Positive Prognostic Overall Survival Impacts of Methylated TGFB2 and MGMT in Adult Glioblastoma Patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. AI-Augmented Summaries of PubMed Abstracts
2.2. Demethylation Impact on OS for Glioblastoma Patients (GBM) of the Target Genes
2.3. Comparing mRNA Expression in GBM Tumors and Normal Brain Samples
2.4. Hazard Ratio Comparisons for GBM Patients to Determine the Independent Effect of High Levels of TGFB1/2/3, MGMT Methylation Levels, Age, Sex
2.5. Geneset Enrichment Analysis (GSEA) to Identify Negatively Correlated Pathways to TGB2 Methylation Levels
2.6. mRNA Expression Validation of Marker Genes Identified in Bulk mRNA Using Single-Cell Determinations in GBM Samples
3. Results
3.1. Positive Prognostic Impact of High TGFB2 and MGMT Gene Methylation Levels on Overall Survival (OS) in Adult GBM Patients
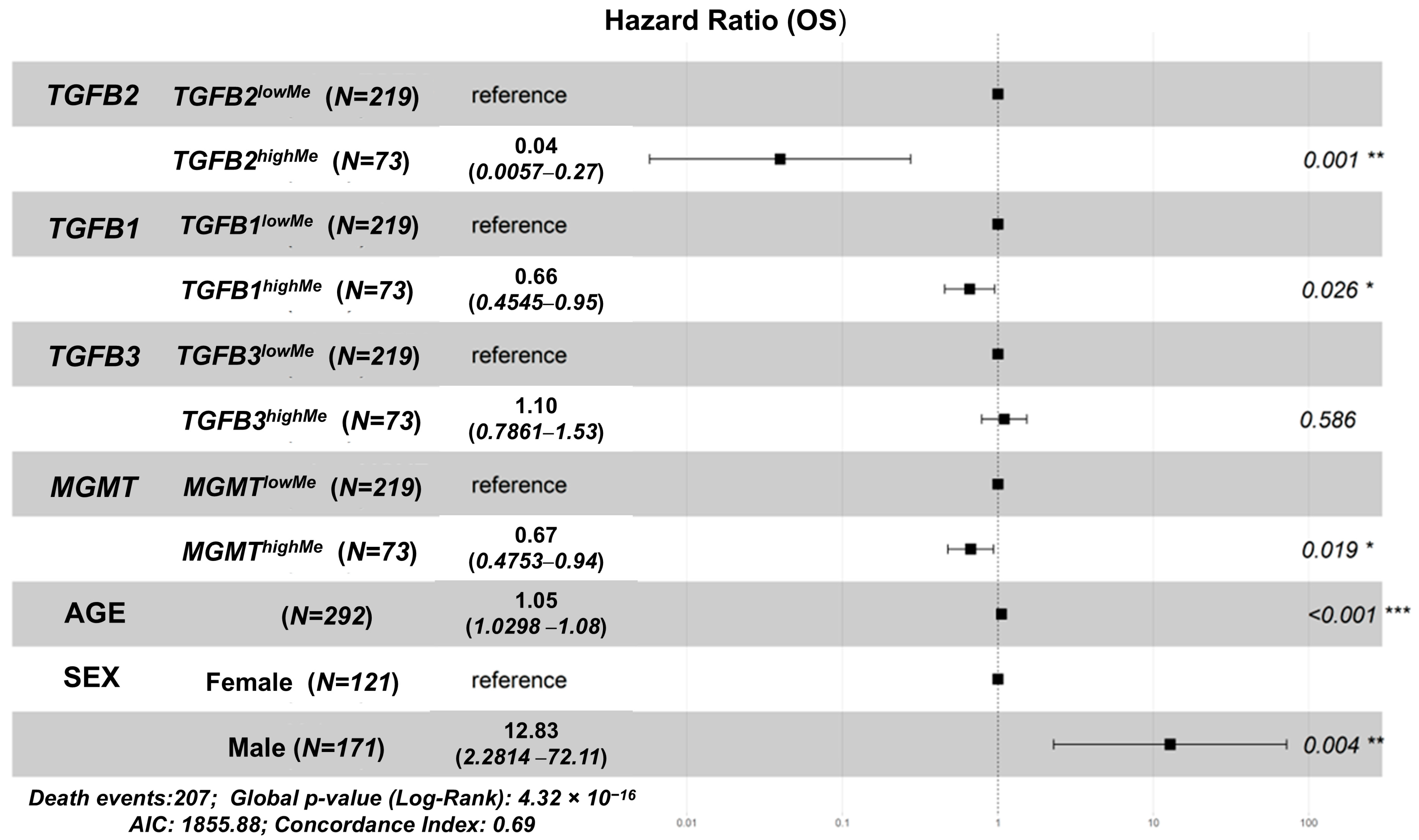
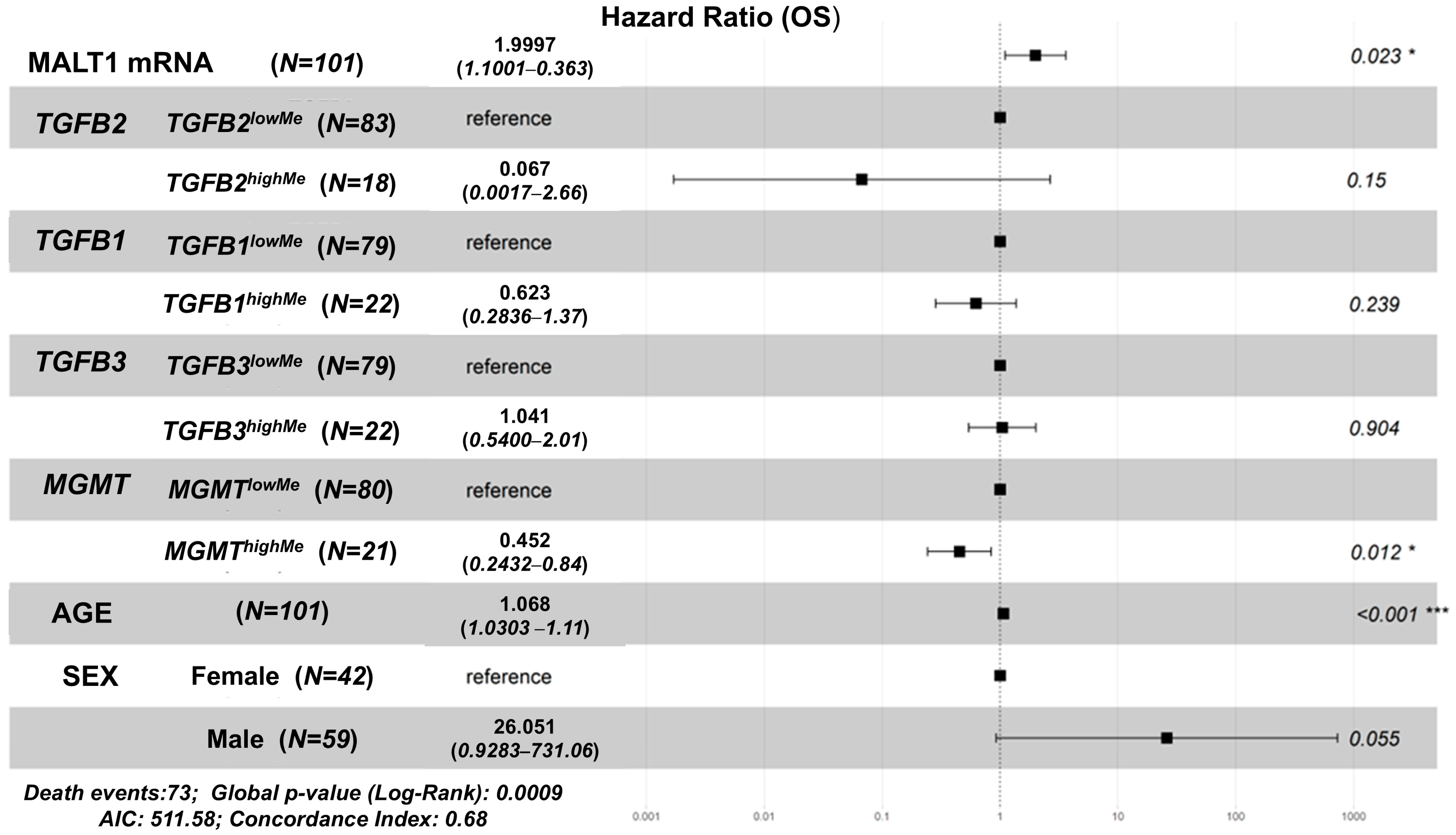
3.2. Independent Prognostic Impacts of TGFB1/2/3, MGMT Gene Methylation, Controlling for Age, Sex, and Determining the Impact of Age Interactions on OS in GBM Patients
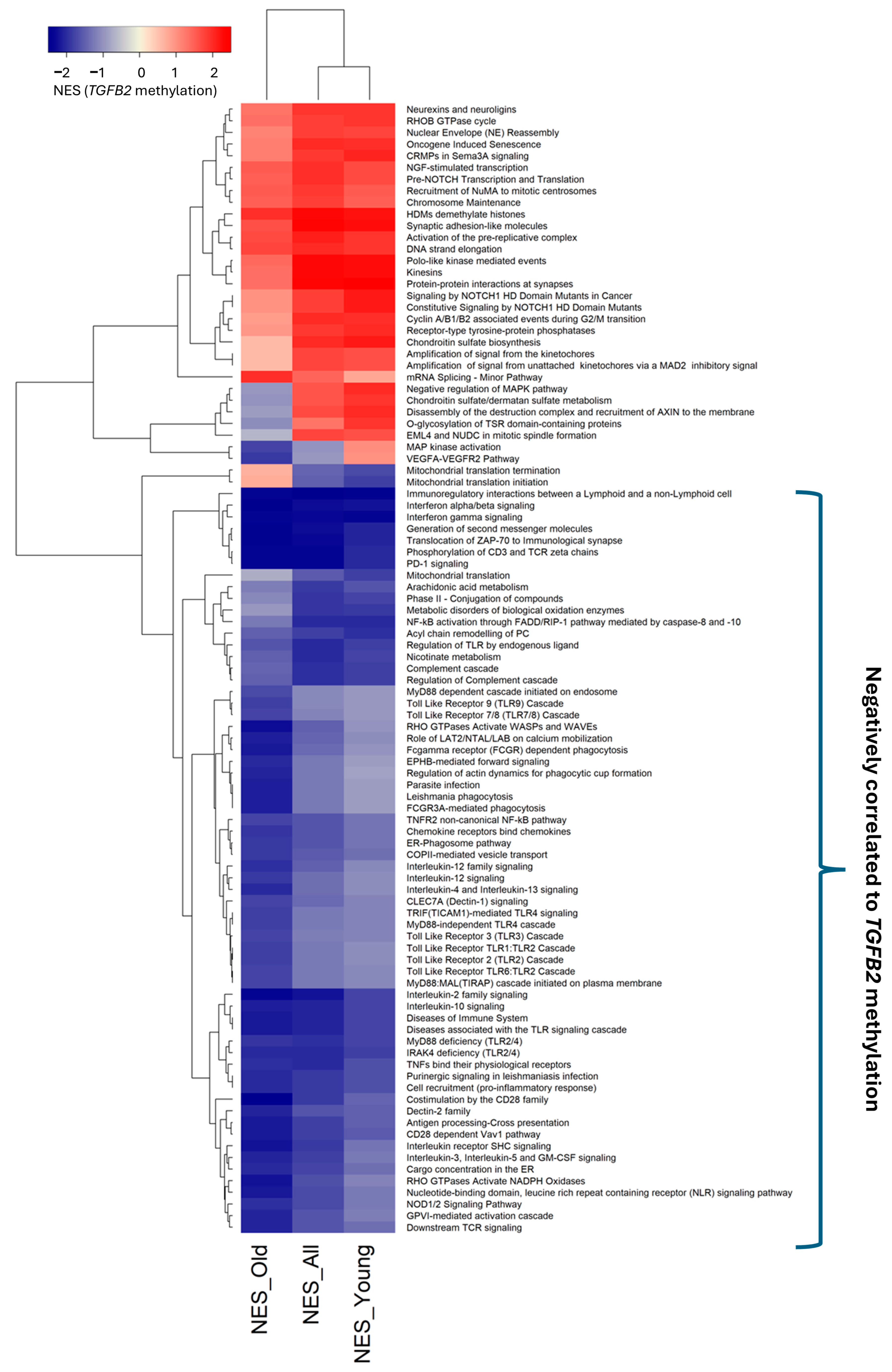
3.3. Age-Independent Identification of Reactome Pathways Enriched with mRNA Expression for Genes Negatively Correlated with TGFB2 Gene Methylation
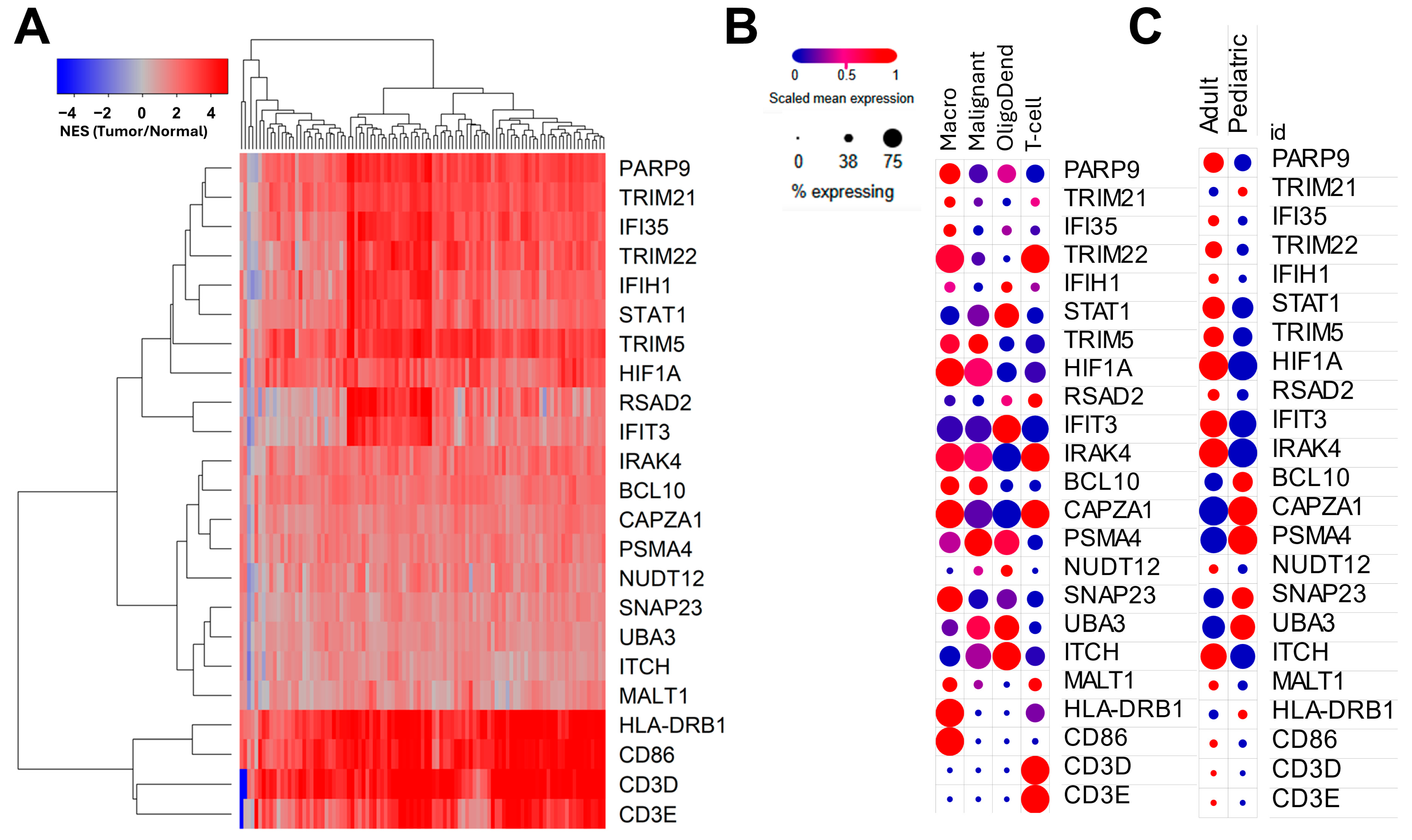
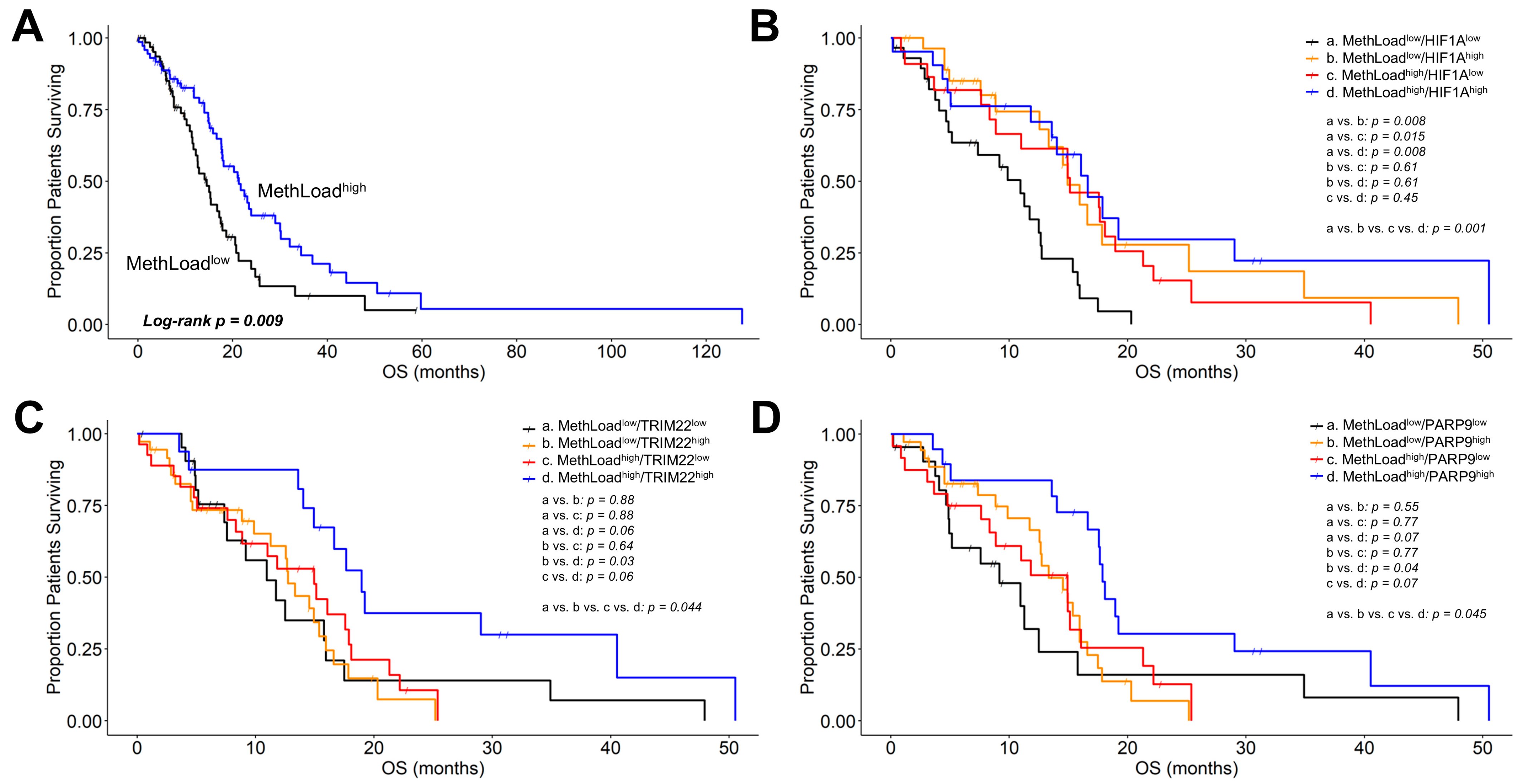
3.4. Genes Negatively Correlated with TGFB2 Methylation Predicted Significant mRNA Increases in Tumor Tissue with Positive Prognostic Impact on OS in GBM Patients
4. Discussion
4.1. Identification of Reactomes Enriched with mRNA Expression of Genes Negatively Correlated with TGFB2 Gene Methylation
