
Leukocyte Trafficking via Lymphatic Vessels in Atherosclerosis


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Atherosclerosis: A Chronic Inflammatory Disease of the Arterial Wall
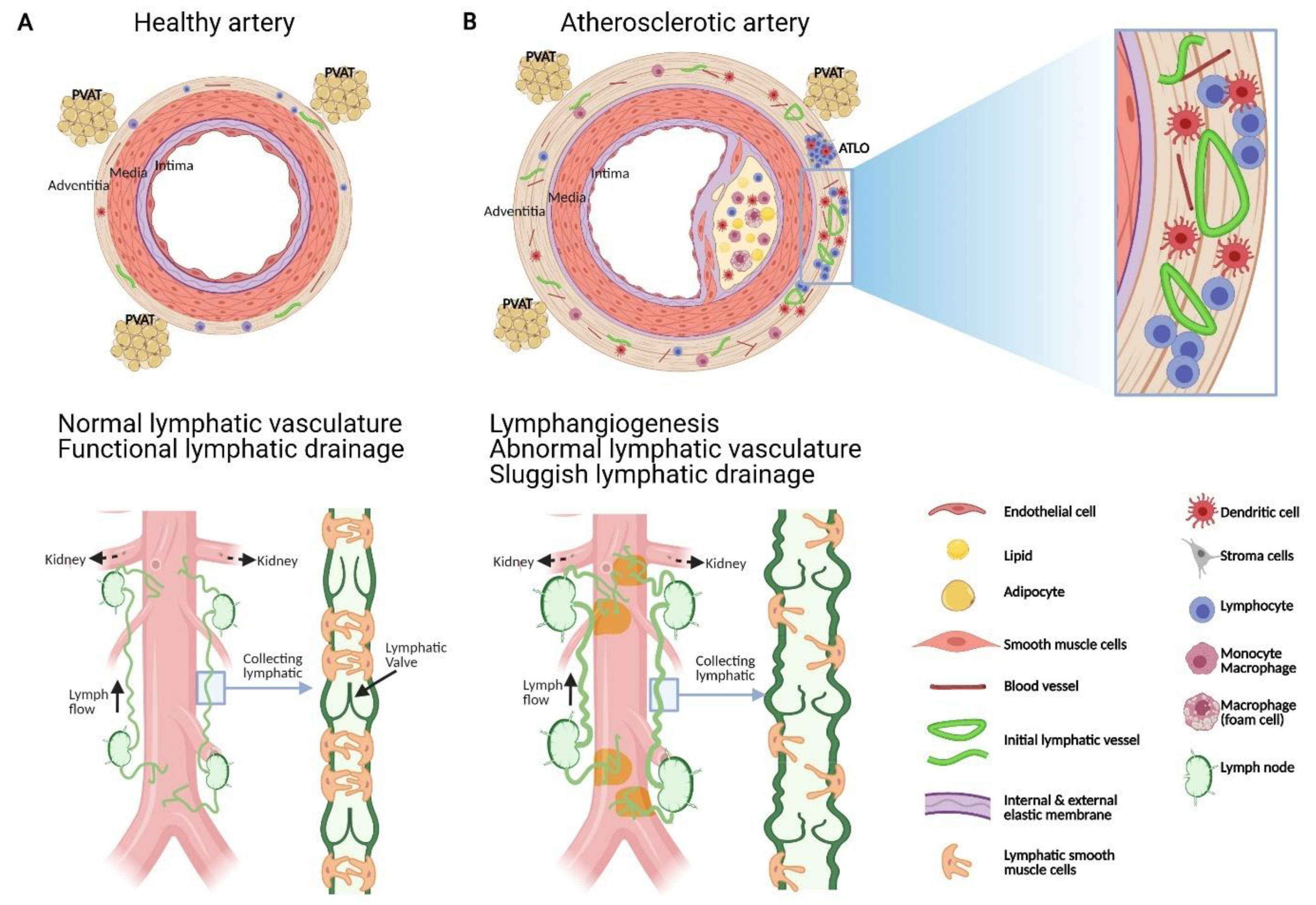
2.1. Intimal Atherosclerotic Plaque Development and Progression
2.2. Adventitial Inflammatory and Immune Response in Atherosclerosis
3. Adventitial Blood and Lymphatic Vasculature
3.1. Adventitial Neovascularization of Vasa Vasorum in Atherosclerosis
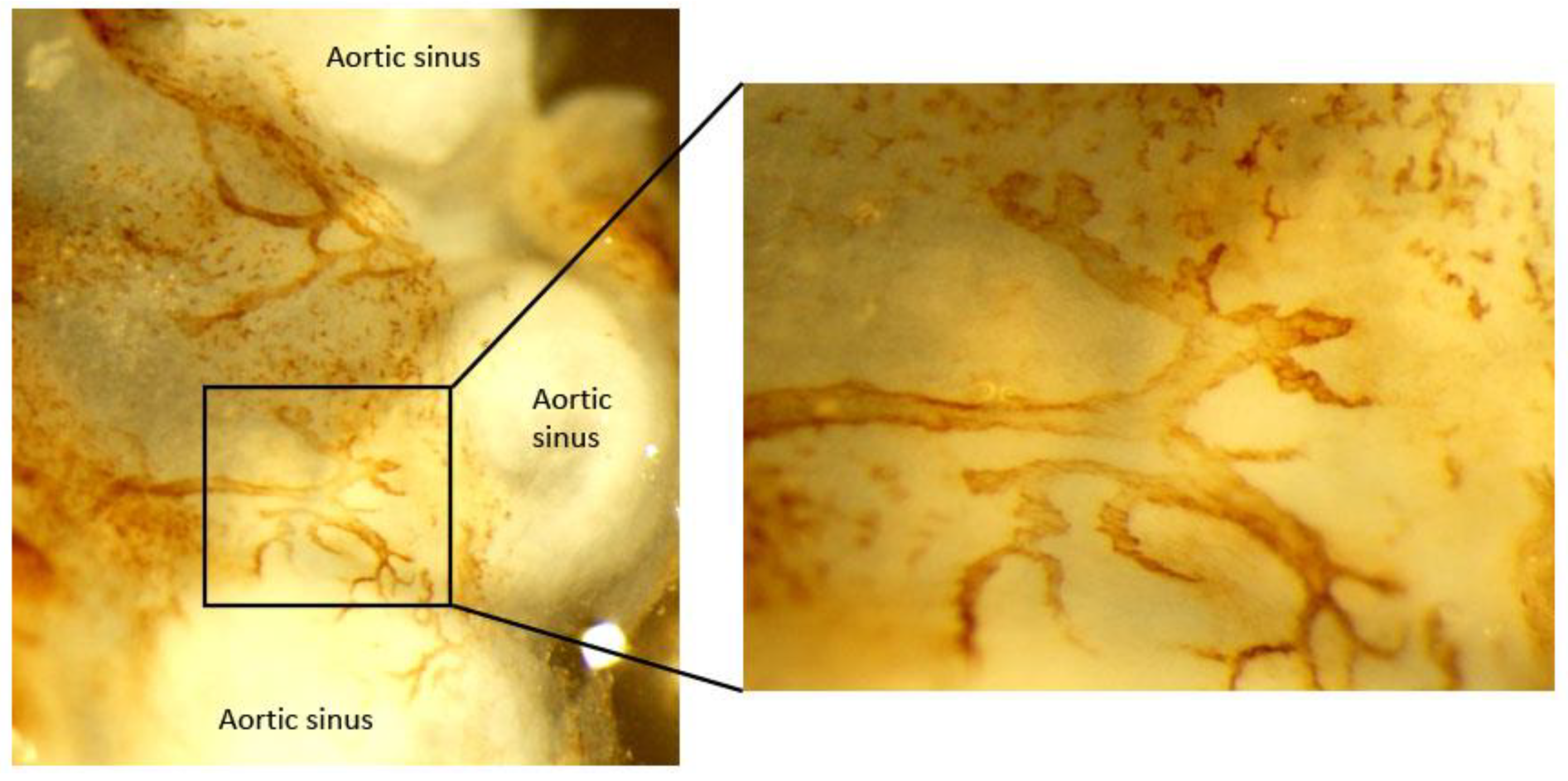
3.2. Adventitial Lymphatic Vessels
3.2.1. Lymphatic Vessel Distribution in Normal Artery
3.2.2. Morphological and Density Alterations in Lymphatic Vessels during Atherosclerosis
4. Functional Roles of Adventitial Lymphatic Vessels in Atherosclerosis
4.1. Emigration of Immune Cells to Lymph Nodes
4.1.1. Emigration of Monocyte, Macrophage, and Dendritic Cell
4.1.2. Emigration of T Lymphocyte
4.2. Lymphoid Neogenesis
4.3. Drainage of Macromolecules
5. Is lymphatic Drainage Defective in Atherosclerosis?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tammela, T.; Alitalo, K. Lymphangiogenesis: Molecular Mechanisms and Future Promise. Cell 2010, 140, 460–476. [Google Scholar] [CrossRef]
- Srinivasan, R.S.; Escobedo, N.; Yang, Y.; Interiano, A.; Dillard, M.E.; Finkelstein, D.; Mukatira, S.; Gil, H.J.; Nurmi, H.; Alitalo, K.; et al. The Prox1–Vegfr3 feedback loop maintains the identity and the number of lymphatic endothelial cell progenitors. Genes Dev. 2014, 28, 2175–2187. [Google Scholar] [CrossRef]
- Tammela, T.; Petrova, T.V.; Alitalo, K. Molecular lymphangiogenesis: New players. Trends Cell Biol. 2005, 15, 434–441. [Google Scholar] [CrossRef]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef]
- Alitalo, K.; Tammela, T.; Petrova, T.V. Lymphangiogenesis in development and human disease. Nature 2005, 438, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Bridenbaugh, E.A.; Gashev, A.A.; Zawieja, D.C. Lymphatic Muscle: A Review of Contractile Function. Lymphat. Res. Biol. 2003, 1, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Sabine, A.; Demir, C.S.; Petrova, T.V. Endothelial Cell Responses to Biomechanical Forces in Lymphatic Vessels. Antioxid. Redox Signal. 2016, 25, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Scallan, J.P.; Zawieja, S.D.; Castorena-Gonzalez, J.; Davis, M.J. Lymphatic pumping: Mechanics, mechanisms and malfunction. J. Physiol. 2016, 594, 5749–5768. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.; Alitalo, K. The lymphatic vasculature: Recent Progress and Paradigms. Annu. Rev. Cell Dev. Biol. 2005, 21, 457–483. [Google Scholar] [CrossRef]
- Petrova, T.V.; Koh, G.Y. Organ-specific lymphatic vasculature: From development to pathophysiology. J. Exp. Med. 2018, 215, 35–49. [Google Scholar] [CrossRef]
- Oliver, G. Lymphatic vasculature development. Nat. Rev. Immunol. 2004, 4, 35–45. [Google Scholar] [CrossRef]
- Oliver, G.; Kipnis, J.; Randolph, G.J.; Harvey, N.L. The Lymphatic Vasculature in the 21st Century: Novel Functional Roles in Homeostasis and Disease. Cell 2020, 182, 270–296. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in Atherosclerosis—No Longer a Theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Rocha, V.Z.; Libby, P. Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 2009, 6, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Pentikainen, M.O.; Öörni, K.; Ala-Korpela, M.; Kovanen, P.T. Modified LDL—Trigger of atherosclerosis and inflammation in the arterial intima. J. Intern. Med. 2000, 247, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Fayad, Z.A.; Swirski, F.K.; Calcagno, C.; Robbins, C.S.; Mulder, W.; Kovacic, J.C. Monocyte and Macrophage Dynamics in the Cardiovascular System: JACC Macrophage in CVD Series (Part 3). J. Am. Coll. Cardiol. 