1. Introduction
It has been well established through many in vivo and in vitro studies that estrogen (E2) is critical for normal development, maintenance, and function of the cardiovascular (CVS) system [
1]. E2 as a steroid hormone acts in development as a signaling molecule to affect differentiation by its ability to modulate cell death and development, among other similar actions [
1]. There is a vast and complex response system throughout cardiac, blood vessel, and smooth muscle tissues all of which have E2 receptors (ERs). E2 is synthesized from androgens by the enzyme aromatase in both male and female species [
1]. A key area of study for applications in medicine for E2 is in therapies for postmenopausal women. Women generally have a lower risk for cardiovascular disease than men. This changes after menopause, and therefore E2 is implicated to be a CVS protectant. Past attempts at E2 replacement therapies, such as the one done by the Women’s Health Initiative in the 90s, was met with unexpected results which reported increased risk of CVS disease and stroke [
1]. Therefore, more research is necessary to understand the effects of E2 and its actions on the mature and developing body to better understand possible medicinal applications and implications.
Part of the mechanism by which E2 exerts control over the CVS is through its influence on the expression of nitric oxide synthase (NOS), the enzyme that produces nitric oxide (NO). NO is a gas that acts as a signaling molecule itself in several pathways and genomic mechanisms [
2,
4]. By virtue of its gaseous state, NO can diffuse across cellular membranes without the aid of membrane bound transport proteins and can interact directly with its end targets either in the cell in which it was synthesized or in surrounding cells. In turn, its actions are precisely controlled due to its very short half-life [
3,
5]. At high concentrations, NO acts as a free radical in some situations or binds to superoxide anion (O
2.), causing pathophysiological effects that act to harm the body [
6]. Conversely, at lower levels NO has been shown to exhibit protective effects within the CVS [
6]. In addition, NO acts to regulate cardiac adaption on a spatio-temporal dimension by affecting the heart through the Frank–Starling mechanism, cardiac contractility at the biochemical level, and altering gene expression leading to long term effects [
7]. There are four isoforms of NOS that synthesize NO from L-arginine: neuronal (nNOS or NOS1), inducible (iNOS or NOS2), endothelial (eNOS or NOS3), and mitochondrial (mtNOS). E2 is implicated in the expression of nNOS and eNOS in the CVS [
1]. nNOS is expressed throughout the body in various tissues, but most notably in nervous tissues and cardiomyocytes and plays physiological roles in neurogenesis, learning, memory, and regulation of blood pressure [
6]. iNOS does not have much support from the literature to suggest it has a significant role in the function of the CVS [
8].
eNOS acts as a potent regulator of the cardiovascular system, most notably in the vasculature as a potent vasodilator, but also in cardiomyocytes. eNOS derived NO acts to increase the amount of sGC and thereby increasing cellular cGMP which leads to vasodilation [
6]. All four isoforms are found throughout the cardiovascular system [
1]. Normal physiological function of the heart and vascular tissues is balanced by NO expressed by all isoforms that work together to regulate contractility and peripheral and central nervous system control of the heart itself [
5,
9,
10].
NO acting at the cellular level interacts with either soluble guanylyl cyclase (sGC) to produce cyclic GMP (cGMP) or causes
S-nitrosylation of cysteine residues, as well as interacting with other pathways in the cell [
7,
11]. sGC in cardiomyocytes can be activated in an autocrine manner by NO produced in the myocytes, or in a paracrine manner by NO produced in non-myocyte cells surrounding the cardiomyocyte such as endothelial cells, fibroblasts, smooth muscles, and neurons [
5]. sGC’s heme group has a higher binding affinity for NO than for molecular O
2 in the cytoplasm, which is critical for the NO/cGMP dependent-pathway since NO is present in much lower concentrations in the cell than O
2 [
12]. Once cGMP is produced in this pathway, it acts to modulate phosphodiesterases, ion-gated channels, or cGMP-dependent protein kinases (PKG), each of which continues to act physiologically in the cardiovascular system and throughout the body in vasodilation, platelet aggregation, and neurotransmission [
12].