4.2. Identification of Reactomes Enriched with mRNA Expression of Genes Negatively Correlated with MGMT Gene Methylation
4.3. Genes Correlated with TGFB2 Methylation and with Positive Prognostic Impact on OS for GBM Patients
4.4. MALT1 mRNA Expression Showed a Negative Prognostic Impact on OS in GBM Patients
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yıldırım, Ö.; Uçar, E. The Molecular Landscape of Glioblastoma: Implications for Diagnosis and Therapy. Eur. J. Biol. 2024, 83, 232–246. [Google Scholar] [CrossRef]
- Smith, H.L.; Wadhwani, N.; Horbinski, C. Major Features of the 2021 WHO Classification of CNS Tumors. Neurotherapeutics 2022, 19, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Lan, Z.; Li, X.; Zhang, X. Glioblastoma: An Update in Pathology, Molecular Mechanisms and Biomarkers. Int. J. Mol. Sci. 2024, 25, 3040. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, S.; Song, C.; Zha, Y.; Li, L. The Prognostic Value of MGMT Promoter Status by Pyrosequencing Assay for Glioblastoma Patients’ Survival: A Meta-Analysis. World J. Surg. Oncol. 2016, 14, 261. [Google Scholar] [CrossRef]
- Szylberg, M.; Sokal, P.; Śledzińska, P.; Bebyn, M.; Krajewski, S.; Szylberg, Ł.; Szylberg, A.; Szylberg, T.; Krystkiewicz, K.; Birski, M.; et al. MGMT Promoter Methylation as a Prognostic Factor in Primary Glioblastoma: A Single-Institution Observational Study. Biomedicines 2022, 10, 2030. [Google Scholar] [CrossRef]
- Trieu, V.; Maida, A.E.; Qazi, S. Transforming Growth Factor Beta 2 (TGFB2) MRNA Levels, in Conjunction with Interferon-Gamma Receptor Activation of Interferon Regulatory Factor 5 (IRF5) and Expression of CD276/B7-H3, Are Therapeutically Targetable Negative Prognostic Markers in Low-Grade Gliomas. Cancers 2024, 16, 1202. [Google Scholar] [CrossRef]