2018, 72, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and Inflammatory Mechanisms of Atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Zernecke, A. Dendritic cells in atherosclerosis: Evidence in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.E.; Zhu, S.-N.; Chen, M.; Nurmohamed, S.; Jongstra-Bilen, J.; Cybulsky, M.I. Resident Intimal Dendritic Cells Accumulate Lipid and Contribute to the Initiation of Atherosclerosis. Circ. Res. 2010, 106, 383–390. [Google Scholar] [CrossRef]
- Major, A.S.; Fazio, S.; Linton, M.F. B-Lymphocyte Deficiency Increases Atherosclerosis in LDL Receptor–Null Mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1892–1898. [Google Scholar] [CrossRef]
- Douna, H.; Kuiper, J. Novel B-cell subsets in atherosclerosis. Curr. Opin. Lipidol. 2016, 27, 493–498. [Google Scholar] [CrossRef]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef]
- Ketelhuth, D.F.; Hansson, G.K. Adaptive Response of T and B Cells in Atherosclerosis. Circ. Res. 2016, 118, 668–678. [Google Scholar] [CrossRef]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Schiopu, A.; Bengtsson, E.; Goncalves, I.; Nilsson, J.; Fredrikson, G.N.; Björkbacka, H. Associations Between Macrophage Colony-Stimulating Factor and Monocyte Chemotactic Protein 1 in Plasma and First-Time Coronary Events: A Nested Case-Control Study. J. Am. Heart Assoc. 2016, 5, e002851. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.E.; Ljungcrantz, I.; Andersson, L.; Bryngelsson, C.; Hedblad, B.; Fredrikson, G.N.; Nilsson, J.; Björkbacka, H. Elevated CD14++ CD16− Monocytes Predict Cardiovascular Events. Circ. Cardiovasc. Genet. 2012, 5, 122–131. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.; Sjouke, B.; Choque, B.; Kastelein, J.J.P.; Hovingh, G.K. The PCSK9 decade. J. Lipid Res. 2012, 53, 2515–2524. [Google Scholar] [CrossRef]
- Mohler, E.R., 3rd; Ballantyne, C.M.; Davidson, M.H.; Hanefeld, M.; Ruilope, L.M.; Johnson, J.L.; Zalewski, A.; Darapladib Investigators. The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: The results of a multicenter, randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol. 2008, 51, 1632–1641. [Google Scholar] [PubMed]
- Serruys, P.W.; García-García, H.M.; Buszman, P.; Erne, P.; Verheye, S.; Aschermann, M.; Duckers, H.; Bleie, O.; Dudek, D.; Bøtker, H.E.; et al. Effects of the Direct Lipoprotein-Associated Phospholipase A 2 Inhibitor Darapladib on Human Coronary Atherosclerotic Plaque. Circulation 2008, 118, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.A.; Lipinski, M.J.; Doran, A.C.; Skaflen, M.D.; Fuster, V.; McNamara, C.A. Lymphocytes and the Adventitial Immune Response in Atherosclerosis. Circ. Res. 2012, 110, 889–900. [Google Scholar] [CrossRef]
- Maiellaro, K.; Taylor, W.R. The role of the adventitia in vascular inflammation. Cardiovasc. Res. 2007, 75, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Moos, M.P.; John, N.; Gräbner, R.; Noßmann, S.; Guünther, B.; Vollandt, R.; Funk, C.D.; Kaiser, B.; Habenicht, A.J. The Lamina Adventitia Is the Major Site of Immune Cell Accumulation in Standard Chow-Fed Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2386–2391. [Google Scholar] [CrossRef]
- Barker, S.G.; Tilling, L.C.; Miller, G.C.; Beesley, J.E.; Fleetwood, G.; Stavri, G.T.; Baskerville, P.A.; Martin, J.F. The adventitia and atherogenesis: Removal initiates intimal proliferation in the rabbit which regresses on generation of a ‘neoadventitia’. Atherosclerosis 1994, 105, 131–144. [Google Scholar] [CrossRef]