The other major pathway activated by NO is that of
S-nitrosylation, which is the posttranscriptional modification of cysteine residues by covalently bonding of NO replacing hydrogen on the sulfur group and changing the configuration of a protein [
2,
11]. These proteins then act as second messengers and signal effectors [
2]. In the vasculature,
S-nitrosylation has been implicated in downstream NO vasodilatory signaling, as well as the onset of atherosclerosis [
2]. In cardiomyocytes
S-nitrosylation seems to have protective functions against the development of arrhythmias by way of the nNOS derived NO pathway [
2]. This action involves the inhibition of L-type Ca
2+ channels by
S-nitrosylation.
S-nitrosylation has also been shown to act on ryanodine receptors of the sarcoplasmic reticulum of cardiomyocytes to regulate intracellular Ca
2+ and thus heart contractility [
2]. All these functions help show the importance of
S-nitrosylation in the function of the CVS and the body as a whole.
Zebrafish are commonly accepted model organisms for use in research labs. They exhibit a very quick developmental life cycle, and can reproduce in large numbers. They are also relatively translucent, making study of internal organs and blood vessels easily visible. They also use many of the same genetic and biochemical pathways that are conserved in mammals and other higher organisms. The zebrafish also exhibits a robust NO response system. Specifically, nNOS is expressed early in zebrafish development at 16 to 19 h post fertilization (hpf). Zebrafish are also responsive to nNOS inhibitors (nNOSI), as well as many of the dependent and independent pathway agonists and antagonists [
13]. The nNOS isoform is also expressed in the developing zebrafish medulla, a region of the brain that is known to control heart rate [
14]. Also, blood vessel development occurs very quickly in the zebrafish and blood begins to circulate in the blood vessels after 24 hpf [
15]. Overall, nNOS plays an important role in the zebrafish cardiovascular system [
11].
Previous work in our lab has described the “listless” model’ which is initiated by E2 and NO deprivation [
13,
16,
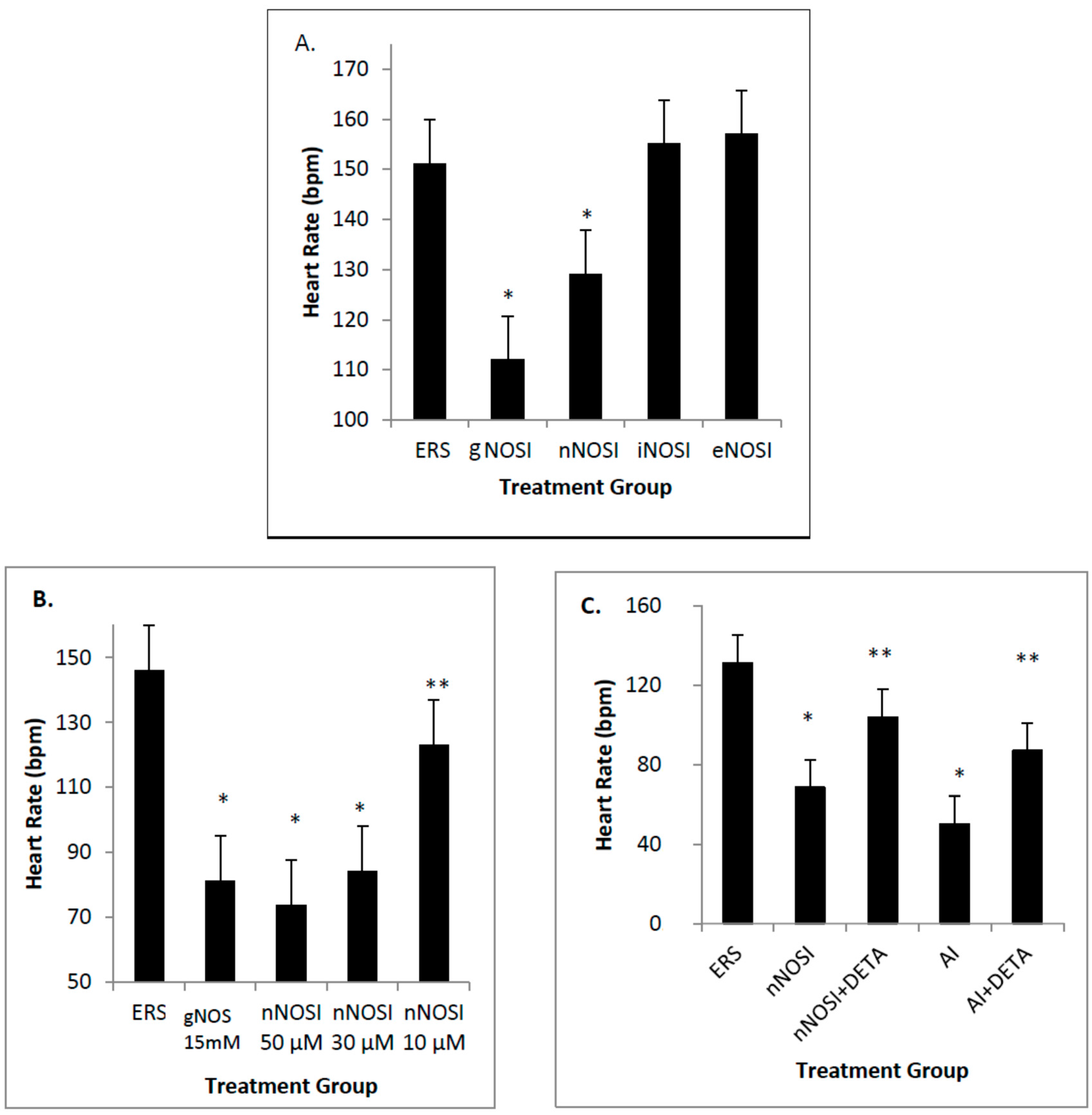
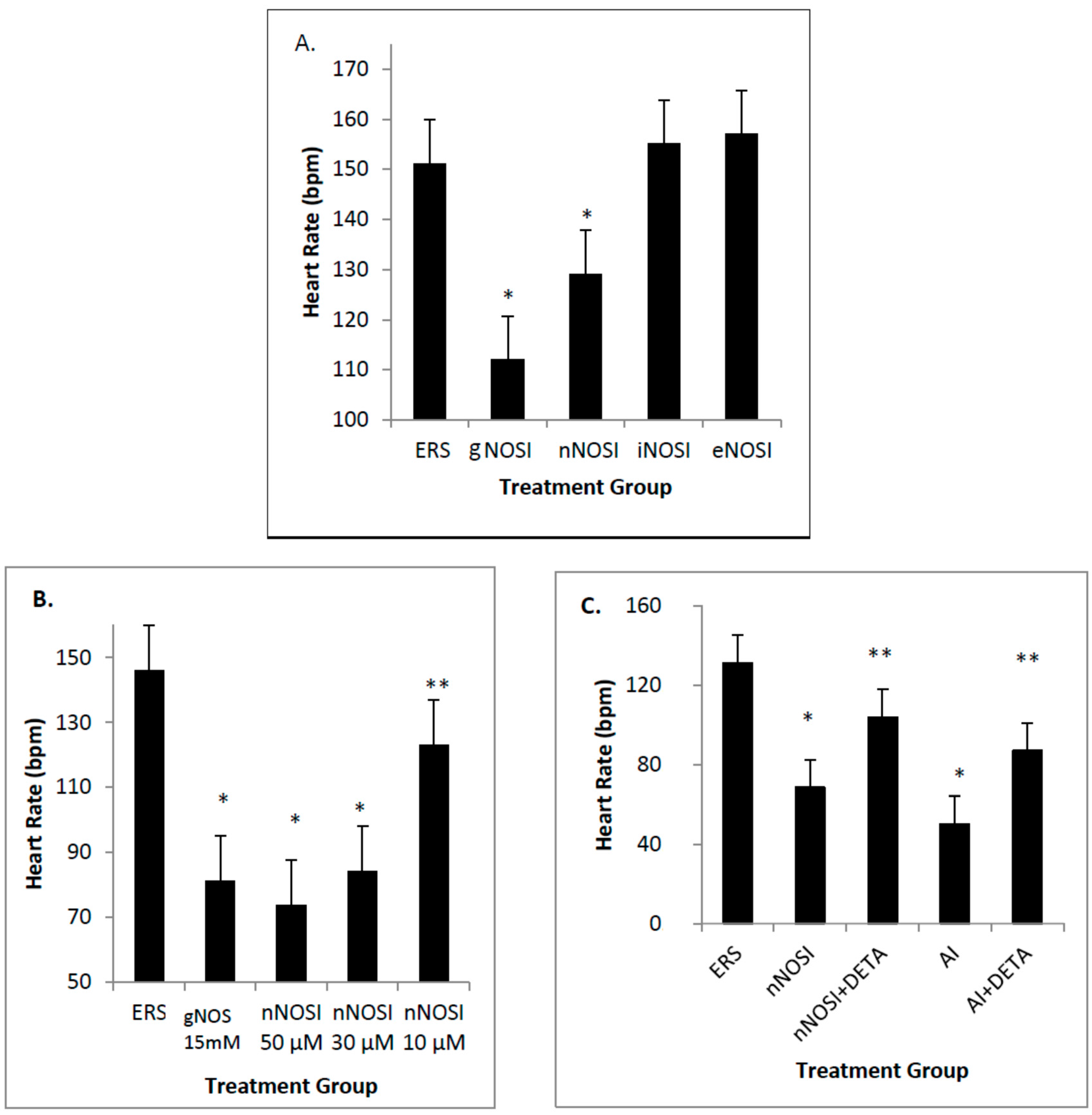
17]. Specifically, both E2 and NO deficiencies cause reduced heart rates and increased arrhythmias, as well as initiating severe sensory-motor deficits. From these earlier observations, it is hypothesized that it is the nNOS isoform that is more effective in regulating heart rate and preventing arrhythmias in the developing zebrafish CVS. In addition, it is also hypothesized that eNOS plays the most prominent role in vascular bed development and health in the developing zebrafish. Results from this study validate these hypotheses. Specifically, nNOS was found to be the prominent isoform regulator of heart health preventing decreased heart rates with accompanying arrhythmias, and is a downstream effector of E2 stimulation. In contrast, eNOS is the isoform which has the most influence on fish blood vessel development.
In summary, this study will help further our knowledge of how the developing heart and blood vessels utilize the various NO signaling pathways in both health and disease. These findings will also aid in medical research and improve our understanding of the use of more novel pharmacological approaches in heart patients suffering for congestive heart failure (CHF) and arrhythmic symptoms.