- Cuny, E.; Loiseau, H.; Penchet, G.; Ellie, E.; Arsaut, J.; Vital, A.; Vincendeau, P.; Demotes-Mainard, J. Association of Elevated Glial Expression of Interleukin-1β with Improved Survival in Patients with Glioblastomas Multiforme. J. Neurosurg. 2002, 96, 294–301. [Google Scholar] [CrossRef]
- Papachristodoulou, A.; Silginer, M.; Weller, M.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P. Therapeutic Targeting of TGFβ Ligands in Glioblastoma Using Novel Antisense Oligonucleotides Reduces the Growth of Experimental Gliomas. Clin. Cancer Res. 2019, 25, 7189–7201. [Google Scholar] [CrossRef]
- Uckun, F.M.; Qazi, S.; Hwang, L.; Trieu, V.N. Recurrent or Refractory High-Grade Gliomas Treated by Convection-Enhanced Delivery of a TGFβ2-Targeting RNA Therapeutic: A Post-Hoc Analysis with Long-Term Follow-Up. Cancers 2019, 11, 1892. [Google Scholar] [CrossRef]
- Qazi, S.; Talebi, Z.; Trieu, V. Transforming Growth Factor Beta 2 (TGFB2) and Interferon Gamma Receptor 2 (IFNGR2) MRNA Levels in the Brainstem Tumor Microenvironment (TME) Significantly Impact Overall Survival in Pediatric DMG Patients. Biomedicines 2024, 12, 191. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S.; Trieu, V. High Intra-Tumor Transforming Growth Factor Beta 2 Level as a Predictor of Poor Treatment Outcomes in Pediatric Diffuse Intrinsic Pontine Glioma. Cancers 2023, 15, 1676. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil Enables Reproducible, Open Source, Big Biomedical Data Analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res 2024, 52, D672–D678. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Lu, K.H.-N.; Michel, J.; Kilian, M.; Aslan, K.; Qi, H.; Kehl, N.; Jung, S.; Sanghvi, K.; Lindner, K.; Zhang, X.-W.; et al. T Cell Receptor Dynamic and Transcriptional Determinants of T Cell Expansion in Glioma-Infiltrating T Cells. Neurooncol. Adv. 2022, 4, vdac140. [Google Scholar] [CrossRef]