- Barker, S.; Beesley, J.; Baskerville, P.; Martin, J. The influence of the adventitia on the presence of smooth muscle cells and macrophages in the arterial intima. Eur. J. Vasc. Endovasc. Surg. 1995, 9, 222–227. [Google Scholar] [CrossRef]
- Higuchi, M.L.; Gutierrez, P.S.; Bezerra, H.G.; Palomino, S.A.; Aiello, V.D.; Silvestre, J.M.L.; Libby, P.; Ramires, J.A.F. Comparison between Adventitial and Intimal Inflammation of Ruptured and Nonruptured Atherosclerotic Plaques in Human Coronary Arteries. Arq. Bras. Cardiol. 2002, 79, 20–24. [Google Scholar] [CrossRef]
- Houtkamp, M.A.; De Boer, O.J.; Van Der Loos, C.M.; Van Der Wal, A.C.; Becker, A.E. Adventitial infiltrates associated with advanced atherosclerotic plaques: Structural organization suggests generation of local humoral immune responses. J. Pathol. 2001, 193, 263–269. [Google Scholar] [CrossRef]
- Tinajero, M.G.; Gotlieb, A.I. Recent Developments in Vascular Adventitial Pathobiology: The Dynamic Adventitia as a Complex Regulator of Vascular Disease. Am. J. Pathol. 2020, 190, 520–534. [Google Scholar] [CrossRef]
- Gerlis, L.M. The significance of adventitial infiltrations in coronary atherosclerosis. Br. Heart J. 1956, 18, 166–172. [Google Scholar] [CrossRef]
- Schwartz, C.J.; Mitchell, J.R.A. Cellular Infiltration of the Human Arterial Adventitia Associated with Atheromatous Plaques. Circulation 1962, 26, 73–78. [Google Scholar] [CrossRef]
- Ramshaw, A.; Parums, D. Immunohistochemical characterization of inflammatory cells associated with advanced atherosclerosis. Histopathology 1990, 17, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Ni, Z.; Tan, Y.-Q.; Deng, J.; Zhang, S.-J.; Lv, Z.-C.; Wang, X.-J.; Chen, T.; Zhang, Z.; Hu, Y.; et al. Adventitial Cell Atlas of wt (Wild Type) and ApoE (Apolipoprotein E)-Deficient Mice Defined by Single-Cell RNA Sequencing. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1055–1071. [Google Scholar] [CrossRef]
- Galkina, E.; Kadl, A.; Sanders, J.; Varughese, D.; Sarembock, I.J.; Ley, K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J. Exp. Med. 2006, 203, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Kratz, A.; Campos-Neto, A.; Hanson, M.S.; Ruddle, N.H. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J. Exp. Med. 1996, 183, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Drayton, D.L.; Liao, S.; Mounzer, R.H.; Ruddle, N.H. Lymphoid organ development: From ontogeny to neogenesis. Nat. Immunol. 2006, 7, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Mohanta, S.; Srikakulapu, P.; Weber, C.; Habenicht, A. Artery Tertiary Lymphoid Organs: Powerhouses of Atherosclerosis Immunity. Front. Immunol. 2016, 7, 387. [Google Scholar] [CrossRef]
- Milutinović, A.; Šuput, D.; Zorc-Pleskovič, R. Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: An updated review. Bosn. J. Basic Med. Sci. 2020, 20, 21–30. [Google Scholar] [CrossRef]
- Mohanta, S.; Yin, C.; Peng, L.; Srikakulapu, P.; Bontha, V.; Hu, D.; Weih, F.; Weber, C.; Gerdes, N.; Habenicht, A.J. Artery Tertiary Lymphoid Organs Contribute to Innate and Adaptive Immune Responses in Advanced Mouse Atherosclerosis. Circ. Res. 2014, 114, 1772–1787. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R.; Purushothaman, K.R.; Fuster, V.; O’Connor, W.N. Intimomedial interface damage and adventitial inflammation is increased beneath disrupted atherosclerosis in the aorta: Implications for plaque vulnerability. Circulation 2002, 105, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Romen, M.; Amberger, A.; Metzler, B.; Mayr, M.; Falkensammer, G.; Xu, Q. Atherosclerosis, autoimmunity, and vascular-associated lymphoid tissue. FASEB J. 1997, 11, 1199–1207. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, F.; Bai, P.; Dong, N.; Chu, C. Identification of key genes and pathways contributing to artery tertiary lymphoid organ development in advanced mouse atherosclerosis. Mol. Med. Rep. 2019, 19, 3071–3086. [Google Scholar] [CrossRef] [PubMed]
