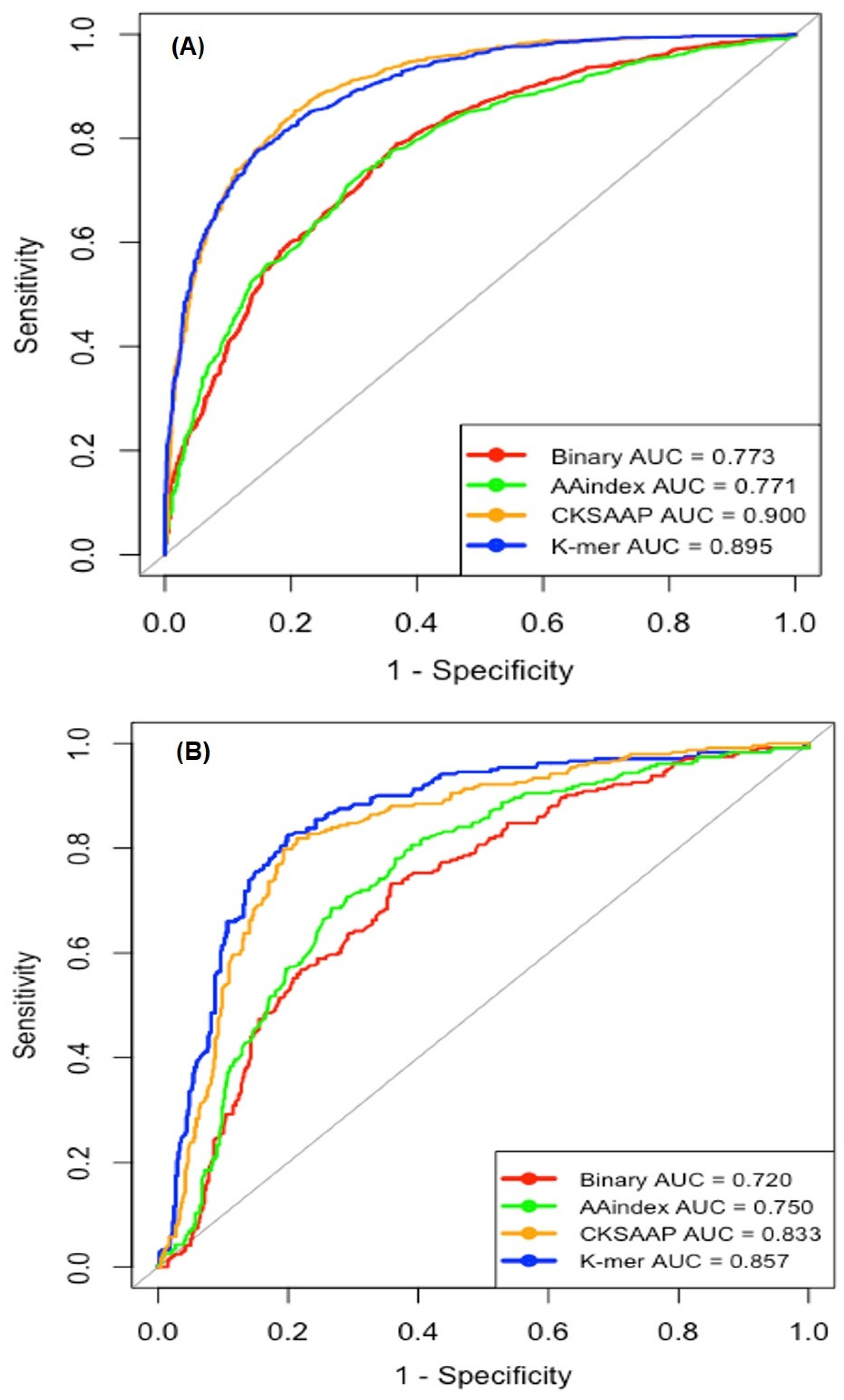
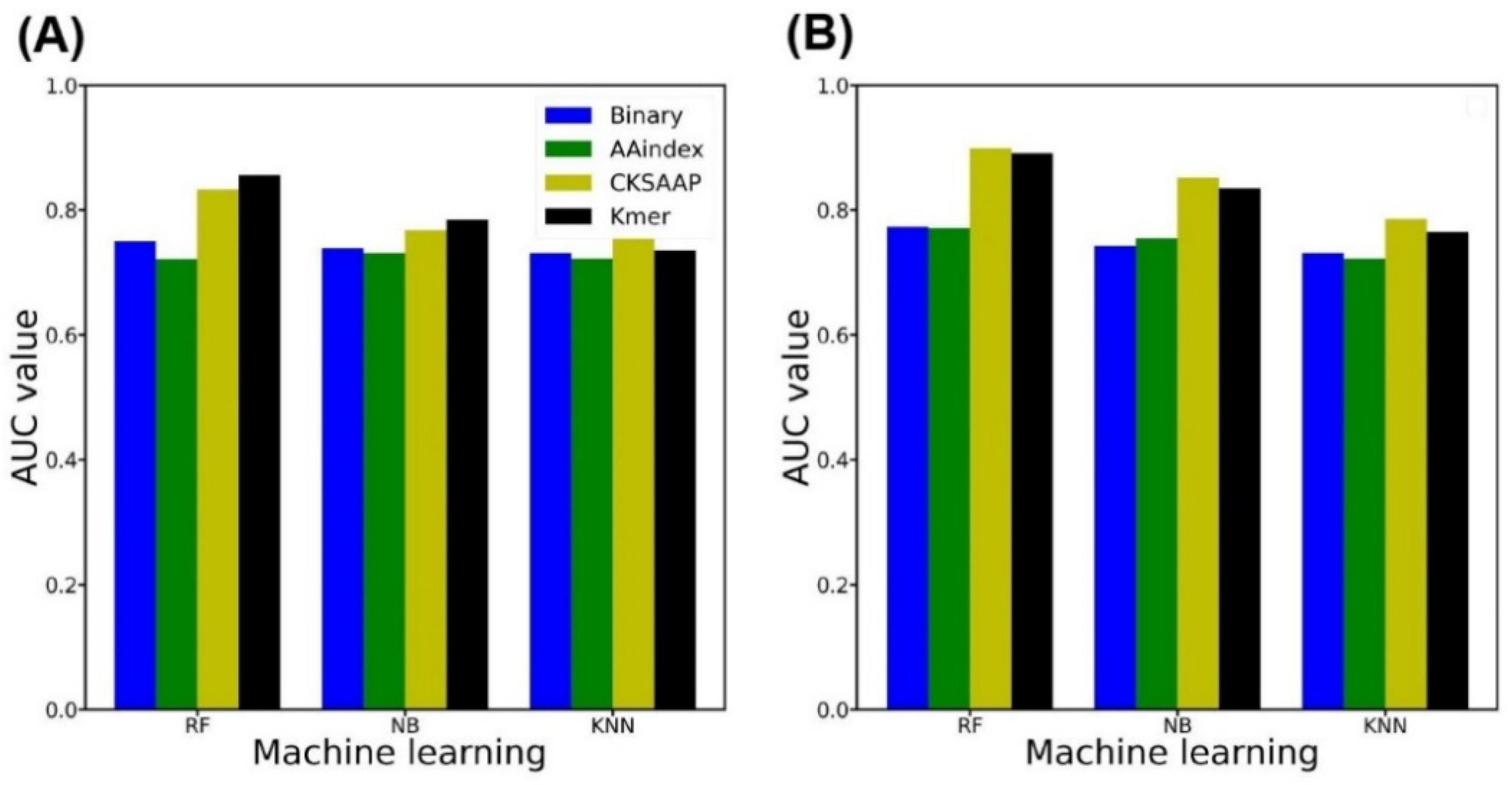
2.2. Single Encoding-Employing RF Model on the Training Dataset
2.3. Single Encoding-Employing RF Model on the Independent Dataset
2.4. Prediction Performance of PredNTS
2.5. Comparison of PredNTS with Other Existing Predictors
2.6. Web Server Implementation
3. Materials and Methods
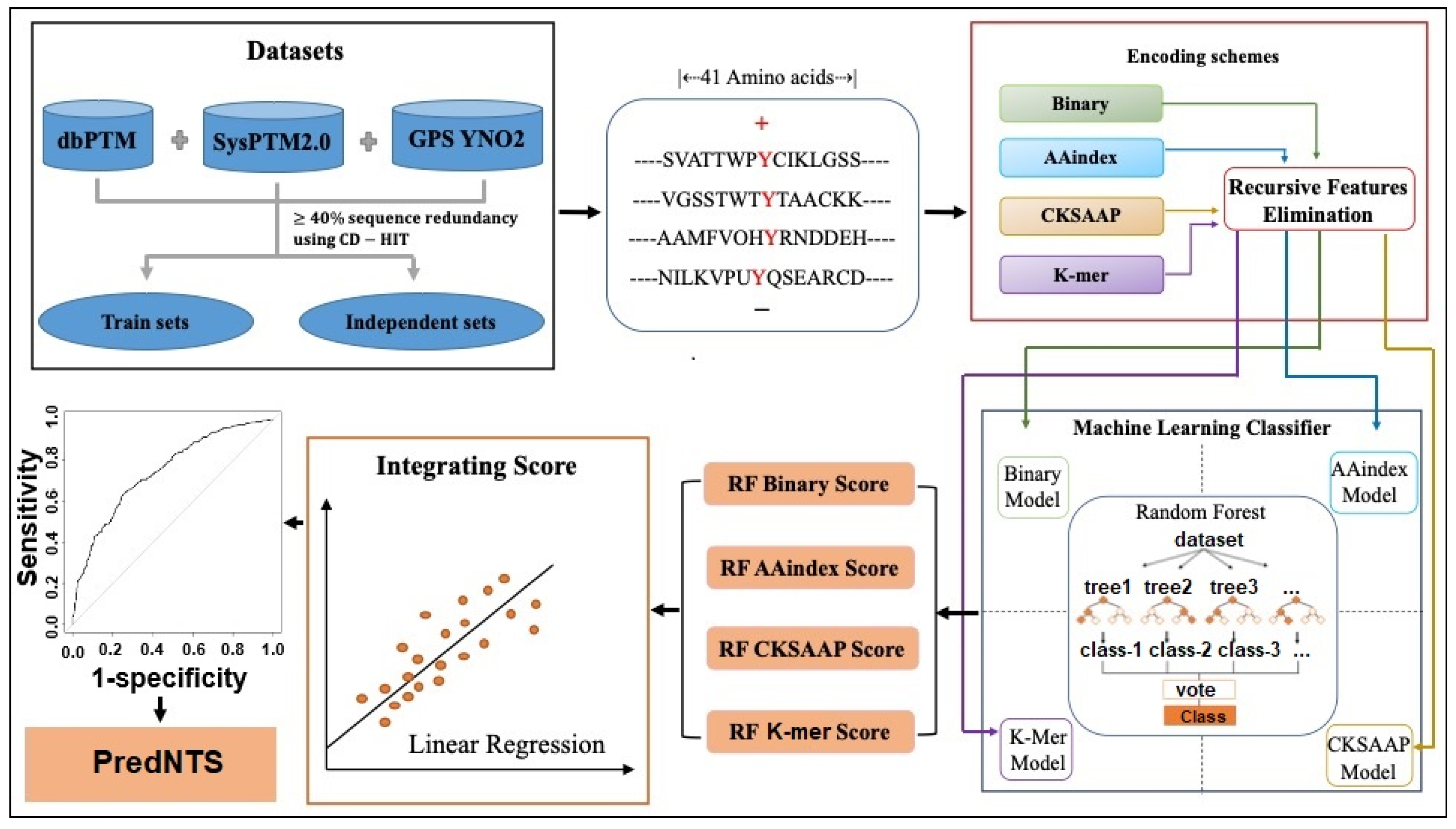
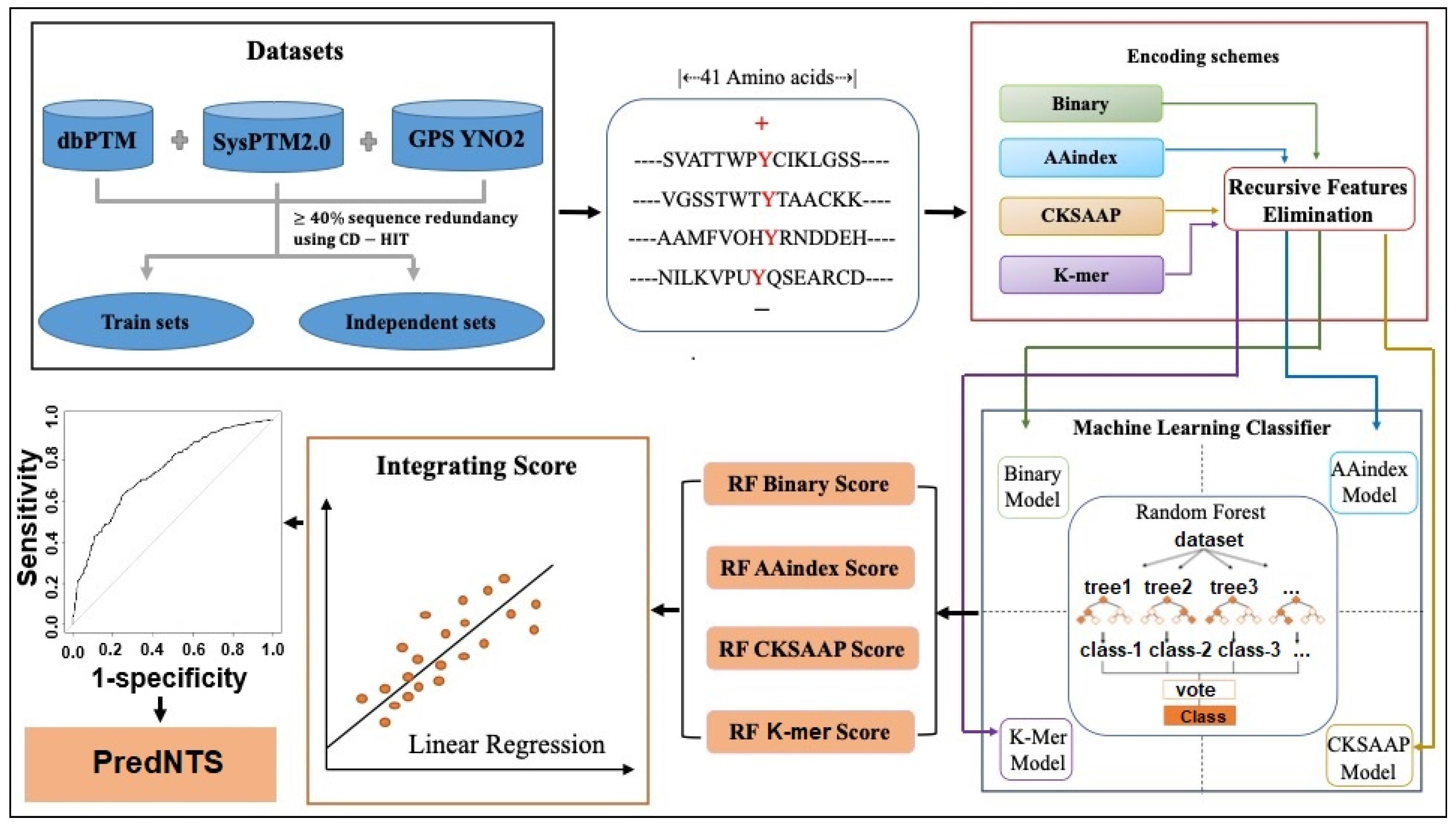
3.1. Dataset Construction
3.2. Sequence Encoding Scheme
3.3. Feature Selection
3.4. Machine Learning Algorithm
3.5. Evaluation Measure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, Y.; Zhang, Y.; Sun, H.; Maroto, R.; Brasier, A.R. Selective Affinity Enrichment of Nitrotyrosine-Containing Peptides for Quantitative Analysis in Complex Samples. J. Proteome Res. 2017, 16, 2983–2992. [Google Scholar] [CrossRef]
- Peng, F.; Li, J.; Guo, T.; Yang, H.; Li, M.; Sang, S.; Li, X.; Desiderio, D.M.; Zhan, X. Nitroproteins in Human Astrocytomas Discovered by Gel Electrophoresis and Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 2062–2076. [Google Scholar] [CrossRef]
- Nuriel, T.; Deeb, R.S.; Hajjar, D.P.; Gross, S.S. Protein 3-nitrotyrosine in complex biological samples: Quantification by high-pressure liquid chromatography/electrochemical detection and emergence of proteomic approaches for unbiased identification of modification sites. Methods Enzym. 2008, 441, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Seeley, K.W.; Fertig, A.R.; Dufresne, C.P.; Pinho, J.P.; Stevens, S.M., Jr. Evaluation of a method for nitrotyrosine site identification and relative quantitation using a stable isotope-labeled nitrated spike-in standard and high resolution fourier transform MS and MS/MS analysis. Int. J. Mol. Sci. 2014, 15, 6265–6285. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Lee, J.R.; Kim, Y.H.; Park, Y.S.; Park, S.I.; Park, H.S.; Kim, K.P. Investigation of tyrosine nitration and nitrosylation of angiotensin II and bovine serum albumin with electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2797–2804. [Google Scholar] [CrossRef]