- Schaettler, M.O.; Desai, R.; Wang, A.Z.; Livingstone, A.J.; Kobayashi, D.K.; Coxon, A.T.; Bowman-Kirigin, J.A.; Liu, C.J.; Li, M.; Bender, D.E.; et al. TCR-Engineered Adoptive Cell Therapy Effectively Treats Intracranial Murine Glioblastoma. J. Immunother. Cancer 2023, 11, e006121. [Google Scholar] [CrossRef]
- Wang, A.F.; Hsueh, B.; Choi, B.D.; Gerstner, E.R.; Dunn, G.P. Immunotherapy for Brain Tumors: Where We Have Been, and Where Do We Go From Here? Curr. Treat. Options Oncol. 2024, 25, 628–643. [Google Scholar] [CrossRef]
- Wolf, S.P.; Anastasopoulou, V.; Drousch, K.; Diehl, M.I.; Engels, B.; Yew, P.Y.; Kiyotani, K.; Nakamura, Y.; Schreiber, K.; Schreiber, H.; et al. One CD4+TCR and One CD8+TCR Targeting Autochthonous Neoantigens Are Essential and Sufficient for Tumor Eradication. Clin. Cancer Res. 2024, 30, 1642–1654. [Google Scholar] [CrossRef] [PubMed]
- Bou-Gharios, J.; Noël, G.; Burckel, H. Preclinical and Clinical Advances to Overcome Hypoxia in Glioblastoma Multiforme. Cell Death Dis. 2024, 15, 503. [Google Scholar] [CrossRef] [PubMed]
- Lo Dico, A.; Martelli, C.; Diceglie, C.; Lucignani, G.; Ottobrini, L. Hypoxia-Inducible Factor-1α Activity as a Switch for Glioblastoma Responsiveness to Temozolomide. Front. Oncol. 2018, 8, 249. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.-J.; Fu, X.-L.; Guang, R.; To, S.-S.T. Advances in the Targeting of HIF-1α and Future Therapeutic Strategies for Glioblastoma Multiforme. Oncol. Rep. 2017, 37, 657–670. [Google Scholar] [CrossRef]