- Gräbner, R.; Lötzer, K.; Döpping, S.; Hildner, M.; Radke, D.; Beer, M.; Spanbroek, R.; Lippert, B.; Reardon, C.A.; Getz, G.S.; et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J. Exp. Med. 2009, 206, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Mohanta, S.K.; Yin, C.; Peng, L.; Ma, Z.; Srikakulapu, P.; Grassia, G.; MacRitchie, N.; Dever, G.; Gordon, P.; et al. Artery Tertiary Lymphoid Organs Control Aorta Immunity and Protect against Atherosclerosis via Vascular Smooth Muscle Cell Lymphotoxin beta Receptors. Immunity 2015, 42, 1100–1115. [Google Scholar] [CrossRef]
- Ritman, E.L.; Lerman, A. The dynamic vasa vasorum. Cardiovasc. Res. 2007, 75, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Heistad, D.D.; Marcus, M.L.; Larsen, G.E.; Armstrong, M.L. Role of vasa vasorum in nourishment of the aortic wall. Am. J. Physiol. 1981, 240, H781–H787. [Google Scholar] [CrossRef]
- Nakata, Y.; Shionoya, S. Vascular Lesions due to Obstruction of the Vasa Vasorum. Nature 1966, 212, 1258–1259. [Google Scholar] [CrossRef]
- Mann, F.D. Vasa vasorum and coronary atherosclerosis. Lancet 1978, 1, 1319–1320. [Google Scholar] [CrossRef]
- Barger, A.C.; Beeuwkes, R.; Lainey, L.L.; Silverman, K.J. Hypothesis: Vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N. Engl. J. Med. 1984, 310, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.G.; Talbert, A.; Cottam, S.; Baskerville, P.A.; Martin, J.F. Arterial intimal hyperplasia after occlusion of the adventitial vasa vasorum in the pig. Arterioscler. Thromb. 1993, 13, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.M.; Sangiorgi, G.; Ritman, E.L.; McKenna, C.; Holmes, D.R.; Schwartz, R.S.; Lerman, A. Enhanced coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J. Clin. Investig. 1998, 101, 1551–1556. [Google Scholar] [CrossRef]
- Herrmann, J.; Lerman, L.O.; Rodriguez-Porcel, M.; Holmes, D.R.; Richardson, D.M.; Ritman, E.L.; Lerman, A. Coronary vasa vasorum neovascularization precedes epicardial endothelial dysfunction in experimental hypercholesterolemia. Cardiovasc. Res. 2001, 51, 762–766. [Google Scholar] [CrossRef]
- Marcus, M.L.; Heistad, D.D.; Armstrong, M.L.; Abboud, F.M. Effects of chronic hypertension on vasa vasorum in the thoracic aorta. Cardiovasc. Res. 1985, 19, 777–781. [Google Scholar] [CrossRef]
- Kai, H.; Kuwahara, F.; Tokuda, K.; Shibata, R.; Kusaba, K.; Niiyama, H.; Tahara, N.; Nagata, T.; Imaizumi, T. Coexistence of Hypercholesterolemia and Hypertension Impairs Adventitial Vascularization. Hypertension 2002, 39, 455–459. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Shibata, R.; Kusaba, K.; Tahara, N.; Niiyama, H.; Nagata, T.; Imaizumi, T. Hypoxia-Inducible Factor-1α/Vascular Endothelial Growth Factor Pathway for Adventitial Vasa Vasorum Formation in Hypertensive Rat Aorta. Hypertension 2002, 39, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Booth, R.; Martin, J.; Honey, A.; Hassall, D.; Beesley, J.; Moncada, S. Rapid development of atherosclerotic lesions in the rabbit carotid artery induced by perivascular manipulation. Atherosclerosis 1989, 76, 257–268. [Google Scholar] [CrossRef]