4. Discussion
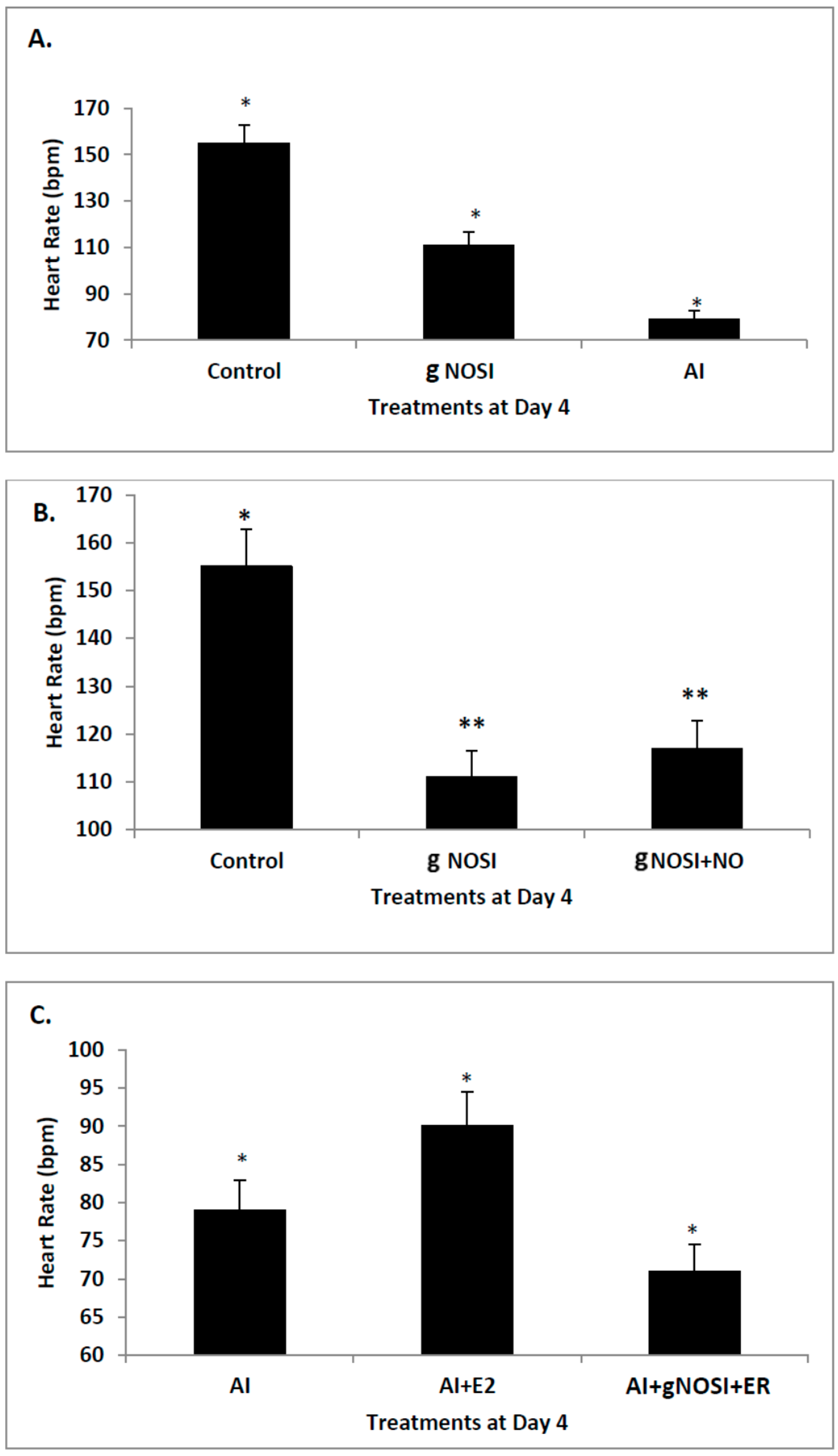
Data from the present study suggests that E2 is an upstream regulator of NO-mediated effects on HR and arrhythmias in the developing zebrafish. Specifically, cardiac arrhythmias could be mimicked by inhibition of E2 synthesis with an AI in a manner similar to that of nNOSI. In both scenarios, by using an NO donor (DETA-NO) in either NO + nNOSI or E2 + AI co-treatments, fish could be significantly rescued from decreased HR and increased arrhythmias. However, the addition of an NOS inhibitor (L-NAME) to the E2 + AI co-treatment fish prevented the rescue of low heart rates and arrhythmias, indicating that E2 is an upstream regulator of these events. There is an abundance of molecular, cellular, biochemical, animal model and human patient literature to support the concept that E2 impacts the CVS in significant ways that may involve both genomic and non-genomic mechanisms [
1]. Indeed, a robust E2 response system exists within heart, vessel endothelial, and vessel smooth muscle cells that express E2 receptors, as well as the enzyme aromatase, which is responsible for the synthesis of E2 from androgens [
1]. Consequently, many prominent CVS physiological effects have also been attributed to the actions of E2 including vasodilation, anti-oxidant properties, decreased post-ischemic inflammation, and anti-artherogenesis effects [
23]. As confirmed in the present study, part of the mechanism by which E2 exerts control over the CVS is through its role in regulating the release of NO through increasing the expression of NOS [
24].