- Ghesquiere, B.; Goethals, M.; Van Damme, J.; Staes, A.; Timmerman, E.; Vandekerckhove, J.; Gevaert, K. Improved tandem mass spectrometric characterization of 3-nitrotyrosine sites in peptides. Rapid Commun. Mass Spectrom. 2006, 20, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Gerding, H.R.; Karreman, C.; Daiber, A.; Delp, J.; Hammler, D.; Mex, M.; Schildknecht, S.; Leist, M. Reductive modification of genetically encoded 3-nitrotyrosine sites in alpha synuclein expressed in E. coli. Redox Biol. 2019, 26, 101251. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Desiderio, D.M. Mass spectrometric identification of in vivo nitrotyrosine sites in the human pituitary tumor proteome. Methods Mol. Biol. 2009, 566, 137–163. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.B.; Maes, M.; Roomruangwong, C.; Bonifacio, K.L.; Barbosa, D.S.; Vargas, H.O.; Anderson, G.; Kubera, M.; Carvalho, A.F.; Nunes, S.O.V. In major affective disorders, early life trauma predict increased nitro-oxidative stress, lipid peroxidation and protein oxidation and recurrence of major affective disorders, suicidal behaviors and a lowered quality of life. Metab. Brain Dis. 2018, 33, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Zhang, S.; Cheng, N.; Ge, S.; Wen, J.; Xiao, J.; Wu, X. Oxidored-nitro domain-containing protein 1 promotes liver fibrosis by activating the Wnt/beta-catenin signaling pathway in vitro. Mol. Med. Rep. 2017, 16, 5050–5054. [Google Scholar] [CrossRef]
- Ha, M.N.; Delpeut, S.; Noyce, R.S.; Sisson, G.; Black, K.M.; Lin, L.T.; Bilimoria, D.; Plemper, R.K.; Prive, G.G.; Richardson, C.D. Mutations in the Fusion Protein of Measles Virus That Confer Resistance to the Membrane Fusion Inhibitors Carbobenzoxy-d-Phe-l-Phe-Gly and 4-Nitro-2-Phenylacetyl Amino-Benzamide. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Luo, X.; Li, Y.; Chen, L.; Ma, W.; Huang, J.; Cui, J.; Zhao, Y.; Xue, Y.; Zuo, Z.; et al. DeepNitro: Prediction of Protein Nitration and Nitrosylation Sites by Deep Learning. Genom. Proteom. Bioinform. 