Genetic Counseling for Hereditary Gastric and Pancreatic Cancer in High-Risk Gastrointestinal Cancer Clinics: An Effective Strategy

 ,
,  ,
,  , ,
, ,
Abstract
1. Introduction
2. Material and Methods
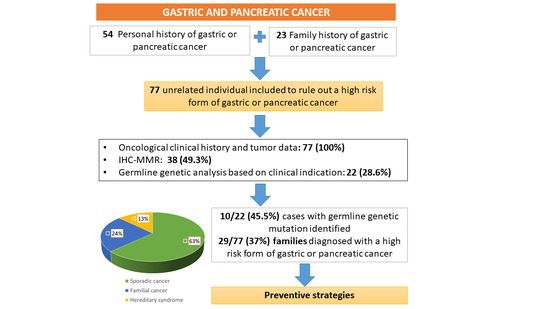
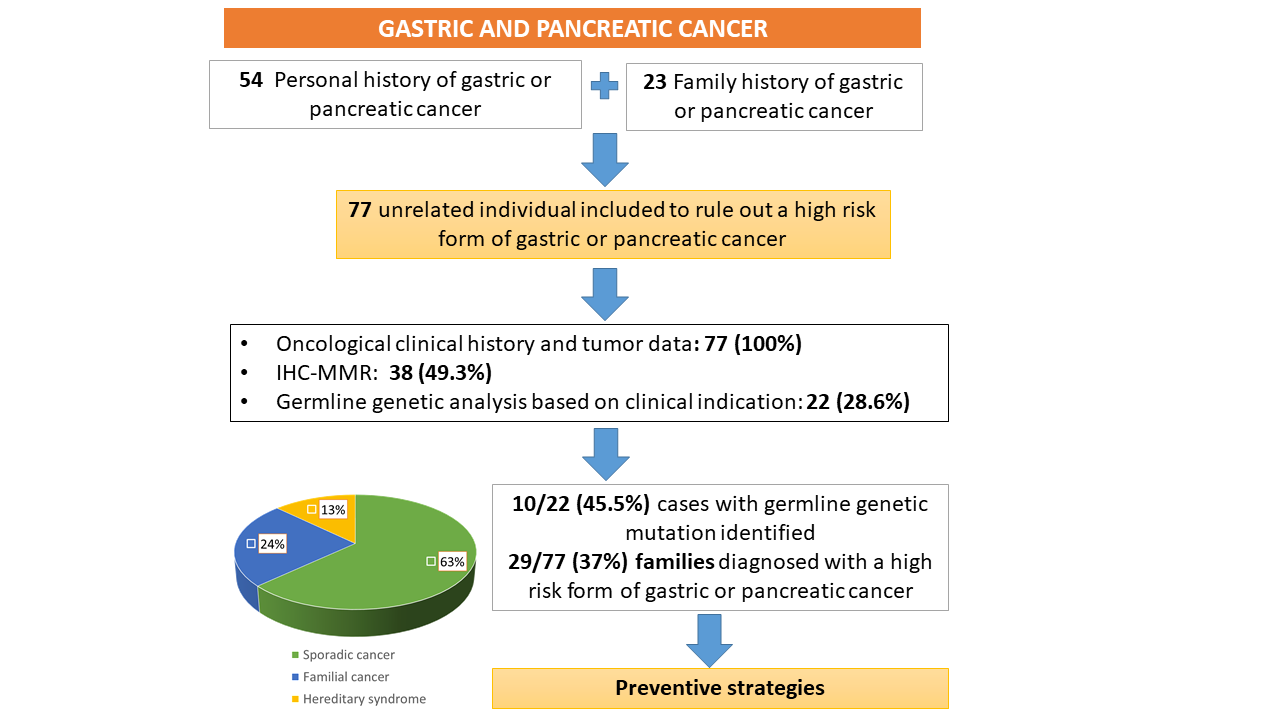
3. Results
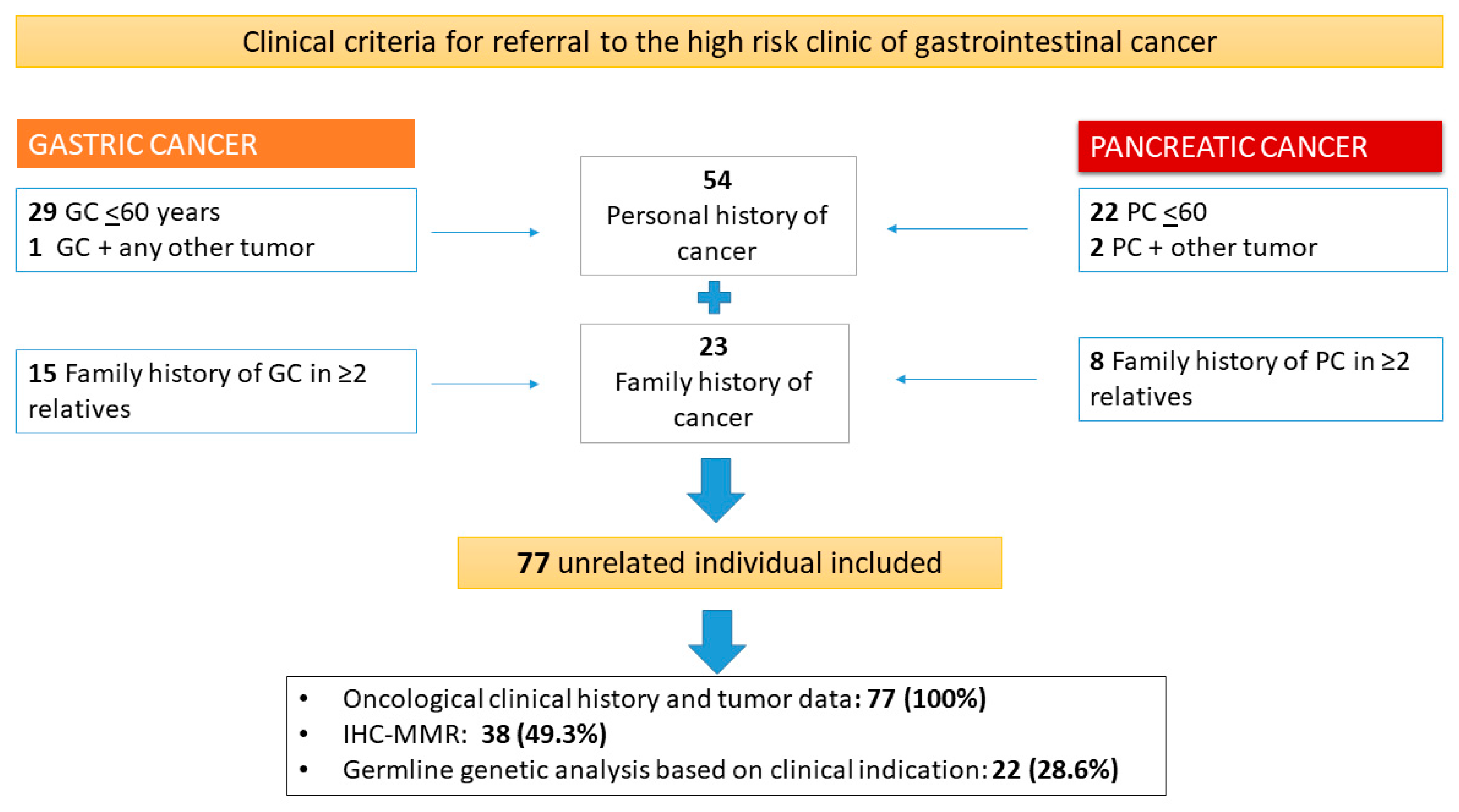
3.1. Characteristics of the Studied Population
3.2. Mismatch Repair Deficiency and Germline Genetic Analysis
3.3. Performance of Clinical Criteria for the Detection of High-Risk Forms
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Serra Sutton, V.; Espallargues, M.; Balaguer, F.; Castells, A. Development of indicators to evaluate colorectal cancer prevention programs in the high-risk population. The experience of a high-risk colorectal cancer clinic. Gastroenterol. Hepatol. 2012, 35, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Pérez Segura, P.; Balaguer, F. Need to implement a coordinated and multidisciplinary care in the Spanish population at increased risk for colorectal cancer. Clin. Transl. Oncol. 2012, 14, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Cubiella, J.; Marzo-Castillejo, M.; Mascort-Roca, J.J.; Amador-Romero, F.J.; Bellas-Beceiro, B.; Clofent-Vilaplana, J.; Carballal, S.; Ferrándiz-Santos, J.; Gimeno-García, A.Z.; Jover, R.; et al. Clinical practice guideline. Diagnosis and prevention of colorectal cancer. 2018 Update. Gastroenterol. Hepatol. 2018, 41, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLof Incidence and Mortality World in 185 Countries. CA Cancer J. Clin. Anticancer Res. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Canto, M.I.; Almario, J.A.; Schulick, R.D.; Yeo, C.J.; Klein, A.; Blackford, A.; Shin, E.J.; Sanyal, A.; Yenokyan, G.; Lennon, A.M.; et al. Risk of Neoplastic Progression in Individuals at High Risk for Pancreatic Cancer Undergoing Long-term Surveillance. Gastroenterology 2018, 155, 740–751. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef]
