Platelets Are Critical Key Players in Sepsis

Abstract
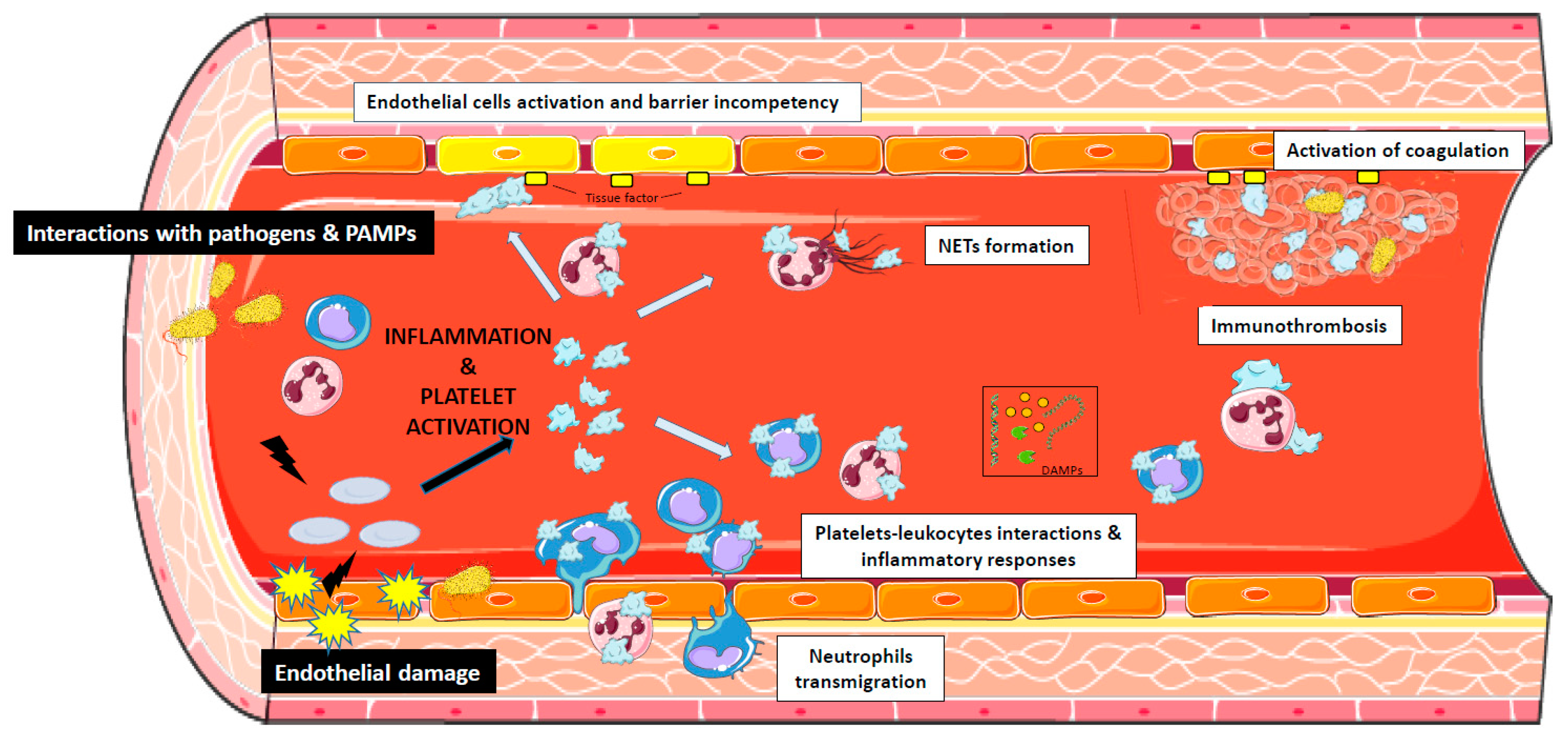
1. Introduction
2. Thrombocytopenia is Common in Sepsis and is Correlated to Mortality
3. Mechanisms that Contribute to Thrombocytopenia in Sepsis
4. The Role of Platelets in Sepsis
5. Platelets and Endothelial Cells Interactions During Sepsis
6. The Interaction of Bacterial Pathogens with Platelets
7. Therapeutic Implications
8. Conclusions
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Iskander, K.N.; Osuchowski, M.F.; Stearns-Kurosawa, D.J.; Kurosawa, S.; Stepien, D.; Valentine, C.; Remick, D.G. Sepsis: Multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol. Rev. 2013, 93, 1247–1288. [Google Scholar] [CrossRef] [PubMed]
- Pfeiler, S.; Massberg, S.; Engelmann, B. Biological basis and pathological relevance of microvascular thrombosis. Thromb. Res. 2014, 133, S35–S37. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2012, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hui, P.; Cook DJ, M.E.; Lim, W.; Fraser, G.A.; Arnold, D.M. The Frequency and Clinical Significance of Thrombocytopenia Complicating Critical Illness. Chest 2011, 139, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Selleng, K. Thrombocytopenia in the intensive care unit patient. Hematol. Am. Soc. Hematol Educ Program. 2010, 2010, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Schultz, M. Hematologic failure. Semin. Respir. Crit. Care Med. 2011, 32, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.; Wehler, M.; Mehler, K.; Kreutzer, D.; Koebnick, C.; Hahn, E.G. Thrombocytopenia in patients in the medical intensive care unit: Bleeding prevalence, transfusion requirements, and outcome*. Critical Care Medicine 2002, 30, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Moreno, R.; Takala, J.; Willatts, S.; De Mendonça, A.; Bruining, H.; Reinhart, C.K.; Suter, P.M.; Thijs, L.G. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 1996, 22, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Lower, E.E.; Flessa, H.C.; Tollerud, D.J. Thrombocytopenia in the intensive care unit. Chest 1993, 104, 1243–1247. [Google Scholar] [CrossRef]
- Housinger, T.A.; Brinkerhoff, C.; Warden, G.D. The relationship between platelet count, sepsis, and survival in pediatric burn patients. Arch. Surg. 1993, 128. [Google Scholar] [CrossRef] [PubMed]
- Stéphan, F.; Hollande, J.; Richard, O.; Cheffi, A.; Maier-Redelsperger, M.; Flahault, A. Thrombocytopenia in a surgical ICU. Chest 1999, 115, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Vanderschueren, S.; De Weerdt, A.; Malbrain, M.; Vankersschaever, D.; Frans, E.; Wilmer, A.; Bobbaers, H. Thrombocytopenia and prognosis in intensive care. Critical Care Medicine 2000, 28, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Akca, S.; Haji-Michael, P.; de Mendonça, A.; Suter, P.; Levi, M.; Vincent, J.-L. Time course of platelet counts in critically ill patients. Critical Care Medicine 2002, 30, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Moreau, D.; Timsit, J.-F.; Vesin, A.; Garrouste-Orgeas, M.; de Lassence, A.; Zahar, J.