2018, 16, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Mollah, M.N.H.; Yong, C.; Dianjing, G. NTyroSite: Computational Identification of Protein Nitrotyrosine Sites Using Sequence Evolutionary Features. Molecules 2018, 23, 1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wen, X.; Wen, L.S.; Wu, L.Y.; Deng, N.Y.; Chou, K.C. iNitro-Tyr: Prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS ONE 2014, 9, e105018. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Cao, J.; Ma, Q.; Gao, X.; Ren, J.; Xue, Y. GPS-YNO2: Computational prediction of tyrosine nitration sites in proteins. Mol. Biosyst. 2011, 7, 1197–1204. [Google Scholar] [CrossRef]
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Hu, J.; Li, F.; Song, J.; Su, R.; Zou, Q. Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief. Bioinform. 2018. [Google Scholar] [CrossRef]
- Hasan, M.M.; Alam, M.A.; Shoombuatong, W.; Kurata, H. IRC-Fuse: Improved and robust prediction of redox-sensitive cysteine by fusing of multiple feature representations. J. Comput. Aided Mol. 2021. [Google Scholar] [CrossRef]
- Hasan, M.M.; Rashid, M.M.; Khatun, M.S.; Kurata, H. Computational identification of microbial phosphorylation sites by the enhanced characteristics of sequence information. Sci. Rep. 2019, 9, 8258. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.M.; Zhou, Y.; Lu, X.; Li, J.; Song, J.; Zhang, Z. Computational Identification of Protein Pupylation Sites by Using Profile-Based Composition of k-Spaced Amino Acid Pairs. PLoS ONE 2015, 10, e0129635. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhou, Y.; Zhang, Z.; Song, J. Towards more accurate prediction of ubiquitination sites: A comprehensive review of current methods, tools and features. Brief. Bioinform. 2015, 16, 640–657. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Hasan, M.M.; Schaduangrat, N.; Basith, S.; Lee, G.; Shoombuatong, W.; Manavalan, B. HLPpred-Fuse: Improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 2020, 36, 3350–3356. [Google Scholar] [CrossRef] [PubMed]