- Feldman, L. Hypoxia within the Glioblastoma Tumor Microenvironment: A Master Saboteur of Novel Treatments. Front. Immunol. 2024, 15, 1384249. [Google Scholar] [CrossRef]
- Begagić, E.; Bečulić, H.; Džidić-Krivić, A.; Kadić Vukas, S.; Hadžić, S.; Mekić-Abazović, A.; Šegalo, S.; Papić, E.; Muchai Echengi, E.; Pugonja, R.; et al. Understanding the Significance of Hypoxia-Inducible Factors (HIFs) in Glioblastoma: A Systematic Review. Cancers 2024, 16, 2089. [Google Scholar] [CrossRef]
- Fei, X.; Dou, Y.; Sun, K.; Wei, J.; Guo, Q.; Wang, L.; Wu, X.; Lv, W.; Jiang, X.; Fei, Z. TRIM22 Promotes the Proliferation of Glioblastoma Cells by Activating MAPK Signaling and Accelerating the Degradation of Raf-1. Exp. Mol. Med. 2023, 55, 1203–1217. [Google Scholar] [CrossRef]
- Ji, J.; Ding, K.; Luo, T.; Zhang, X.; Chen, A.; Zhang, D.; Li, G.; Thorsen, F.; Huang, B.; Li, X.; et al. TRIM22 Activates NF-ΚB Signaling in Glioblastoma by Accelerating the Degradation of IκBα. Cell Death Differ. 2021, 28, 367–381. [Google Scholar] [CrossRef]
- Li, X.; Li, G.; Li, L.; Gao, B.; Niu, X.; Wang, Y.; Wang, Z. SP140 Inhibitor Suppressing TRIM22 Expression Regulates Glioma Progress through PI3K/AKT Signaling Pathway. Brain Behav. 2024, 14, e3465. [Google Scholar] [CrossRef]
- Lu, J.; Liang, K.; Zou, R.; Peng, Y.; Wang, H.; Huang, R.; Zeng, Z.; Feng, Z.; Fan, Y.; Zhang, S.; et al. Comprehensive Analysis of the Prognostic and Immunological Signature of Eight Tripartitemotif (TRIM) Family Molecules in Human Gliomas. Aging 2023, 15, 5798–5825. [Google Scholar] [CrossRef]
