Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mechanisms of Disulfide Bond Formation during Protein Folding
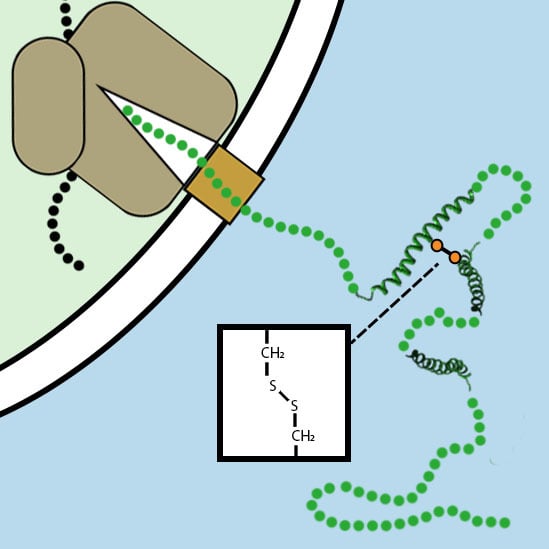
3. Disulfide Bond Formation in Nascent Polypeptides during Translocation
3.1. The Folding of Proteins at the Co-Translational Stage
3.2. Capturing Disulfide Bond Formation during Folding in Cells
3.3. Evidence for Disulfide Bond Formation in Translation Intermediates
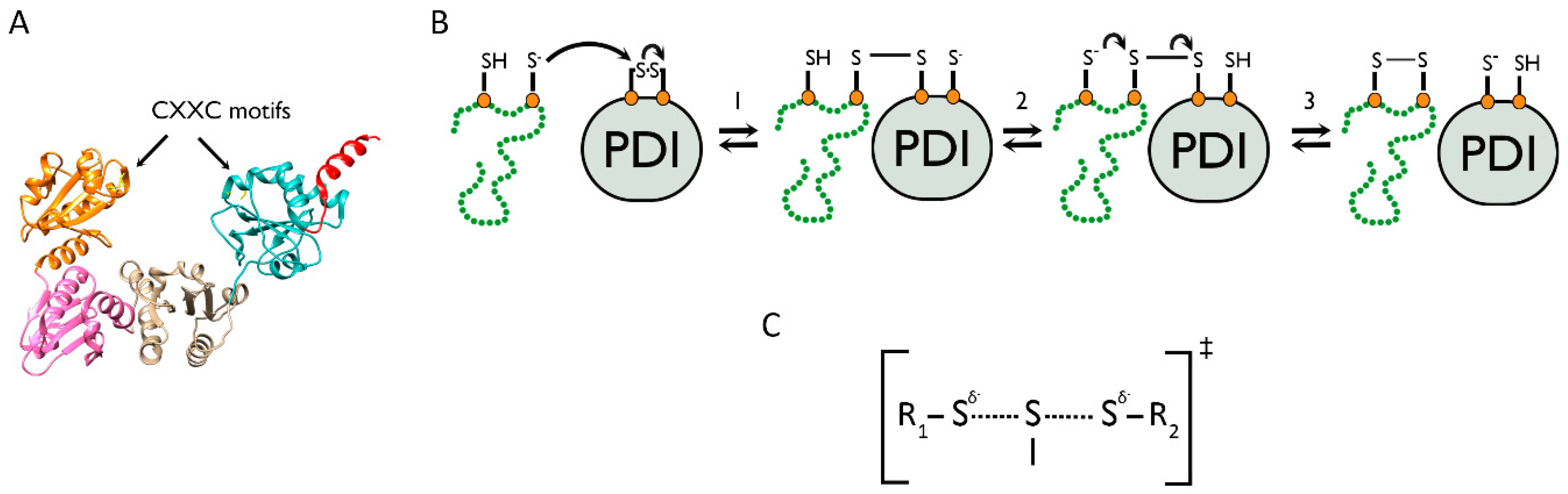
4. The Role of PDI Family Members in Nascent Polypeptide Folding
4.1. The PDI Family Acts via the Mechanism of Thiol–Disulfide Exchange
4.2. Oxidation Pathways to Re-Activate PDI Family Members Following Catalysis of Disulfide Bond Formation
4.3. PDI Preferentially Interacts with Unfolded Substrates to Catalyse Disulfide Bond Formation
4.4. Evidence for Disulfide Reduction and Rearrangements at the Co-Translational Stage
5. Conclusions and Future Directions
5.1. What Are the Structures of Translation Intermediates and How Do They Change throughout Translocation?
5.2. How Does Our Understanding of Co-Translational Folding and Disulfide Bond Formation Apply across the Proteome?
5.3. How and When Do Folding Factors Interact with Nascent Polypeptides?
Author Contributions
Funding
Conflicts of Interest
References
- Aviram, N.; Schuldiner, M. Targeting and translocation of proteins to the endoplasmic reticulum at a glance. J. Cell Sci. 2017, 130, 4079–4085. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.; Thorpe, C. Chemistry and Enzymology of Disulfide Cross-Linking in Proteins. Chem. Rev. 2018, 118, 1169–1198. [Google Scholar] [CrossRef]
- Bošnjak, I.; Bojović, V.; Šegvić-Bubić, T.; Bielen, A. Occurrence of protein disulfide bonds in different domains of life: A comparison of proteins from the Protein Data Bank. Protein Eng. Des. Sel. 2014, 27, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Berndt, C.; Lillig, C.H.; Holmgren, A. Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim. Biophys. Acta 2008, 1783, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef]
- Bulleid, N.J.; Ellgaard, L. Multiple ways to make disulfides. Trends Biochem. Sci. 2011, 36, 485–492. [Google Scholar] [CrossRef]
- Ellgaard, L.; Sevier, C.S.; Bulleid, N.J. How Are Proteins Reduced in the Endoplasmic Reticulum? Trends Biochem. Sci. 2018, 43, 32–43. [Google Scholar] [CrossRef]
- Montero, D.; Tachibana, C.; Rahr Winther, J.; Appenzeller-Herzog, C. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol. 2013, 1, 508–513. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef]
- Creighton, T.E. Disulfide bonds as probes of protein folding pathways. Methods Enzymol. 1986, 131, 83–106. [Google Scholar] [CrossRef]
- Arolas, J.L.; Aviles, F.X.; Chang, J.Y.; Ventura, S. Folding of small disulfide-rich proteins: Clarifying the puzzle. Trends Biochem. Sci. 2006, 31, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, B.; Chung, E.W.; Robinson, C.V.; Mateo, P.L.; Dobson, C.M. The oxidative refolding of hen lysozyme and its catalysis by protein disulfide isomerase. EMBO J. 1999, 18, 4794–4803. [Google Scholar] [CrossRef] [PubMed]
- Narayan, M. The Structure-Forming Juncture in Oxidative Protein Folding: What Happens in the ER? Adv. Exp. Med. Biol. 2017, 966, 163–179. [Google Scholar] [CrossRef]
- Robinson, P.J.; Pringle, M.A.; Woolhead, C.A.; Bulleid, N.J. Folding of a single domain protein entering the endoplasmic reticulum precedes disulfide formation. J. Biol. Chem. 2017, 292, 6978–6986. [Google Scholar] [CrossRef]