- Zhang, Y.; Cliff, W.J.; Schoefl, G.I.; Higgins, G. Immunohistochemical study of intimal microvessels in coronary atherosclerosis. Am. J. Pathol. 1993, 143, 164–172. [Google Scholar]
- Langheinrich, A.C.; Michniewicz, A.; Sedding, D.G.; Walker, G.; Beighley, P.E.; Rau, W.S.; Bohle, R.M.; Ritman, E.L. Correlation of vasa vasorum neovascularization and plaque progression in aortas of apolipoprotein E(−/−)/low-density lipoprotein(−/−) double knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, M.; Nakashima, Y.; Sueishi, K. Intimal neovascularization in human coronary atherosclerosis: Its origin and pathophysiological significance. Hum. Pathol. 1995, 26, 450–456. [Google Scholar] [CrossRef]
- Moreno, P.R.; Purushothaman, K.R.; Fuster, V.; Echeverri, D.; Truszczynska, H.; Sharma, S.K.; Badimon, J.J.; O’Connor, W.N. Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: Implications for plaque vulnerability. Circulation 2004, 110, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis Inhibitors Endostatin or TNP-470 Reduce Intimal Neovascularization and Plaque Growth in Apolipoprotein E-Deficient Mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef]
- Moulton, K.S.; Vakili, K.; Zurakowski, D.; Soliman, M.; Butterfield, C.; Sylvin, E.; Lo, K.-M.; Gillies, S.; Javaherian, K.; Folkman, J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 4736–4741. [Google Scholar] [CrossRef]
- Bates, D.; Harper, S. Regulation of vascular permeability by vascular endothelial growth factors. Vasc. Pharmacol. 2002, 39, 225–237. [Google Scholar] [CrossRef]
- Nielsen, L.B. Atherogenecity of lipoprotein(a) and oxidized low density lipoprotein: Insight from in vivo studies of arterial wall influx, degradation and efflux. Atherosclerosis 1999, 143, 229–243. [Google Scholar] [CrossRef]
- Hoggan, G.; Hoggan, F.E. The Lymphatics of the Walls of the Larger Blood-Vessels and Lymphatics. J. Anat. Physiol. 1882, 17 Pt 1, 1–23. [Google Scholar]
- Johnson, R.A. Lymphatics of blood vessels. Lymphology 1969, 2, 44–56. [Google Scholar]
- Sacchi, G.; Weber, E.; Comparini, L. Histological framework of lymphatic vasa vasorum of major arteries: An experimental study. Lymphology 1990, 23, 135–139. [Google Scholar] [PubMed]
- Xu, X.; Lu, H.; Lin, H.; Li, X.; Ni, M.; Sun, H.; Li, C.; Jiang, H.; Li, F.; Zhang, M.; et al. Aortic adventitial angiogenesis and lymphangiogenesis promote intimal inflammation and hyperplasia. Cardiovasc. Pathol. 2009, 18, 269–278. [Google Scholar] [CrossRef]
- Drozdz, K.; Janczak, D.; Dzięgiel, P.; Podhorska, M.; Patrzałek, D.; Ziółkowski, P.; Andrzejak, R.; Szuba, A. Adventitial lymphatics of internal carotid artery in healthy and atherosclerotic vessels. Folia Histochem. Cytobiol. 2008, 46, 433–436. [Google Scholar] [CrossRef]
- Eliska, O.; Eliskova, M.; Miller, A.J. The absence of lymphatics in normal and atherosclerotic coronary arteries in man: A morphologic study. Lymphology 2006, 39, 76–83. [Google Scholar]
- Nakano, T.; Nakashima, Y.; Yonemitsu, Y.; Sumiyoshi, S.; Chen, Y.-X.; Akishima, Y.; Ishii, T.; Iida, M.; Sueishi, K. Angiogenesis and lymphangiogenesis and expression of lymphangiogenic factors in the atherosclerotic intima of human coronary arteries. Hum. Pathol. 2005, 36, 330–340. [Google Scholar] [CrossRef]
- Yeo, K.P.; Lim, H.Y.; Thiam, C.H.; Azhar, S.H.; Tan, C.; Tang, Y.; See, W.Q.; Koh, X.H.; Zhao, M.H.; Phua, M.L.; et al. Efficient aortic lymphatic drainage is necessary for atherosclerosis regression induced by ezetimibe. Sci. Adv. 2020, 6, eabc2697. [Google Scholar] [CrossRef]