- Kluijt, I.; Sijmons, R.H.; Hoogerbrugge, N.; Plukker, J.T.; De Jong, D.; Van Krieken, J.H.; Van Hillegersberg, R.; Ligtenberg, M.; Bleiker, E.; Cats, A. Familial gastric cancer: Guidelines for diagnosis, treatment and periodic surveillance. Fam. Cancer 2012, 11, 363–369. [Google Scholar] [CrossRef]
- Stjepanovic, N.; Moreira, L.; Carneiro, F.; Balaguer, F.; Cervantes, A.; Balmaña, J.; Martinelli, E. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1–34. [Google Scholar] [CrossRef]
- Fitzgerald, R.C.; Caldas, C. Clinical implications of E-cadherin associated hereditary diffuse gastric cancer. Gut 2004, 53, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Leoz, M.L.; Sánchez, A.; Carballal, S.; Ruano, L.; Ocaña, T.; Pellisé, M.; Castells, A.; Balaguer, F.; Moreira, L. Hereditary gastric and pancreatic cancer predisposition syndromes. Gastroenterol. Hepatol. (English Ed.) 2016. [Google Scholar] [CrossRef]
- Assumpção, P.; Araújo, T.; Khayat, A.; Santos, S.; Barra, W.; Assumpção, P.; Ishak, G.; Felipe Acioli, J.; Rossi, B. Hereditary gastric cancer: Three rules to reduce missed diagnoses. World J. Gastroenterol. 2020, 26, 1382. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, R.; Lott, P.; Bohorquez, M.; Toal, T.; Estrada, A.P.; Suarez, J.J.; Brea-Fernández, A.; Cameselle-Teijeiro, J.; Pinto, C.; Ramos, I.; et al. Germline Mutations in PALB2, BRCA1, and RAD51C, Which Regulate DNA Recombination Repair, in Patients With Gastric Cancer. Gastroenterology 2017, 152, 983–986.e6. [Google Scholar] [CrossRef] [PubMed]
- Ohmoto, A.; Yachida, S.; Morizane, C. Genomic features and clinical management of patients with hereditary pancreatic cancer syndromes and familial pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 561. [Google Scholar] [CrossRef]
- Llach, J.; Carballal, S.; Moreira, L. Familial pancreatic cancer: Current perspectives. Cancer Manag. Res. 2020, 12, 743–758. [Google Scholar] [CrossRef]
- Stoffel, E.M.; McKernin, S.E.; Khorana, A.A. Evaluating susceptibility to pancreatic cancer: ASCO clinical practice provisional clinical opinion summary. J. Oncol. Pract. 2019, 15, 108–111. [Google Scholar] [CrossRef]