-R.; Adrie, C.; Vincent, F.; Cohen, Y.; Schlemmer, B.; et al. Platelet count decline: An early prognostic marker in critically ill patients with prolonged ICU stays. Chest 2007, 131, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Opal, S.M. Coagulation abnormalities in critically ill patients. Crit Care 2006, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Tsirigotis, P.; Chondropoulos, S.; Frantzeskaki, F.; Stamouli, M.; Gkirkas, K.; Bartzeliotou, A.; Papanikolaou, N.; Atta, M.; Papassotiriou, I.; Dimitriadis, G.; et al. Thrombocytopenia in critically ill patients with severe sepsis/septic shock: Prognostic value and association with a distinct serum cytokine profile. J. Crit Care 2016, 32, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Claushuis, T.A.M.; van Vught, L.A.; Scicluna, B.P.; Wiewel, M.A.; Klein Klouwenberg, P.M.C.; Hoogendijk, A.J.; Ong, D.S.Y.; Cremer, O.L.; Horn, J.; Franitza, M.; et al. Molecular Diagnosis and Risk Stratification of Sepsis Consortium Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood 2016, 127, 3062–3072. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Ten Cate, H. Disseminated intravascular coagulation. New England Journal of Medicine 1999, 341, 586–592. [Google Scholar] [CrossRef]
- Halacli, B.; Unver, N.; Halacli, S.O.; Canpinar, H.; Ersoy, E.O.; Ocal, S.; Guc, D.; Buyukasik, Y.; Topeli, A. Investigation of hemophagocytic lymphohistiocytosis in severe sepsis patients. J. Crit Care 2016, 35, 185–190. [Google Scholar] [CrossRef]
- Janka, G.E. Hemophagocytic lymphohistiocytosis. Hematology 2005, 10, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Grommes, J.; Alard, J.-E.; Drechsler, M.; Wantha, S.; Mörgelin, M.; Kuebler, W.M.; Jacobs, M.; von Hundelshausen, P.; Markart, P.; Wygrecka, M.; et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am. J. Respir. Crit. Care Med. 2012, 185, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.C.; Kubes, P. Platelets, neutrophils, and neutrophil extracellular traps (NETs) in sepsis. J. Thromb. Haemost. 2008, 6, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Alhamdi, Y.; Toh, C.-H. The role of extracellular histones in haematological disorders. Br. J. Haematol. 2016, 173, 805–811. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Kelton, J.G.; Neame, P.B.; Gauldie, J.; Hirsh, J. Elevated platelet-associated IgG in the thrombocytopenia of septicemia. New Engl. J. Med. 1979, 300, 760–764. [Google Scholar] [CrossRef]
- Stéphan, F.; Cheffi, M.A.; Kaplan, C.; Maillet, J.; Novara, A.; Fagon, J.; Bonnet, F. Autoantibodies against platelet glycoproteins in critically ill patients with thrombocytopenia. Am. J. Med. 2000, 108, 554–560. [Google Scholar] [CrossRef]
- de Stoppelaar, S.F.; van t Veer, C.; van der Poll, T. The role of platelets in sepsis. Thromb. Haemost. 2014, 112, 666–677. [Google Scholar] [PubMed]
- Crawley, J.T.B.; Zanardelli, S.; Chion, C.K.N.K.; Lane, D.A. The central role of thrombin in hemostasis. J. Thromb. Haemost. 2007, 5, 95–101. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef]
- Aggrey, A.A.; Srivastava, K.; Ture, S.; Field, D.J.; Morrell, C.N. Platelet induction of the acute-phase response is protective in murine experimental cerebral malaria. J. Immunol. 2013, 190, 4685–4691. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, G.A. Two by two: The pairings of P-selectin and P-selectin glycoprotein ligand 1. Proc. Natl. Acad. Sci. USA 2001, 98, 10023–10024. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Polanowska-Grabowska, R.K.; Ley, K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2007, 21, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Joshi, M.B.; Baipadithaya, G.; Balakrishnan, A.; Hegde, M.; Vohra, M.; Ahamed, R.; Nagri, S.K.; Ramachandra, L.; Satyamoorthy, K. Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci. Rep. 2016, 6, 36362. [Google Scholar] [CrossRef] [PubMed]