- Kunstfeld, R.; Hirakawa, S.; Hong, Y.-K.; Schacht, V.; Lange-Asschenfeldt, B.; Velasco, P.; Lin, C.; Fiebiger, E.; Wei, X.; Wu, Y.; et al. Induction of cutaneous delayed-type hypersensitivity reactions in VEGF-A transgenic mice results in chronic skin inflammation associated with persistent lymphatic hyperplasia. Blood 2004, 104, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Tammela, T.; Ator, E.; Lyubynska, N.; Achen, M.; Hicklin, D.J.; Jeltsch, M.; Petrova, T.V.; Pytowski, B.; Stacker, S.; et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Investig. 2005, 115, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Kholová, I.; Dragneva, G.; Čermáková, P.; Laidinen, S.; Kaskenpää, N.; Hazes, T.; Čermáková, E.; Šteiner, I.; Ylä-Herttuala, S. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur. J. Clin. Investig. 2011, 41, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Kutkut, I.H.Y.; Meens, M.J.; McKee, T.; Bochaton-Piallat, M.-L.; Kwak, B.R. Lymphatic vessels: An emerging actor in atherosclerotic plaque development. Eur. J. Clin. Investig. 2015, 45, 100–108. [Google Scholar] [CrossRef]
- Martel, C.; Li, W.; Fulp, B.; Platt, A.M.; Gautier, E.L.; Westerterp, M.; Bittman, R.; Tall, A.R.; Chen, S.-H.; Thomas, M.J.; et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J. Clin. Investig. 2013, 123, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, T.; Van Der Vorst, E.P.C.; Daissormont, I.T.M.N.; Otten, J.J.T.; Theodorou, K.; Theelen, T.L.; Gijbels, M.; Anisimov, A.; Nurmi, H.; Lindeman, J.H.N.; et al. Adventitial lymphatic capillary expansion impacts on plaque T cell accumulation in atherosclerosis. Sci. Rep. 2017, 7, 45263. [Google Scholar] [CrossRef]
- Singla, B.; Lin, H.-P.; Ahn, W.; White, J.; Csányi, G. Oxidatively Modified LDL Suppresses Lymphangiogenesis via CD36 Signaling. Antioxidants 2021, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.H.D.; Lim, S.Y.J.; Leong, Y.F.I.; Thiam, C.H.; Tan, K.W.; Torta, F.T.; Narayanaswamy, P.; Wenk, M.; Angeli, V. Halted Lymphocyte Egress via Efferent Lymph Contributes to Lymph Node Hypertrophy During Hypercholesterolemia. Front. Immunol. 2019, 10, 575. [Google Scholar] [CrossRef]
- Randolph, G.J.; Ivanov, S.; Zinselmeyer, B.H.; Scallan, J.P. The Lymphatic System: Integral Roles in Immunity. Annu. Rev. Immunol. 2017, 35, 31–52. [Google Scholar] [CrossRef]
- Martens, R.; Permanyer, M.; Werth, K.; Yu, K.; Braun, A.; Halle, O.; Halle, S.; Patzer, G.E.; Bošnjak, B.; Kiefer, F.; et al. Efficient homing of T cells via afferent lymphatics requires mechanical arrest and integrin-supported chemokine guidance. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Johnson, L.A.; Jackson, D.G. Inflammation-induced secretion of CCL21 in lymphatic endothelium is a key regulator of integrin-mediated dendritic cell transmigration. Int. Immunol. 2010, 22, 839–849. [Google Scholar] [CrossRef]
- Johnson, L.A.; Clasper, S.; Holt, A.P.; Lalor, P.; Baban, D.; Jackson, D.G. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J. Exp. Med. 2006, 203, 2763–2777. [Google Scholar] [CrossRef] [PubMed]
- Tal, O.; Lim, H.Y.; Gurevich, I.; Milo, I.; Shipony, Z.; Ng, L.G.; Angeli, V.; Shakhar, G. DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J. Exp. Med. 2011, 208, 2141–2153. [Google Scholar] [CrossRef]
- Debes, G.F.; Arnold, C.N.; Young, A.J.; Krautwald, S.; Lipp, M.; Hay, J.B.; Butcher, E.C. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol. 2005, 6, 889–894. [Google Scholar] [CrossRef]
- Bromley, S.K.; Thomas, S.Y.; Luster, A.D. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol. 2005, 6, 895–901. [Google Scholar] [CrossRef]