- Vasen, H.; Ibrahim, I.; Robbers, K.; Van Mil, A.M.; Potjer, T.; Bonsing, B.A.; Bergman, W.; Wasser, M.; Morreau, H.; De Vos Tot Nederveen Cappel, W.H.; et al. Benefit of surveillance for pancreatic cancer in high-risk individuals: Outcome of long-term prospective follow-up studies from three European expert centers. J. Clin. Oncol. 2016, 34, 2010–2019. [Google Scholar] [CrossRef]
- Areia, M.; Spaander, M.C.W.; Kuipers, E.J.; Dinis-Ribeiro, M. Endoscopic screening for gastric cancer: A cost-utility analysis for countries with an intermediate gastric cancer risk. United Eur. Gastroenterol. J. 2018, 6, 192–202. [Google Scholar] [CrossRef]
- Saumoy, M.; Schneider, Y.; Shen, N.; Kahaleh, M.; Sharaiha, R.Z.; Shah, S.C. Cost Effectiveness of Gastric Cancer Screening According to Race and Ethnicity. Gastroenterology 2018, 155, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Vangala, D.B.; Cauchin, E.; Balmaña, J.; Wyrwicz, L.; van Cutsem, E.; Güller, U.; Castells, A.; Carneiro, F.; Hammel, P.; Ducreux, M.; et al. Screening and surveillance in hereditary gastrointestinal cancers: Recommendations from the European Society of Digestive Oncology (ESDO) expert discussion at the 20th European Society for Medical Oncology (ESMO)/World Congress on Gastrointestinal Cancer. Eur. J. Cancer 2018, 104, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kim, Y.I.; Kook, M.C.; Park, B.; Joo, J. Family history of gastric cancer and Helicobacter pylori treatment. N. Engl. J. Med. 2020, 382, 427–436. [Google Scholar] [CrossRef]
- Pérez-Carbonell, L.; Ruiz-Ponte, C.; Guarinos, C.; Alenda, C.; Payá, A.; Brea, A.; Egoavil, C.M.; Castillejo, A.; Barberá, V.M.; Bessa, X.; et al. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut 2012, 61, 865–872. [Google Scholar] [CrossRef]
- Vasen, H.F.A.; Gruis, N.A.; Frants, R.R.; Van Der Velden, P.A.; Hille, E.T.M.; Bergman, W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int. J. Cancer 2000, 87, 809–811. [Google Scholar] [CrossRef]