- Khatun, M.S.; Hasan, M.M.; Kurata, H. PreAIP: Computational Prediction of Anti-inflammatory Peptides by Integrating Multiple Complementary Features. Front. Genet. 2019, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Yang, S.; Zhou, Y.; Mollah, M.N. SuccinSite: A computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol. Biosyst. 2016, 12, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2008, 36, D202–D205. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.M.; Kurata, H. GPSuc: Global Prediction of Generic and Species-specific Succinylation Sites by aggregating multiple sequence features. PLoS ONE 2018, 13, e0200283. [Google Scholar] [CrossRef] [PubMed]
- Khatun, M.S.; Hasan, M.M.; Shoombuatong, W.; Kurata, H. ProIn-Fuse: Improved and robust prediction of proinflammatory peptides by fusing of multiple feature representations. J. Comput. Aided Mol. Des. 2020, 34, 1229–1236. [Google Scholar] [CrossRef]
- Hasan, M.M.; Manavalan, B.; Shoombuatong, W.; Khatun, M.S.; Kurata, H. i6mA-Fuse: Improved and robust prediction of DNA 6 mA sites in the Rosaceae genome by fusing multiple feature representation. Plant Mol. Biol. 2020, 103, 225–234. [Google Scholar] [CrossRef]
- Hasan, M.M.; Manavalan, B.; Khatun, M.S.; Kurata, H. i4mC-ROSE, a bioinformatics tool for the identification of DNA N4-methylcytosine sites in the Rosaceae genome. Int. J. Biol. Macromol. 2020, 157, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Basith, S.; Khatun, M.S.; Lee, G.; Manavalan, B.; Kurata, H. Meta-i6mA: An interspecies predictor for identifying DNA N6-methyladenine sites of plant genomes by exploiting informative features in an integrative machine-learning framework. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Peng, C.; Wu, X.; Yuan, W.; Zhang, X.; Li, Y. MGRFE: Multilayer recursive feature elimination based on an embedded genetic algorithm for cancer classification. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef]