- Rother, N.; Pieterse, E.; Lubbers, J.; Hilbrands, L.; van der Vlag, J. Acetylated Histones in Apoptotic Microparticles Drive the Formation of Neutrophil Extracellular Traps in Active Lupus Nephritis. Front. Immunol. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High Level of Neutrophil Extracellular Traps Correlates With Poor Prognosis of Severe Influenza A Infection. The Journal of Infectious Diseases 2018, 217, 428–437. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Davis, R.P.; Kim, S.-J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef] [PubMed]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinflammatory IL-1β-rich microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular Neutrophil Extracellular Traps Capture Bacteria from the Bloodstream during Sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Camicia, G.; Pozner, R.; de Larrañaga, G. Neutrophil extracellular traps in sepsis. Shock 2014, 42, 286–294. [Google Scholar] [CrossRef]
- von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Zinsser, H.; Pryde, AW. Experimental study of physical factors, including fibrin formation, influencing the spread of fluids and small particles within and from the peritoneal cavity of the dog. Ann. Surg. 1952, 136, 818–827. [Google Scholar] [CrossRef]
- Engelmann, B. Extracellular DNA and histones as thrombus stabilizer. Thromb. Haemost. 2015, 113, 1164. [Google Scholar] [PubMed]
- Xu, J.; Zhang, X.; Monestier, M.; Esmon, N.L.; Esmon, C.T. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 2011, 187, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; De La Motte, C.; Reyes, B.M.R.; Sans, M.; Levine, A.D.; Fiocchi, C. Cutting edge: T cells trigger CD40-dependent platelet activation and granular RANTES release: A novel pathway for immune response amplification. J. Immunol. 2004, 172, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.S.S.; Andre, P.; He, M.; Bao, M.; Manganello, J.; Phillips, D.R. Soluble CD40 ligand induces beta3 integrin tyrosine phosphorylation and triggers platelet activation by outside-in signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 12367–12371. [Google Scholar] [CrossRef] [PubMed]
- Inwald, D.P.; McDowall, A.; Peters, M.J.; Callard, R.E.; Klein, N.J. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ. Res. 2003, 92, 1041–1048. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.M.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Khakpour, S.; Wilhelmsen, K.; Hellman, J. Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun. 2015, 21, 827–846. [Google Scholar] [CrossRef]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascón, G.A.; Hernandez, G.; Murray, P.; De Backer, D. ADQI XIV Workgroup THE ENDOTHELIUM IN SEPSIS. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Derangement of the endothelial glycocalyx in sepsis. J. Thromb. Haemost. 2019, 17, 283–294. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T.; Schultz, M. Systemic versus localized coagulation activation contributing to organ failure in critically ill patients. Semin. Immunopathol. 2012, 34, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Levi, M. Platelets in Critical Illness. Semin. Thromb. Hemost. 2016, 42, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Kerrigan, S.W.; Watson, S.P. Platelets and the innate immune system: Mechanisms of bacterial-induced platelet activation. J. Thromb. Haemost. 2011, 9, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Arvand, M.; Bhakdi, S.; Dahlbäck, B.; Preissner, K.T. Staphylococcus aureus alpha-toxin attack on human platelets promotes assembly of the prothrombinase complex. J. Biol. Chem. 1990, 265, 14377–14381. [Google Scholar] [PubMed]