- Brose, M.S.; Rebbeck, T.R.; Calzone, K.A.; Stopfer, J.E.; Nathanson, K.L.; Weber, B.L. Cancer risk estimates for BCRA1 mutation carriers identified in a risk evaluation program. J. Natl. Cancer Inst. 2002, 94, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Offerhaus, G.J.A.; Lee, D.H.; Krush, A.J.; Tersmette, A.C.; Booker, S.V.; Kelley, N.C.; Hamilton, S.R. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut 1993, 34, 1394–1396. [Google Scholar] [CrossRef]
- Kastrinos, F.; Mukherjee, B.; Tayob, N.; Wang, F.; Sparr, J.; Raymond, V.M.; Bandipalliam, P.; Stoffel, E.M.; Gruber, S.B.; Syngal, S. Risk of pancreatic cancer in families with Lynch syndrome. JAMA J. Am. Med. Assoc. 2009, 302, 1790–1795. [Google Scholar] [CrossRef]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef]
- Moreira, L.; Balaguer, F.; Lindor, N.; De La Chapelle, A.; Hampel, H.; Aaltonen, L.A.; Hopper, J.L.; Le Marchand, L.; Gallinger, S.; Newcomb, P.A.; et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA J. Am. Med. Assoc. 2012, 308, 1555–1565. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Characteristic | Gastric Cancer (45) | Pancreatic Cancer (32) | All (77) |
|---|---|---|---|
| Age at cancer diagnosis1; median (IQR) | 47 (40–55) | 51.5 (45.5–59) | 49 (41.5–58) |
| Gender (women); number (%) | 29 (64.4) | 17 (53.1) | 46 (59.7) |
| Race | |||
| Caucasian, number (%) | 43 (95.6) | 33 (100) | 75 (97.4) |
| Asian, number (%) | 2 (4.4) | 0 | 2 (2-6) |
| Referral criteria, number (%) | |||
| -Age ≤60 years old | 29 (64.4) | 22 (68.5) | 51 (66.2) |
| -Multiplicity (PH GC/PC and any other tumor) | 1 (2.3) | 2 (6.5) | 3 (3.9) |
| -Family history | 15 (33.3) | 8 (25) | 23 (29.9) |
| PH of any other neoplasm, number (%) | 6 (13) | 3 (8.8) | 9 (11.7) |
| Age; median (IQR) | 49 (41.5–58) | 53 (35–53) | 53 (39.5–58) |
| Neoplasm type, number (%) | |||
| Breast | 3 (50) | 2 (66%) | 5 (55.5) |
| Endometrium | 2 (33.4) | - | 2 (22.2) |
| Colon | 1 (16.6) | - | 1 (11.1) |
| Lymphoma | - | 1 (34%) | 1 (11.1) |
| Family history of the same tumor2 | |||
| (FDR or SDR), number (%) | 20 (44.4) | 10 (31.2) | 30 (38.9) |
| Number of affected relatives; median (IQR) | 2 (1.2–3) | 2 (1–2.7) | ---- |