- Wei, L.; He, W.; Malik, A.; Su, R.; Cui, L.; Manavalan, B. Computational prediction and interpretation of cell-specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Hwan Shin, T.; Lee, G. Machine intelligence in peptide therapeutics: A next-generation tool for rapid disease screening. Med. Res. Rev. 2020, 40, 1276–1314. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. Meta-4mCpred: A Sequence-Based Meta-Predictor for Accurate DNA 4mC Site Prediction Using Effective Feature Representation. Mol. Ther. Nucleic Acids 2019, 16, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.M.; Manavalan, B.; Khatun, M.S.; Kurata, H. Prediction of S-nitrosylation sites by integrating support vector machines and random forest. Mol. Omics 2019, 15, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. mAHTPred: A sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 2019, 35, 2757–2765. [Google Scholar] [CrossRef]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. SDM6A: A Web-Based Integrative Machine-Learning Framework for Predicting 6mA Sites in the Rice Genome. Mol. Ther. Nucleic Acids 2019, 18, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Tasmia, S.A.; Faisal, F.F.; Mosharaf, M.P.; Hasan, M.M.; Mollah, M.N.H. An Improved Computational Prediction Model for Lysine Succinylation Sites Mapping on Homo sapiens by Fusing Two Sequence Encoding Schemes with the Random Forest Classifier. Curr. Genom. 2021. [Google Scholar] [CrossRef]
- Auliah, F.N.; Nilamyani, A.N.; Shoombuatong, W.; Alam, M.A.; Hasan, M.M.; Kurata, H. PUP-Fuse: Prediction of Protein Pupylation Sites by Integrating Multiple Sequence Representations. Int. J. Mol. Sci. 2021, 22, 2120. [Google Scholar] [CrossRef]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, D.Y.; Lee, G. Evolution of Machine Learning Algorithms in the Prediction and Design of Anticancer Peptides. Curr. Protein. Pept. Sci. 2020, 21, 1242–1250. [Google Scholar] [CrossRef]
- Khatun, S.; Hasan, M.; Kurata, H. Efficient computational model for identification of antitubercular peptides by integrating amino acid patterns and properties. FEBS Lett. 2019, 593, 3029–3039. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. AIPpred: Sequence-Based Prediction of Anti-inflammatory Peptides Using Random Forest. Front. Pharm. 2018, 9, 276. [Google Scholar] [CrossRef]