- Qin, M.; Wang, W.; Thirumalai, D. Protein folding guides disulfide bond formation. Proc. Natl. Acad. Sci. USA 2015, 112, 11241–11246. [Google Scholar] [CrossRef]
- Kosuri, P.; Alegre-Cebollada, J.; Feng, J.; Kaplan, A.; Ingles-Prieto, A.; Badilla, C.L.; Stockwell, B.R.; Sanchez-Ruiz, J.M.; Holmgren, A.; Fernandez, J.M. Protein folding drives disulfide formation. Cell 2012, 151, 794–806. [Google Scholar] [CrossRef]
- Wallis, A.K.; Freedman, R.B. Assisting oxidative protein folding: How do protein disulphide-isomerases couple conformational and chemical processes in protein folding? Top. Curr. Chem. 2013, 328, 1–34. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Camacho, C.J.; Thirumalai, D. Modeling the role of disulfide bonds in protein folding: Entropic barriers and pathways. Proteins 1995, 22, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Fass, D. Disulfide bonding in protein biophysics. Annu. Rev. Biophys. 2012, 41, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Welker, E.; Wedemeyer, W.J.; Narayan, M.; Scheraga, H.A. Coupling of conformational folding and disulfide-bond reactions in oxidative folding of proteins. Biochemistry 2001, 40, 9059–9064. [Google Scholar] [CrossRef] [PubMed]
- Feige, M.J.; Braakman, I.; Hendershot, L.M. CHAPTER 1.1 Disulfide Bonds in Protein Folding and Stability. In Oxidative Folding of Proteins: Basic Principles, Cellular Regulation and Engineering; The Royal Society of Chemistry: London, UK, 2018; pp. 1–33. [Google Scholar] [CrossRef]
- Chang, J.Y. Diverse pathways of oxidative folding of disulfide proteins: Underlying causes and folding models. Biochemistry 2011, 50, 3414–3431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ignatova, Z. Folding at the birth of the nascent chain: Coordinating translation with co-translational folding. Curr. Opin. Struct. Biol. 2011, 21, 25–31. [Google Scholar] [CrossRef]
- Chen, W.; Helenius, A. Role of ribosome and translocon complex during folding of influenza hemagglutinin in the endoplasmic reticulum of living cells. Mol. Biol. Cell 2000, 11, 765–772. [Google Scholar] [CrossRef][Green Version]
- Ellgaard, L.; McCaul, N.; Chatsisvili, A.; Braakman, I. Co- and Post-Translational Protein Folding in the ER. Traffic 2016, 17, 615–638. [Google Scholar] [CrossRef]
- Haran, G. How, when and why proteins collapse: The relation to folding. Curr. Opin. Struct. Biol. 2012, 22, 14–20. [Google Scholar] [CrossRef]
- Krishna, M.M.; Englander, S.W. The N-terminal to C-terminal motif in protein folding and function. Proc. Natl. Acad. Sci. USA 2005, 102, 1053–1058. [Google Scholar] [CrossRef]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Walsh, I.M.; Bowman, M.A.; Soto Santarriaga, I.F.; Rodriguez, A.; Clark, P.L. Synonymous codon substitutions perturb cotranslational protein folding in vivo and impair cell fitness. Proc. Natl. Acad. Sci. USA 2020, 117, 3528–3534. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A. Unraveling co-translational protein folding: Concepts and methods. Methods 2018, 137, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, L.D.; Hsu, S.T.; Launay, H.; Dobson, C.M.; Christodoulou, J. Probing ribosome-nascent chain complexes produced in vivo by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 22239–22244. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, L.D.; Cassaignau, A.M.E.; Launay, H.M.M.; Waudby, C.A.; Wlodarski, T.; Camilloni, C.; Karyadi, M.E.; Robertson, A.L.; Wang, X.; Wentink, A.S.; et al. A structural ensemble of a ribosome-nascent chain complex during cotranslational protein folding. Nat. Struct. Mol. Biol. 2016, 23, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.; Küng, S.; Martoglio, B.; Helenius, A. Protein folding during cotranslational translocation in the endoplasmic reticulum. Mol. Cell 2002, 10, 769–778. [Google Scholar] [CrossRef]