| Family history of the same tumor in FDR, number (%) | 18 (40) | 8 (25) | 26 (33.7) |
| Younger age at diagnosis; median (IQR) | 55 (44.5–67.5) | 62.5 (52.5–69.7) | ----- |
| Family history of other neoplasms (FDR or SDR) | 29 (64.4) | 24 (75) | 53 (68.8%) |
| Number of affected relatives; median (IQR) | 1.5 (1–3) | 2 (1–3) | 2 (1–3) |
| Total registered tumors | 65 | 56 | 121 |
| Most frequent tumors, number (%) | |||
| Breast | 14 (21.5) | 4 (7.1) | 18 (14.8) |
| Colon | 7 (10.7) | 5 (8.9) | 13 (10.7) |
| Lung | 2 (3.1) | 3 (5.3) | 5 (4.1) |
| Prostate | 3 (4.6) | 3 (5.3) | 6 (4.9) |
| Endometrium | 1 (1.5) | 3 (5.3) | 4 (3.3) |
| Pancreas | 5 (7.7) | - | 5 (4.1) |
| Leukemia | 1 (1.5) | 3 (5.3) | 4 (3.3) |
| IHC-MMR tumor, number (%) | 29 (64.4) | 9 (28.1) | 38 (49.3) |
| Altered IHC-MMR, number (%) | 1 (3.4) | 0 | 1 (2.6) |
| Germline genetic study, number (%) | 16 (35.6) | 6 (18.8) | 22 (28.6) |
| Gene-guided study (Sanger) | 8 (50) | 1 (16.7) | 9 (40.1) |
| Multi-gen panel | 8 (50) | 5 (83.3) | 13 (59.1) |
| Germline genetic mutations identified, number (%) | 7 (43.7) | 3 (18.8) | 10 (30.3) |
| Familial cancer criteria, number (%) | 19 (24.7) | ||
| Criteria of FGC | 17 (37.8) | - | |
| ≥3 FDR or SDR with GC | 5 (29.4) | - | |
| ≥2 FDR or SDR with GC, 1 < 50 | 7 (70.6) | - | |
| Criteria of FPC | - | 7 (21.8) | |
| ≥3 FDR/SDR/TDR with PC | - | 2 (28.5) | |
| ≥2 FDR with PC | - | 5 (71.5) | |
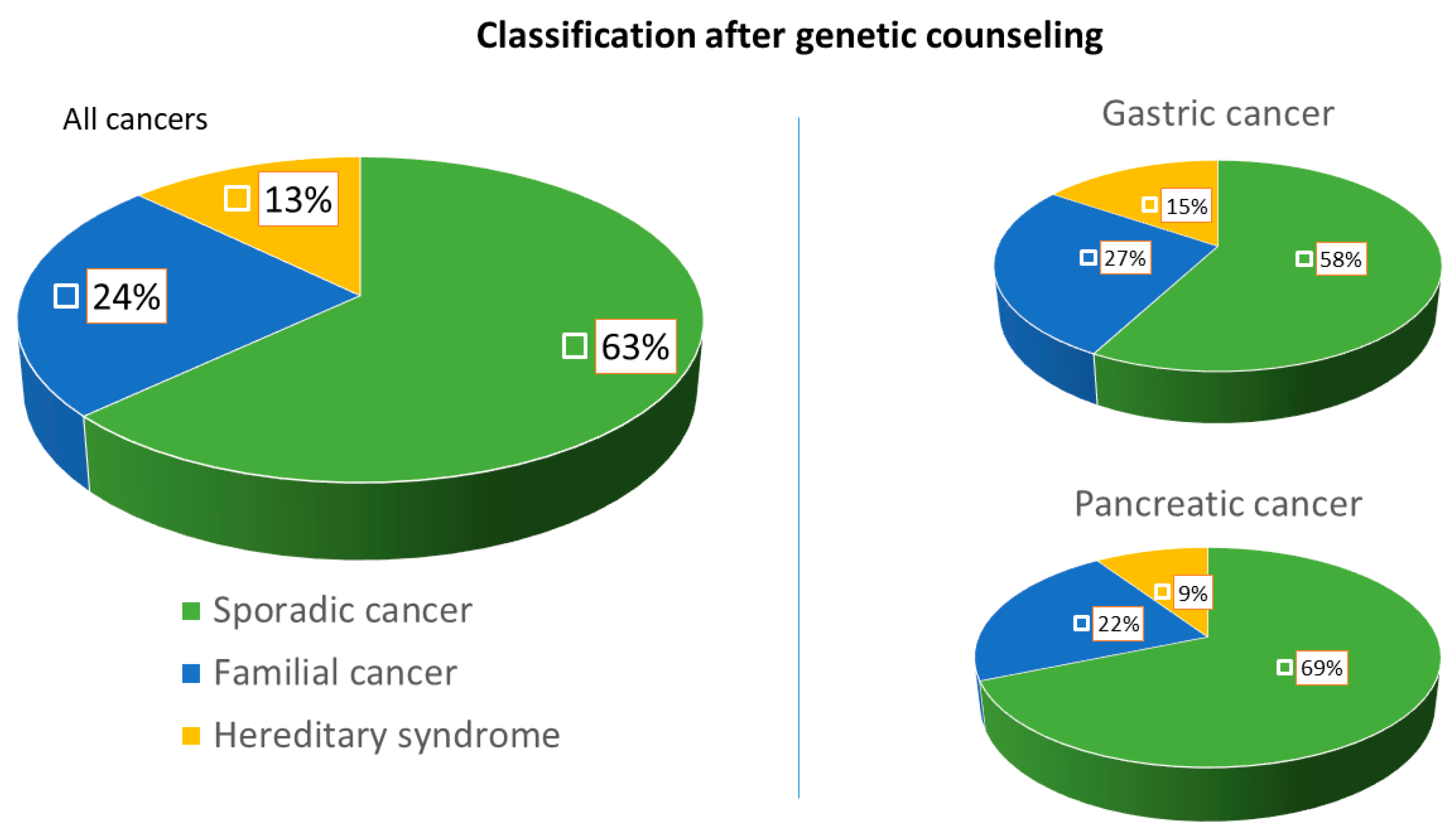
| Final classification after genetic counseling, number (%) | 7 (26.7) | 3 (9.4) | 10 (13) |
| (a) Hereditary-cancer associated syndrome | |||
| (b) Familial form of cancer | 12 (15.5) | 7 (21.9) | 19 (24.6) |
| (c) Sporadic cancer | 26 (57.8) | 22 (68.7) | 48 (62.4) |
| P. | G. | Age 1 | Gender | Referral Criteria | Personal History of Other Cancer | Family History of Cancer * | IHC-MMR | GeneticsTechnique | Gene Affected (Pathogenic Variant) | Hereditary Syndrome |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GC | 75 | Female | GC+ other | Endometrium | No | MSH2/MSH6 | Single-gene testing | MSH2 (c.602dupT p.Leu201Phe*31) | Lynch S |