- Randolph, G.J. Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr. Opin. Lipidol. 2008, 19, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J. The fate of monocytes in atherosclerosis. J. Thromb. Haemost. 2009, 7 (Suppl. S1), 28–30. [Google Scholar] [CrossRef]
- Bellingan, G.J.; Caldwell, H.; Howie, S.E.; Dransfield, I.; Haslett, C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: Inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J. Immunol. 1996, 157, 2577–2585. [Google Scholar] [PubMed]
- Williams, K.J.; Feig, J.E.; Fisher, E.A. Rapid regression of atherosclerosis: Insights from the clinical and experimental literature. Nat. Clin. Pract. Cardiovasc. Res. 2008, 5, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Llodra, J.; Angeli, V.; Liu, J.; Trogan, E.; Fisher, E.A.; Randolph, G.J. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl. Acad. Sci. USA 2004, 101, 11779–11784. [Google Scholar] [CrossRef]
- Trogan, E.; Feig, J.E.; Dogan, S.; Rothblat, G.H.; Angeli, V.; Tacke, F.; Randolph, G.J.; Fisher, E.A. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef]
- Potteaux, S.; Gautier, E.L.; Hutchison, S.B.; Van Rooijen, N.; Rader, D.J.; Thomas, M.J.; Sorci-Thomas, M.G.; Randolph, G.J. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J. Clin. Investig. 2011, 121, 2025–2036. [Google Scholar] [CrossRef]
- Dal Canto, A.J.; Swanson, P.E.; O’Guin, A.K.; Speck, S.H.; Virgin, H.W. IFN-gamma action in the media of the great elastic arteries, a novel immunoprivileged site. J. Clin. Investig. 2001, 107, R15–R22. [Google Scholar] [CrossRef] [PubMed]
- Waldo, S.W.; Li, Y.; Buono, C.; Zhao, B.; Billings, E.M.; Chang, J.; Kruth, H.S. Heterogeneity of Human Macrophages in Culture and in Atherosclerotic Plaques. Am. J. Pathol. 2008, 172, 1112–1126. [Google Scholar] [CrossRef] [PubMed]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-Phosphate and Lymphocyte Egress from Lymphoid Organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef]
- Lo, C.G.; Xu, Y.; Proia, R.; Cyster, J.G. Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. J. Exp. Med. 2005, 201, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Baluk, P.; Xu, Y.; Grigorova, I.; Bankovich, A.J.; Pappu, R.; Coughlin, S.R.; McDonald, D.M.; Schwab, S.R.; Cyster, J.G. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 2010, 207, 17–27. [Google Scholar] [CrossRef]
- Angeli, V.; Llodrá, J.; Rong, J.X.; Satoh, K.; Ishii, S.; Shimizu, T.; Fisher, E.A.; Randolph, G.J. Dyslipidemia Associated with Atherosclerotic Disease Systemically Alters Dendritic Cell Mobilization. Immunity 2004, 21, 561–574. [Google Scholar] [CrossRef]
- Ludewig, B.; Jäggi, M.; Dumrese, T.; Brduscha-Riem, K.; Odermatt, B.; Hengartner, H.; Zinkernagel, R.M. Hypercholesterolemia Exacerbates Virus-Induced Immunopathologic Liver Disease Via Suppression of Antiviral Cytotoxic T Cell Responses. J. Immunol. 2001, 166, 3369–3376. [Google Scholar] [CrossRef]
- Netea, M.G.; Demacker, P.N.; de Bont, N.; Boerman, O.C.; Stalenhoef, A.F.; Van der Meer, J.W.; Kullberg, B.J. Hyperlipoproteinemia enhances susceptibility to acute disseminated Candida albicans infection in low-density-lipoprotein-receptor-deficient mice. Infect. Immun. 1997, 65, 2663–2667. [Google Scholar] [CrossRef]
- Roselaar, S.E.; Daugherty, A. Apolipoprotein E-deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J. Lipid Res. 1998, 39, 1740–1743. [Google Scholar] [CrossRef]