- Lourbakos, A.; Yuan, Y.P.; Jenkins, A.L.; Travis, J.; Andrade-Gordon, P.; Santulli, R.; Potempa, J.; Pike, R.N. Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation: A new trait in microbial pathogenicity. Blood 2001, 97, 3790–3797. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.P.; Loughman, A.; Devocelle, M.; Arasu, S.; Chubb, A.J.; Foster, T.J.; Cox, D. Elucidating the role of Staphylococcus epidermidis serine-aspartate repeat protein G in platelet activation. J. Thromb. Haemost. 2009, 7, 1364–1372. [Google Scholar] [CrossRef]
- Miajlovic, H.; Zapotoczna, M.; Geoghegan, J.A.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. Direct interaction of iron-regulated surface determinant IsdB of Staphylococcus aureus with the GPIIb/IIIa receptor on platelets. Microbiology 2010, 156, 920–928. [Google Scholar] [CrossRef]
- Petersen, H.J.; Keane, C.; Jenkinson, H.F.; Vickerman, M.M.; Jesionowski, A.; Waterhouse, J.C.; Cox, D.; Kerrigan, S.W. Human platelets recognize a novel surface protein, PadA, on Streptococcus gordonii through a unique interaction involving fibrinogen receptor GPIIbIIIa. Infect. Immun. 2010, 78, 413–422. [Google Scholar] [CrossRef]
- Hamzeh-Cognasse, H.; Damien, P.; Chabert, A.; Pozzetto, B.; Cognasse, F.; Garraud, O. Platelets and infections—complex interactions with bacteria. Front. Immunol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Siboo, I.R.; Chambers, H.F.; Sullam, P.M. Role of SraP, a Serine-Rich Surface Protein of Staphylococcus aureus, in binding to human platelets. Infect. Immun. 2005, 73, 2273–2280. [Google Scholar] [CrossRef]
- McDevitt, D.; Francois, P.; Vaudaux, P.; Foster, T.J. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol. Microbiol. 1994, 11, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.R.; Loughman, A.; Keane, F.; Brennan, M.; Knobel, M.; Higgins, J.; Visai, L.; Speziale, P.; Cox, D.; Foster, T.J. Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor. Mol. Microbiol. 2006, 59, 212–230. [Google Scholar] [CrossRef] [PubMed]
- Winning, J.; Reichel, J.; Eisenhut, Y.; Hamacher, J.; Kohl, M.; Deigner, H.P.; Claus, R.A.; Bauer, M.; Lösche, W. Anti-platelet drugs and outcome in severe infection: Clinical impact and underlying mechanisms. Platelets 2009, 20, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Zhang, S.; Chew, M.; Syk, I.; Jeppsson, B.; Thorlacius, H. Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J. Thromb. Haemost. 2013, 11, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Halushka, P.V.; Wise, W.C.; Cook, J.A. Studies on the beneficial effects of aspirin in endotoxic shock. Relationship to inhibition of arachidonic acid metabolism. Am. J. Med. 1983, 74, 91–96. [Google Scholar] [CrossRef]
- Liverani, E.; Rico, M.C.; Tsygankov, A.Y.; Kilpatrick, L.E.; Kunapuli, S.P. P2Y12 Receptor Modulates Sepsis-Induced Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 961–971. [Google Scholar] [CrossRef]
- Jackson, S.P.; Schoenwaelder, S.M.; Goncalves, I.; Nesbitt, W.S.; Yap, C.L.; Wright, C.E.; Kenche, V.; Anderson, K.E.; Dopheide, S.M.; Yuan, Y.; et al. PI 3-kinase p110β: A new target for antithrombotic therapy. Nat. Med. 2005, 11, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Kull, B.; Björkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skärby, T.; Inghardt, T.; Fjellström, O.; et al. Human target validation of phosphoinositide 3-kinase (PI3K)β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Barlaam, B.; Berglund, S.; Edman, K.; Karlsson, O.; Lindberg, J.; Nylander, S.; Inghardt, T. Discovery of 9-(1-phenoxyethyl)-2-morpholino-4-oxo-pyrido[1,2-a]pyrimidine-7-carboxamides as oral PI3Kβ inhibitors, useful as antiplatelet agents. Bioorg. Med. Chem. Lett. 2014, 24, 3936–3943. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Guillermet-Guibert, J.; Chicanne, G.; Cabou, C.; Jandrot-Perrus, M.; Plantavid, M.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.-P. Deletion of the p110beta isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood 2010, 115, 2008–2013. [Google Scholar] [CrossRef] [PubMed]