- Kumar, D.M.; Patil, V.; Ramachandran, B.; Nila, M.V.; Dharmalingam, K.; Somasundaram, K. Temozolomide-Modulated Glioma Proteome: Role of Interleukin-1 Receptor-Associated Kinase-4 (IRAK4) in Chemosensitivity. Proteomics 2013, 13, 2113–2124. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, B.; Yao, J.; Liu, Z.; Wang, H.; Zhang, B.; Lian, X.; Ren, Z.; Liu, L.; Gao, Y. Interleukin-1 Receptor-associated Kinase 4 as a Potential Biomarker: Overexpression Predicts Poor Prognosis in Patients with Glioma. Oncol. Lett. 2021, 21, 254. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Platzbecker, U.; Lim, K.-H.; Nowakowski, G.; Abdel-Wahab, O.; Kantarjian, H.; Verma, A.; Starczynowski, D.T. Research and Clinical Updates on IRAK4 and Its Roles in Inflammation and Malignancy: Themes and Highlights from the 1st Symposium on IRAK4 in Cancer. Front. Hematol. 2024, 3, 1339870. [Google Scholar] [CrossRef]
- Xu, H.; Chai, S.; Wang, Y.; Wang, J.; Xiao, D.; Li, J.; Xiong, N. Molecular and Clinical Characterization of PARP9 in Gliomas: A Potential Immunotherapeutic Target. CNS Neurosci. Ther. 2020, 26, 804–814. [Google Scholar] [CrossRef]
- Iwata, H.; Goettsch, C.; Sharma, A.; Ricchiuto, P.; Goh, W.W.B.; Halu, A.; Yamada, I.; Yoshida, H.; Hara, T.; Wei, M.; et al. PARP9 and PARP14 Cross-Regulate Macrophage Activation via STAT1 ADP-Ribosylation. Nat. Commun. 2016, 7, 12849. [Google Scholar] [CrossRef]
- Lukic, Z.; Campbell, E.M. The Cell Biology of TRIM5α. Curr. HIV/AIDS Rep. 2012, 9, 73–80. [Google Scholar] [CrossRef]
- Nakayama, E.E.; Shioda, T. Role of Human TRIM5α in Intrinsic Immunity. Front. Microbiol. 2012, 3, 21603. [Google Scholar] [CrossRef]