- Jansens, A.; van Duijn, E.; Braakman, I. Coordinated nonvectorial folding in a newly synthesized multidomain protein. Science 2002, 298, 2401–2403. [Google Scholar] [CrossRef]
- Woolhead, C.A.; McCormick, P.J.; Johnson, A.E. Nascent membrane and secretory proteins differ in FRET-detected folding far inside the ribosome and in their exposure to ribosomal proteins. Cell 2004, 116, 725–736. [Google Scholar] [CrossRef]
- Natan, E.; Wells, J.N.; Teichmann, S.A.; Marsh, J.A. Regulation, evolution and consequences of cotranslational protein complex assembly. Curr. Opin. Struct. Biol. 2017, 42, 90–97. [Google Scholar] [CrossRef]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta 2014, 1840, 838–846. [Google Scholar] [CrossRef]
- Braakman, I.; Helenius, J.; Helenius, A. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO J. 1992, 11, 1717–1722. [Google Scholar] [CrossRef]
- Peters, T.; Davidson, L.K. The biosynthesis of rat serum albumin. In vivo studies on the formation of the disulfide bonds. J. Biol. Chem. 1982, 257, 8847–8853. [Google Scholar] [PubMed]
- Bergman, L.W.; Kuehl, W.M. Formation of an intrachain disulfide bond on nascent immunoglobulin light chains. J. Biol. Chem. 1979, 254, 8869–8876. [Google Scholar] [PubMed]
- Braakman, I.; Hoover-Litty, H.; Wagner, K.R.; Helenius, A. Folding of influenza hemagglutinin in the endoplasmic reticulum. J. Cell Biol. 1991, 114, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Helenius, J.; Braakman, I.; Helenius, A. Cotranslational folding and calnexin binding during glycoprotein synthesis. Proc. Natl. Acad. Sci. USA 1995, 92, 6229–6233. [Google Scholar] [CrossRef]
- Tatu, U.; Hammond, C.; Helenius, A. Folding and oligomerization of influenza hemagglutinin in the ER and the intermediate compartment. EMBO J. 1995, 14, 1340–1348. [Google Scholar] [CrossRef]
- Kadokura, H.; Dazai, Y.; Fukuda, Y.; Hirai, N.; Nakamura, O.; Inaba, K. Observing the nonvectorial yet cotranslational folding of a multidomain protein, LDL receptor, in the ER of mammalian cells. Proc. Natl. Acad. Sci. USA 2020, 117, 16401–16408. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- McGinnes, L.W.; Morrison, T.G. Role of cotranslational disulfide bond formation in the folding of the hemagglutinin-neuraminidase protein of Newcastle disease virus. Virology 1996, 224, 465–476. [Google Scholar] [CrossRef]
- Robinson, P.J.; Kanemura, S.; Cao, X.; Bulleid, N.J. Protein secondary structure determines the temporal relationship between folding and disulfide formation. J. Biol. Chem. 2020, 295, 2438–2448. [Google Scholar] [CrossRef]
- Lambert, N.; Freedman, R.B. The latency of rat liver microsomal protein disulphide-isomerase. Biochem. J. 1985, 228, 635–645. [Google Scholar] [CrossRef]