- Khatun, M.S.; Shoombuatong, W.; Hasan, M.M.; Kurata, H. Evolution of Sequence-based Bioinformatics Tools for Protein-protein Interaction Prediction. Curr. Genom. 2020, 21, 454–463. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Yana, J.; Schaduangrat, N.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iBitter-SCM: Identification and characterization of bitter peptides using a scoring card method with propensity scores of dipeptides. Genomics 2020, 112, 2813–2822. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Yana, J.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iUmami-SCM: A Novel Sequence-Based Predictor for Prediction and Analysis of Umami Peptides Using a Scoring Card Method with Propensity Scores of Dipeptides. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iTTCA-Hybrid: Improved and robust identification of tumor T cell antigens by utilizing hybrid feature representation. Anal. Biochem. 2020, 599, 113747. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. Meta-iPVP: A sequence-based meta-predictor for improving the prediction of phage virion proteins using effective feature representation. J. Comput. Aided Mol. Des. 2020, 34, 1105–1116. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Kanthawong, S.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iDPPIV-SCM: A Sequence-Based Predictor for Identifying and Analyzing Dipeptidyl Peptidase IV (DPP-IV) Inhibitory Peptides Using a Scoring Card Method. J. Proteome Res. 2020, 19, 4125–4136. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Kanthawong, S.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iAMY-SCM: Improved prediction and analysis of amyloid proteins using a scoring card method with propensity scores of dipeptides. Genomics 2020. [Google Scholar] [CrossRef]