- Laurent, P.-A.; Laurent, P.A.; Séverin, S.; Severin, S.; Hechler, B.; Hechler, B.; Vanhaesebroeck, B.; Vanhaesebroeck, B.; Payrastre, B.; Payrastre, B.; et al. Platelet PI3K and GSK3 regulate thrombus stability at a high shear rate. Blood 2015, 125, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Vardon-Bounes, F.; Mémier, V.; Marcaud, M.; Jacquemin, A.; Hamzeh-Cognasse, H.; Garcia, C.; Series, J.; Sié, P.; Minville, V.; Gratacap, M.-P.; et al. Platelet activation and prothrombotic properties in a mouse model of peritoneal sepsis. Sci. Rep. 2018, 8, 13536. [Google Scholar] [CrossRef] [PubMed]
- Akinosoglou, K.; Alexopoulos, D. Use of antiplatelet agents in sepsis: A glimpse into the future. Thromb. Res. 2014, 133, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Eisen, D.P.; Reid, D.; McBryde, E.S. Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit. Care Med. 2012, 40, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Otto, G.P.; Sossdorf, M.; Boettel, J.; Kabisch, B.; Breuel, H.; Winning, J.; Lösche, W. Effects of low-dose acetylsalicylic acid and atherosclerotic vascular diseases on the outcome in patients with severe sepsis or septic shock. Platelets 2012, 24, 480–485. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Main Etiologies of Thrombocytopenia during Sepsis |
| Pseudothrombocytopenia |
| Laboratory artefact (in vitro agglutination in EDTA-anticoagulated blood) |
| Decreased platelet production |
| Viral infection (EBV, CMV, HCV, HIV) |
| Bone marrow suppression due to medication (antibiotics, proton pump inhibitor) |
| Hemodilution |
| Massive vascular infusion of fluids |
| Increased platelet consumption/sequestration |
| Thrombin-mediated platelet activation |
| Disseminated Intravascular Coagulation |
| Acquired hemophagocytic lymphohistiocytosis (HLH) |
| Platelet aggregation/adhesion to leukocytes and endothelial cells |
| Thrombus formation in extracellular DNA fibers from neutrophil extracellular traps (NETs) |
| Immune-mediated destruction |
| IgG antibodies associated to platelets (PAIgG) |
| Autoantibodies directed against platelets glycoproteins |
| Heparin-induced thrombocytopenia |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vardon-Bounes, F.; Ruiz, S.; Gratacap, M.-P.; Garcia, C.; Payrastre, B.; Minville, V. Platelets Are Critical Key Players in Sepsis. Int. J. Mol. Sci. 2019, 20, 3494. https://doi.org/10.3390/ijms20143494
Vardon-Bounes F, Ruiz S, Gratacap M-P, Garcia C, Payrastre B, Minville V. Platelets Are Critical Key Players in Sepsis. International Journal of Molecular Sciences. 2019; 20(14):3494. https://doi.org/10.3390/ijms20143494
Chicago/Turabian StyleVardon-Bounes, Fanny, Stéphanie Ruiz, Marie-Pierre Gratacap, Cédric Garcia, Bernard Payrastre, and Vincent Minville. 2019. "Platelets Are Critical Key Players in Sepsis" International Journal of Molecular Sciences 20, no. 14: 3494. https://doi.org/10.3390/ijms20143494
APA StyleVardon-Bounes, F., Ruiz, S., Gratacap, M.-P., Garcia, C., Payrastre, B., & Minville, V. (2019). Platelets Are Critical Key Players in Sepsis. International Journal of Molecular Sciences, 20(14), 3494. https://doi.org/10.3390/ijms20143494