- Lyles, M.M.; Gilbert, H.F. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: Pre-steady-state kinetics and the utilization of the oxidizing equivalents of the isomerase. Biochemistry 1991, 30, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Kadokura, H.; Inaba, K. Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 83, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, B.B.; Holmgren, A.; Jacquot, J.P.; Scheibe, R. Fifty years in the thioredoxin field and a bountiful harvest. Biochim. Biophys. Acta 2012, 1820, 1822–1829. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Thioredoxin. The amino acid sequence of the protein from escherichia coli B. Eur. J. Biochem. 1968, 6, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Edman, J.C.; Ellis, L.; Blacher, R.W.; Roth, R.A.; Rutter, W.J. Sequence of protein disulphide isomerase and implications of its relationship to thioredoxin. Nature 1985, 317, 267–270. [Google Scholar] [CrossRef]
- Bach, R.D.; Dmitrenko, O.; Thorpe, C. Mechanism of thiolate-disulfide interchange reactions in biochemistry. J. Org. Chem. 2008, 73, 12–21. [Google Scholar] [CrossRef]
- Creighton, T.E.; Hillson, D.A.; Freedman, R.B. Catalysis by protein-disulphide isomerase of the unfolding and refolding of proteins with disulphide bonds. J. Mol. Biol. 1980, 142, 43–62. [Google Scholar] [CrossRef]
- Quan, S.; Schneider, I.; Pan, J.; Von Hacht, A.; Bardwell, J.C. The CXXC motif is more than a redox rheostat. J. Biol. Chem. 2007, 282, 28823–28833. [Google Scholar] [CrossRef]
- Oka, O.B.; Bulleid, N.J. Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2425–2429. [Google Scholar] [CrossRef]
- Tavender, T.J.; Bulleid, N.J. Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum. Antioxid. Redox Signal. 2010, 13, 1177–1187. [Google Scholar] [CrossRef]
- Moilanen, A.; Ruddock, L.W. Non-native proteins inhibit the ER oxidoreductin 1 (Ero1)-protein disulfide-isomerase relay when protein folding capacity is exceeded. J. Biol. Chem. 2020, 295, 8647–8655. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Riemer, J.; Christensen, B.; Sørensen, E.S.; Ellgaard, L. A novel disulphide switch mechanism in Ero1alpha balances ER oxidation in human cells. EMBO J. 2008, 27, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Tavender, T.J.; Springate, J.J.; Bulleid, N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010, 29, 4185–4197. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.D.; Saaranen, M.J.; Karala, A.R.; Lappi, A.K.; Wang, L.; Raykhel, I.B.; Alanen, H.I.; Salo, K.E.; Wang, C.C.; Ruddock, L.W. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 2011, 406, 503–515. [Google Scholar] [CrossRef]
- Schulman, S.; Wang, B.; Li, W.; Rapoport, T.A. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc. Natl. Acad. Sci. USA 2010, 107, 15027–15032. [Google Scholar] [CrossRef]
- Oka, O.B.; Yeoh, H.Y.; Bulleid, N.J. Thiol-disulfide exchange between the PDI family of oxidoreductases negates the requirement for an oxidase or reductase for each enzyme. Biochem. J. 2015, 469, 279–288. [Google Scholar] [CrossRef]
- Römer, R.A.; Wells, S.A.; Emilio Jimenez-Roldan, J.; Bhattacharyya, M.; Vishweshwara, S.; Freedman, R.B. The flexibility and dynamics of protein disulfide isomerase. Proteins 2016, 84, 1776–1785. [Google Scholar] [CrossRef]
- Okumura, M.; Noi, K.; Kanemura, S.; Kinoshita, M.; Saio, T.; Inoue, Y.; Hikima, T.; Akiyama, S.; Ogura, T.; Inaba, K. Dynamic assembly of protein disulfide isomerase in catalysis of oxidative folding. Nat. Chem. Biol. 2019, 15, 499–509. [Google Scholar] [CrossRef]