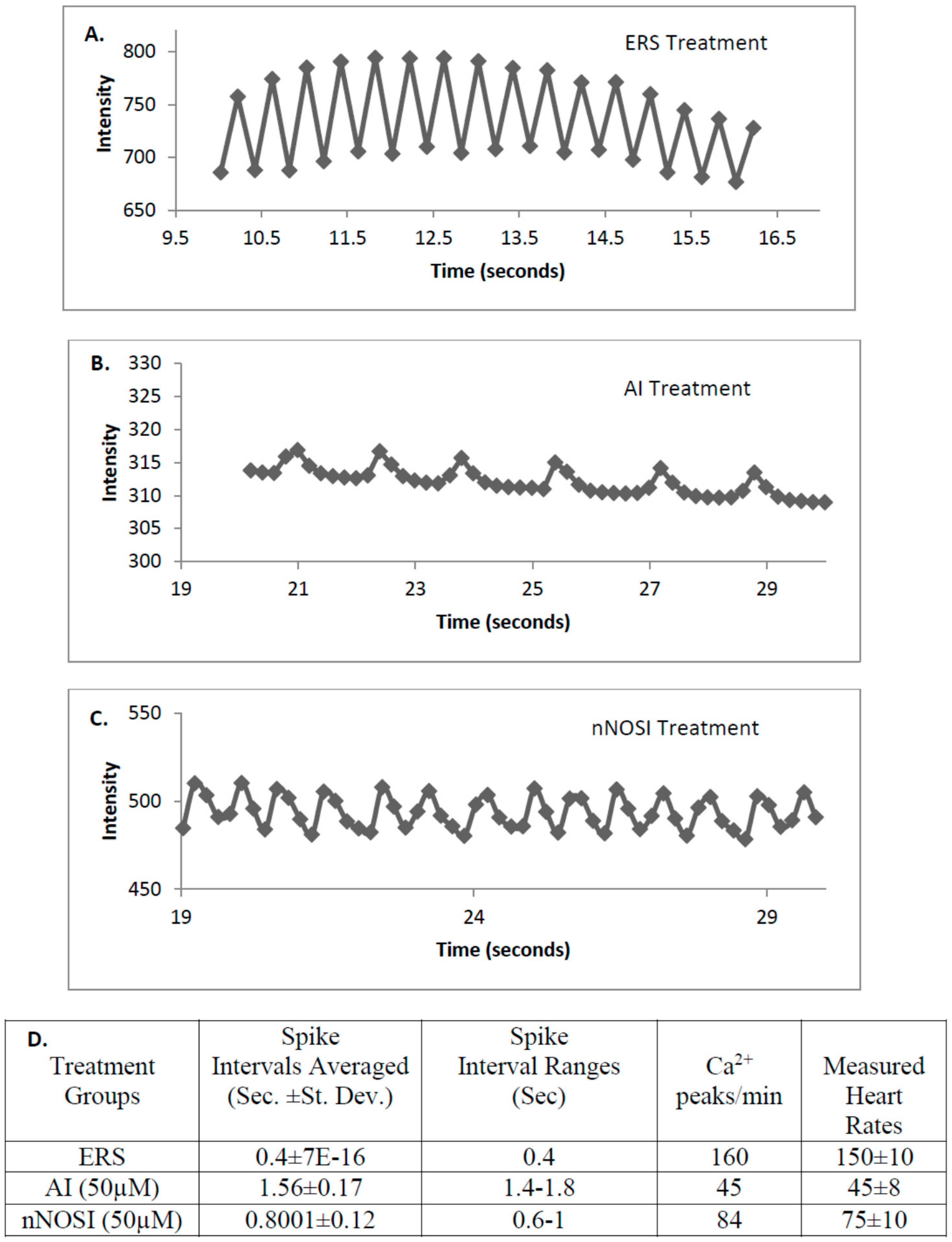
nNOS is the principal NO source for cardiomyocytes and plays vital roles in all cardiac functions that range from contractility to microcirculation, as well as the principle NO source from the autonomic innervation [
4]. Cardiomyocyte contraction is dictated by Ca
2+ signaling. Ca2
+ cycling is crucial for excitation-contraction coupling (ECC), which starts when the membrane of the cardiomyocyte depolarizes via voltage-gated Na
+ channels and causes Ca
2+ to enter the cytosol through L-type Ca
2+ channels [
2]. Increased cytosolic Ca
2+ triggers the release of more Ca
2+ from the SR through ryanodine receptor/Ca
2+ release channels (RyR2), which then leads to Ca
2+ binding to troponin C, activating myosin ATPase which leads to the cardiomyocyte, and therefore the heart, contracting. Elimination of Ca
2+ from the cytosol leads to relaxation of the cardiomyocyte [
2]. nNOS also plays a role in CNS control of the heart, separate from its affects within the cardiomyocytes [
6]. The NOS isoforms, nNOS and eNOS, both act as regulators of cardiomyocyte contractility through Ca
2+ cycling by regulation of L-type Ca