- Ning, Q.; Ma, Z.; Zhao, X.; Yin, M. SSKM_Succ: A novel succinylation sites prediction method incorprating K-means clustering with a new semi-supervised learning algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Ning, Q.; Yu, M.; Ji, J.; Ma, Z.; Zhao, X. Analysis and prediction of human acetylation using a cascade classifier based on support vector machine. BMC Bioinform. 2019, 20, 346. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.M.; Shoombuatong, W.; Kurata, H.; Manavalan, B. Critical evaluation of web-based DNA N6-methyladenine site prediction tools. Brief. Funct. Genom. 2021. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.M.; Khatun, M.S.; Kurata, H. iLBE for Computational Identification of Linear B-cell Epitopes by Integrating Sequence and Evolutionary Features. Genom. Proteom. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Kurata, H. Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites. Cells 2019, 8, 95. [Google Scholar] [CrossRef] [Green Version]
- Charoenkwan, P.; Chiangjong, W.; Lee, V.S.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. Improved prediction and characterization of anticancer activities of peptides using a novel flexible scoring card method. Sci. Rep. 2021, 11, 3017. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Su, R.; Luan, S.; Liao, Z.; Manavalan, B.; Zou, Q.; Shi, X. Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics 2019, 35, 4930–4937. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Nantasenama, C.; Hasan, M.M.; Manavalan, B.; Shoombuatong, W. BERT4Bitter: A bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides in Bioinformatics. Bioinformatics 2021. [Google Scholar] [CrossRef]
- Manavalan, B.; Hasan, M.M.; Basith, S.; Gosu, V.; Shin, T.H.; Lee, G. Empirical Comparison and Analysis of Web-Based DNA N (4)-Methylcytosine Site Prediction Tools. Mol. Ther. Nucleic Acids 2020, 22, 406–420. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Encoding Scheme | Sn | Sp | Acc | MCC |
|---|---|---|---|---|
| Binary | 0.519 (0.19) | 0.808 (0.01) | 0.658 (0.04) | 0.331 (0.14) |
| AAindex | 0.473 (0.24) | 0.803 (0.00) | 0.638 (0.02) | 0.293 (0.16) |
| CKSAAP | 0.731 (0.16) | 0.808 (0.00) | 0.709 (0.11) | 0.519 (0.20) |
| K-mer | 0.785 (0.11) | 0.807 (0.01) | 0.796 (0.06) | 0.593 (0.13) |
| Encoding Scheme | Sn | Sp | Acc | MCC |