| 2 | GC | 55 | Female | Family history | Breast | No | Undone | Multigen panel | ATM (c.4507C>T (p.Gln1503Ter)) | ATM |
| 3 | GC | 35 | Female | Family history | Endometrium | Yes (pancreas and breast) | Undone | Multigen panel | ATM (c.2921+1G>A. 19 intron) | ATM |
| 4 | GC | 41 | Male | GC ≤ 60 years | No | No | Undone | Multigen panel | CDH1 (c.220C>T (p.Arg74 *)) | HDGC |
| 5 | GC | 38 | Male | GC ≤ 60 years | No | No | Undone | Multigen panel | CDH1 c.2164+5G>C) | HDGC |
| 6 | GC | 49 | Female | GC ≤ 60 years | Breast | Yes (breast) | MMR+ | Single-gene testing | BRCA2 (c.3166 C>T)) | HBOC |
| 7 | GC | 34 | Male | GC ≤ 60 years | No | Yes (breast and colon) | MMR+ | Multigen panel | TP53 (c.365_366delTG (p.Val122AspfsTer26)) | Li-Fraumeni S |
| 8 | PC | 45 | Female | Family history | No | Yes (Breast, stomach) | Undone | Directed | BRCA2 c.3264dupT (p. (Gln1089Serfs*10), | HBOC |
| 9 | PC | 33 | Male | PC ≤ 60 years | No | Yes (breast) | Undone | Multigen panel | ATM (c.6711_6715delGGAAA (p.Lys2237Asnfs*10) | ATM |
| 10 | PC | 33 | Male | PC ≤ 60 years | No | Yes (breast) | Undone | Multigene panel | PALB2 (c.3483delT; p. (Phe1161Leufs*2)) | HBOC |
| (a) Factors Associated with High Risk Condition of Cancer (Hereditary or Familial Cancer) | ||||
| Characteristic | Sporadic Cancer (48) | High Risk Form (29) | p-Value (Univariate) | OR (95% CI); p-Value (Multivariate) |
| Age, years; median (IQR) | 53 (41.5–67) | 49 (41.2–55.5) | 0.084 | 0.98 (0.92–1.03); p = 0.46 |
| Gender: women; number (%) | 29 (60.4) | 17 (58.6) | 0.876 | - |
| PH of other cancer | 4 (8.3) | 5 (17.2) | 0.252 | - |
| Referral criteria: | ||||
| Age ≤ 60 years old | 42 (87.5) | 9 (31) | 0.000 | 0.16 (0.008–3.37); p = 0.24 |
| Multiplicity | 2 (4.1) | 1 (3.4) | 0.875 | - |
| Family history | 4 (8.3) | 19 (65.5) | 0.000 | 21 (5.8-79); p = 0.000 |
| (b) Factors Associated with Hereditary Cancer (Only Cases with Pathogenic Mutation Identified) | ||||
| Characteristic | Non-Hereditary Cancer (67) | Hereditary Syndrome (10) | P-Value (univariate) | OR (95% CI); p-Value (multivariate) |