- Mandell, M.A.; Saha, B.; Thompson, T.A. The Tripartite Nexus: Autophagy, Cancer, and Tripartite Motif-Containing Protein Family Members. Front. Pharmacol. 2020, 11, 308. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Q.; Zhang, J.; Gu, R.; Li, K.; Zhao, G.; Yuan, H.; Feng, T.; Ou, D.; Lin, P. Increased TRIM5 Is Associated with a Poor Prognosis and Immune Infiltration in Glioma Patients. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 2020, 37, 469–479. [Google Scholar] [CrossRef]
- Zhang, L.; He, J.; Zhao, W.; Zhou, Y.; Li, J.; Li, S.; Zhao, W.; Zhang, L.; Tang, Z.; Tan, G.; et al. CD2AP Promotes the Progression of Glioblastoma Multiforme via TRIM5-Mediated NF-KB Signaling. Cell Death Dis. 2024, 15, 722. [Google Scholar] [CrossRef]
- Lee, H.-J. The Role of Tripartite Motif Family Proteins in TGF-β Signaling Pathway and Cancer. J. Cancer Prev. 2018, 23, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Demeyer, A.; Skordos, I.; Driege, Y.; Kreike, M.; Hochepied, T.; Baens, M.; Staal, J.; Beyaert, R. MALT1 Proteolytic Activity Suppresses Autoimmunity in a T Cell Intrinsic Manner. Front. Immunol. 2019, 10, 1898. [Google Scholar] [CrossRef]
- McAllister-Lucas, L.M.; Baens, M.; Lucas, P.C. MALT1 Protease: A New Therapeutic Target in B Lymphoma and Beyond? Clin. Cancer Res. 2011, 17, 6623–6631. [Google Scholar] [CrossRef]
- Jaworski, M.; Thome, M. The Paracaspase MALT1: Biological Function and Potential for Therapeutic Inhibition. Cell. Mol. Life Sci. 2016, 73, 459–473. [Google Scholar] [CrossRef]
- O’Neill, T.J.; Tofaute, M.J.; Krappmann, D. Function and Targeting of MALT1 Paracaspase in Cancer. Cancer Treat. Rev. 2023, 117, 102568. [Google Scholar] [CrossRef]
- Liu, X.; Yue, C.; Shi, L.; Liu, G.; Cao, Q.; Shan, Q.; Wang, Y.; Chen, X.; Li, H.; Wang, J.; et al. MALT1 Is a Potential Therapeutic Target in Glioblastoma and Plays a Crucial Role in EGFR-Induced NF-ΚB Activation. J. Cell Mol. Med. 2020, 24, 7550–7562. [Google Scholar] [CrossRef]