| Binary | 0.598 (0.18) | 0.800 (0.00) | 0.699 (0.02) | 0.407 (0.12) |
| AAindex | 0.571 (0.19) | 0.809 (0.01) | 0.690 (0.07) | 0.391 (0.17) |
| CKSAAP | 0.829 (0.09) | 0.809 (0.00) | 0.819 (0.09) | 0.639 (0.14) |
| K-mer | 0.811 (0.06) | 0.808 (0.00) | 0.810 (0.11) | 0.619 (0.08) |
| Encoding Scheme | Sn | Sp | Acc | MCC |
|---|---|---|---|---|
| Binary | 0.384 | 0.806 | 0.736 | 0.170 |
| AAindex | 0.397 | 0.800 | 0.733 | 0.174 |
| CKSAAP | 0.458 | 0.800 | 0.743 | 0.224 |
| K-mer | 0.480 | 0.800 | 0.747 | 0.242 |
| Encoding Scheme | Sn | Sp | Acc | MCC |
|---|---|---|---|---|
| Binary | 0.445 | 0.801 | 0.742 | 0.214 |
| AAindex | 0.438 | 0.801 | 0.741 | 0.209 |
| CKSAAP | 0.504 | 0.805 | 0.755 | 0.268 |
| K-mer | 0.532 | 0.804 | 0.758 | 0.288 |
| Dataset | Predictor | AUC |
|---|---|---|
| Training dataset | Binary + AAindex + CKSAAP + K-mer | 0.910 |
| Independent dataset | Binary + AAindex + CKSAAP + K-mer | 0.860 |
| Encoding Scheme | Sn | Sp | Acc | MCC |
|---|---|---|---|---|
| GPS-YNO2 | 0.334 | 0.801 | 0.724 | 0.122 |
| DeepNitro | 0.339 | 0.803 | 0.726 | 0.128 |
| NTyroSite | 0.440 | 0.793 | 0.744 | 0.196 |
| PredNTS | 0.522 | 0.809 | 0.761 | 0.286 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilamyani, A.N.; Auliah, F.N.; Moni, M.A.; Shoombuatong, W.; Hasan, M.M.; Kurata, H. PredNTS: Improved and Robust Prediction of Nitrotyrosine Sites by Integrating Multiple Sequence Features. Int. J. Mol. Sci. 2021, 22, 2704. https://doi.org/10.3390/ijms22052704
Nilamyani AN, Auliah FN, Moni MA, Shoombuatong W, Hasan MM, Kurata H. PredNTS: Improved and Robust Prediction of Nitrotyrosine Sites by Integrating Multiple Sequence Features. International Journal of Molecular Sciences. 2021; 22(5):2704. https://doi.org/10.3390/ijms22052704
Chicago/Turabian StyleNilamyani, Andi Nur, Firda Nurul Auliah, Mohammad Ali Moni, Watshara Shoombuatong, Md Mehedi Hasan, and Hiroyuki Kurata. 2021. "PredNTS: Improved and Robust Prediction of Nitrotyrosine Sites by Integrating Multiple Sequence Features" International Journal of Molecular Sciences 22, no. 5: 2704. https://doi.org/10.3390/ijms22052704