| Age, years; median (IQR) | 50 (43–59) | 39.5 (33.7–50.5) | 0.041 | |
| Younger case age < 40 | 10 (15) | 5 (50) | 0.009 | 11.3 (1.9–67); 0.007 |
| Gender: women; number (%) | 41 (61.2) | 5 (50) | 0.501 | |
| PH of other cancer | 5 (7.4) | 4 (40) | 0.003 | 17.4 (2.5–119.9); p = 0.004 |
| Referral criteria: | ||||
| Age ≤ 60 years old | 45 (67.2) | 6 (60) | 0.655 | |
| Multiplicity | 2 (2.9) | 1 (10) | 0.285 | |
| Family history | 20 (29.8) | 3 (30) | 0.992 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llach, J.; Moreno, L.; Sánchez, A.; Herrera-Pariente, C.; Ocaña, T.; Cuatrecasas, M.; Rivero-Sánchez, L.; Moreira, R.; Díaz, M.; Jung, G.; et al. Genetic Counseling for Hereditary Gastric and Pancreatic Cancer in High-Risk Gastrointestinal Cancer Clinics: An Effective Strategy. Cancers 2020, 12, 2386. https://doi.org/10.3390/cancers12092386
Llach J, Moreno L, Sánchez A, Herrera-Pariente C, Ocaña T, Cuatrecasas M, Rivero-Sánchez L, Moreira R, Díaz M, Jung G, et al. Genetic Counseling for Hereditary Gastric and Pancreatic Cancer in High-Risk Gastrointestinal Cancer Clinics: An Effective Strategy. Cancers. 2020; 12(9):2386. https://doi.org/10.3390/cancers12092386
Chicago/Turabian StyleLlach, Joan, Lorena Moreno, Ariadna Sánchez, Cristina Herrera-Pariente, Teresa Ocaña, Miriam Cuatrecasas, Liseth Rivero-Sánchez, Rebeca Moreira, Mireia Díaz, Gerhard Jung, and et al. 2020. "Genetic Counseling for Hereditary Gastric and Pancreatic Cancer in High-Risk Gastrointestinal Cancer Clinics: An Effective Strategy" Cancers 12, no. 9: 2386. https://doi.org/10.3390/cancers12092386
APA StyleLlach, J., Moreno, L., Sánchez, A., Herrera-Pariente, C., Ocaña, T., Cuatrecasas, M., Rivero-Sánchez, L., Moreira, R., Díaz, M., Jung, G., Pellisé, M., Castells, A., Balaguer, F., Carballal, S., & Moreira, L. (2020). Genetic Counseling for Hereditary Gastric and Pancreatic Cancer in High-Risk Gastrointestinal Cancer Clinics: An Effective Strategy. Cancers, 12(9), 2386. https://doi.org/10.3390/cancers12092386