- De Bont, N.; Netea, M.G.; Demacker, P.N.; Verschueren, I.; Kullberg, B.J.; Van Dijk, K.W.; Van Der Meer, J.W.; Stalenhoef, A.F. Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J. Lipid Res. 1999, 40, 680–685. [Google Scholar] [CrossRef]
- Thaunat, O.; Kerjaschki, D.; Nicoletti, A. Is defective lymphatic drainage a trigger for lymphoid neogenesis? Trends Immunol. 2006, 27, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Thiam, C.H.; Yeo, K.P.; Bisoendial, R.; Hii, C.S.; McGrath, K.C.; Tan, K.W.; Heather, A.; Alexander, J.S.J.; Angeli, V. Lymphatic Vessels Are Essential for the Removal of Cholesterol from Peripheral Tissues by SR-BI-Mediated Transport of HDL. Cell Metab. 2013, 17, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Randolph, G.J.; Miller, N.E. Lymphatic transport of high-density lipoproteins and chylomicrons. J. Clin. Investig. 2014, 124, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Milasan, A.; Smaani, A.; Martel, C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr(−/−) mice. Atherosclerosis 2019, 283, 106–119. [Google Scholar] [CrossRef]
- Dieterich, L.C.; Seidel, C.D.; Detmar, M. Lymphatic vessels: New targets for the treatment of inflammatory diseases. Angiogenesis 2014, 17, 359–371. [Google Scholar] [CrossRef]
- Rutkowski, J.; Moya, M.; Johannes, J.; Goldman, J.; Swartz, M.A. Secondary lymphedema in the mouse tail: Lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc. Res. 2006, 72, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Rutkowski, J.; Helft, J.; Reddy, S.T.; Swartz, M.A.; Randolph, G.J.; Angeli, V. Hypercholesterolemic Mice Exhibit Lymphatic Vessel Dysfunction and Degeneration. Am. J. Pathol. 2009, 175, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Milasan, A.; Jean, G.; Dallaire, F.; Tardif, J.; Merhi, Y.; Sorci-Thomas, M.; Martel, C. Apolipoprotein A-I Modulates Atherosclerosis Through Lymphatic Vessel-Dependent Mechanisms in Mice. J. Am. Heart Assoc. 2017, 6, e006892. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-H.; Zinselmeyer, B.H.; Chang, C.-H.; Saunders, B.T.; Elvington, A.; Baba, O.; Broekelmann, T.J.; Qi, L.; Rueve, J.S.; Swartz, M.A.; et al. Interleukin-17 Drives Interstitial Entrapment of Tissue Lipoproteins in Experimental Psoriasis. Cell Metab. 2019, 29, 475–487. [Google Scholar] [CrossRef]
- Lemole, G.M. The Role of Lymphstasis in Atherogenesis. Ann. Thorac. Surg. 1981, 31, 290–293. [Google Scholar] [CrossRef]
- Vuorio, T.; Nurmi, H.; Moulton, K.; Kurkipuro, J.; Robciuc, M.R.; Öhman, M.; Heinonen, S.E.; Samaranayake, H.; Heikura, T.; Alitalo, K.; et al. Lymphatic Vessel Insufficiency in Hypercholesterolemic Mice Alters Lipoprotein Levels and Promotes Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1162–1170. [Google Scholar] [CrossRef]
- Kraft, J.; Blomgran, R.; Lundgaard, I.; Quiding-Järbrink, M.; Bromberg, J.; Börgeson, E. Specialized Pro-Resolving Mediators and the Lymphatic System. Int. J. Mol. Sci. 2021, 22, 2750. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, K.P.; Lim, H.Y.; Angeli, V. Leukocyte Trafficking via Lymphatic Vessels in Atherosclerosis. Cells 2021, 10, 1344. https://doi.org/10.3390/cells10061344
Yeo KP, Lim HY, Angeli V. Leukocyte Trafficking via Lymphatic Vessels in Atherosclerosis. Cells. 2021; 10(6):1344. https://doi.org/10.3390/cells10061344
Chicago/Turabian StyleYeo, Kim Pin, Hwee Ying Lim, and Veronique Angeli. 2021. "Leukocyte Trafficking via Lymphatic Vessels in Atherosclerosis" Cells 10, no. 6: 1344. https://doi.org/10.3390/cells10061344
APA StyleYeo, K. P., Lim, H. Y., & Angeli, V. (2021). Leukocyte Trafficking via Lymphatic Vessels in Atherosclerosis. Cells, 10(6), 1344. https://doi.org/10.3390/cells10061344