- Hofstätter Azambuja, J.; Yerneni, S.S.; Maurer, L.M.; Crentsil, H.E.; Debom, G.N.; Klei, L.; Smyers, M.; Sneiderman, C.T.; Schwab, K.E.; Acharya, R.; et al. MALT1 Protease Inhibition Restrains Glioblastoma Progression by Reversing Tumor-Associated Macrophage-Dependent Immunosuppression. bioRxiv 2024. [Google Scholar] [CrossRef]
- Mazi, F.A.; Cakiroglu, E.; Uysal, M.; Kalyoncu, M.; Demirci, D.; Sozeri, P.Y.G.; Yilmaz, G.O.; Ozhan, S.E.; Senturk, S. The Paracaspase MALT1 Is a Downstream Target of Smad3 and Potentiates the Crosstalk between TGF-β and NF-KB Signaling Pathways in Cancer Cells. Cell Signal. 2023, 105, 110611. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qazi, S.; Potts, M.; Myers, S.; Richardson, S.; Trieu, V. Positive Prognostic Overall Survival Impacts of Methylated TGFB2 and MGMT in Adult Glioblastoma Patients. Cancers 2025, 17, 1122. https://doi.org/10.3390/cancers17071122
Qazi S, Potts M, Myers S, Richardson S, Trieu V. Positive Prognostic Overall Survival Impacts of Methylated TGFB2 and MGMT in Adult Glioblastoma Patients. Cancers. 2025; 17(7):1122. https://doi.org/10.3390/cancers17071122
Chicago/Turabian StyleQazi, Sanjive, Michael Potts, Scott Myers, Stephen Richardson, and Vuong Trieu. 2025. "Positive Prognostic Overall Survival Impacts of Methylated TGFB2 and MGMT in Adult Glioblastoma Patients" Cancers 17, no. 7: 1122. https://doi.org/10.3390/cancers17071122
APA StyleQazi, S., Potts, M., Myers, S., Richardson, S., & Trieu, V. (2025). Positive Prognostic Overall Survival Impacts of Methylated TGFB2 and MGMT in Adult Glioblastoma Patients. Cancers, 17(7), 1122. https://doi.org/10.3390/cancers17071122