- Irvine, A.G.; Wallis, A.K.; Sanghera, N.; Rowe, M.L.; Ruddock, L.W.; Howard, M.J.; Williamson, R.A.; Blindauer, C.A.; Freedman, R.B. Protein disulfide-isomerase interacts with a substrate protein at all stages along its folding pathway. PLoS ONE 2014, 9, e82511. [Google Scholar] [CrossRef]
- Molinari, M.; Helenius, A. Glycoproteins form mixed disulphides with oxidoreductases during folding in living cells. Nature 1999, 402, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Kolšek, K.; Aponte-Santamaría, C.; Gräter, F. Accessibility explains preferred thiol-disulfide isomerization in a protein domain. Sci. Rep. 2017, 7, 9858. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Cebollada, J.; Kosuri, P.; Rivas-Pardo, J.A.; Fernández, J.M. Direct observation of disulfide isomerization in a single protein. Nat. Chem. 2011, 3, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Poet, G.J.; Oka, O.B.; van Lith, M.; Cao, Z.; Robinson, P.J.; Pringle, M.A.; Arnér, E.S.; Bulleid, N.J. Cytosolic thioredoxin reductase 1 is required for correct disulfide formation in the ER. EMBO J. 2017, 36, 693–702. [Google Scholar] [CrossRef]
- Cao, X.; Lilla, S.; Cao, Z.; Pringle, M.A.; Oka, O.B.V.; Robinson, P.J.; Szmaja, T.; van Lith, M.; Zanivan, S.; Bulleid, N.J. The mammalian cytosolic thioredoxin reductase pathway acts via a membrane protein to reduce ER-localised proteins. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Oka, O.B.; Pringle, M.A.; Schopp, I.M.; Braakman, I.; Bulleid, N.J. ERdj5 is the ER reductase that catalyzes the removal of non-native disulfides and correct folding of the LDL receptor. Mol. Cell 2013, 50, 793–804. [Google Scholar] [CrossRef]
- Hirano, N.; Shibasaki, F.; Sakai, R.; Tanaka, T.; Nishida, J.; Yazaki, Y.; Takenawa, T.; Hirai, H. Molecular cloning of the human glucose-regulated protein ERp57/GRP58, a thiol-dependent reductase. Identification of its secretory form and inducible expression by the oncogenic transformation. Eur. J. Biochem. 1995, 234, 336–342. [Google Scholar] [CrossRef]
- Daniels, R.; Kurowski, B.; Johnson, A.E.; Hebert, D.N. N-linked glycans direct the cotranslational folding pathway of influenza hemagglutinin. Mol. Cell 2003, 11, 79–90. [Google Scholar] [CrossRef]
- Brar, G.A.; Weissman, J.S. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 651–664. [Google Scholar] [CrossRef]
- Cherepanova, N.A.; Shrimal, S.; Gilmore, R. Oxidoreductase activity is necessary for N-glycosylation of cysteine-proximal acceptor sites in glycoproteins. J. Cell Biol. 2014, 206, 525–539. [Google Scholar] [CrossRef]
- Land, A.; Zonneveld, D.; Braakman, I. Folding of HIV-1 envelope glycoprotein involves extensive isomerization of disulfide bonds and conformation-dependent leader peptide cleavage. FASEB J. 2003, 17, 1058–1067. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, P.J.; Bulleid, N.J. Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells 2020, 9, 1994. https://doi.org/10.3390/cells9091994
Robinson PJ, Bulleid NJ. Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells. 2020; 9(9):1994. https://doi.org/10.3390/cells9091994
Chicago/Turabian StyleRobinson, Philip J., and Neil J. Bulleid. 2020. "Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway" Cells 9, no. 9: 1994. https://doi.org/10.3390/cells9091994
APA StyleRobinson, P. J., & Bulleid, N. J. (2020). Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells, 9(9), 1994. https://doi.org/10.3390/cells9091994