2+ channels and ryanodine receptors (RyR) in the membrane of the SR [
25]. These isoforms use similar substrates and cofactors, and depend on Ca
2+ but exert opposite inotropic effects in cardiomyocytes [
6]. nNOS and eNOS have also been shown to be found in specific subcellular locations, giving insight as to their specific function in a cardiomyocyte [
26]. Therefore, our findings, that of the three NOS isoforms tested nNOS was the one which most prominently affected the zebrafish HR, fits with this concept. Specifically, evidence points to a role for nNOS in regulating heart contraction, relaxation, and arrhythmic bahavior. There is evidence that nNOS actually attenuates the HR in a number of species but in others may have an opposite effect [
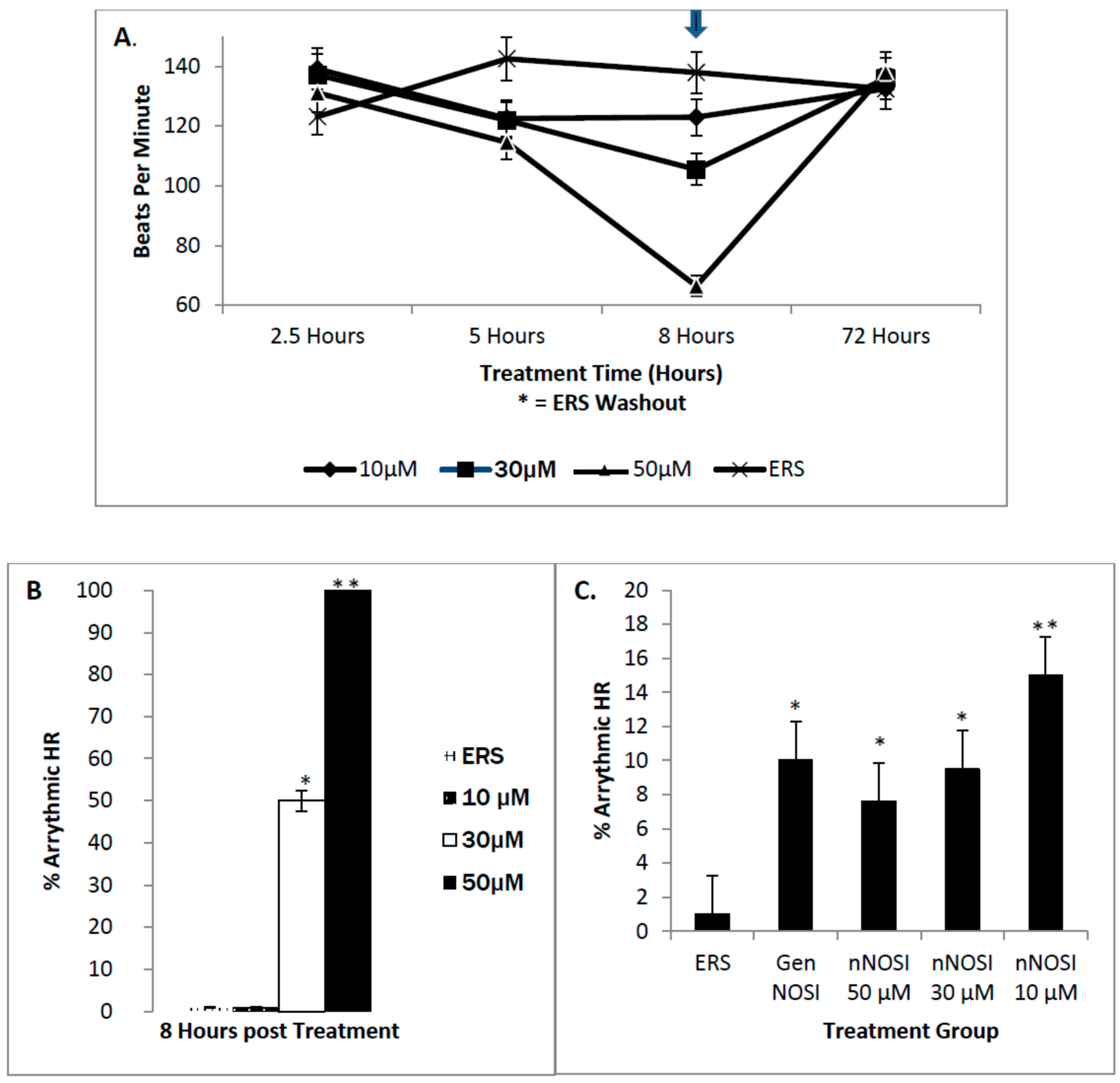
4]. Our findings show that nNOSI treatment significantly lowers the developing zebrafish HR, as well as a dramatic increase in the arrhythmic phenotype, similar to E2 deprivation in our previous study [
18]. This may have to do with our additional findings that over 90% of the nNOSI treated population also express arrhythmias. Indeed, mice deficient in nNOS demonstrate a diastolic Ca
2+ leak [
27], which creates contractile dysfunction and an arrhythmic state [
28,
29] much as we see in our model. Also, nNOS has been found to localize on the sarcoplasmic reticulum (SR) in cardiomyocytes [
11,
25]. nNOS is mainly regulated by Ca
2+ levels in the cytosol, and is inactive at basal concentrations but activated once levels rise, and is specifically activated by allosteric binding of calmodulin [
22]. NO synthesized by NOS has been implicated to increase the open probability of SR Ca
2+ release channels (the ryanodine receptor isoform, RyR2) [
30]. In mammals, NO acts directly by way of
S-nitrosylation of cysteine residues to increase the open probability by activating RyR2 [
2]. This, in turn, enhances contraction of the cardiomyocyte. Increased intracellular levels of calcium lead to increased inotropic activity, which is the force of myocyte contraction, of the cardiomyocyte. Conversely, Wang et al. [
30] used nNOS
−/
− knockout mice to show how
S-nitrosylation levels of RyR2 channels were decreased, leading to decreased open probability, which in turn leads to decreased inotropic activity by way of lower Ca
2+ sparks [
31]. The
S-nitrosylation pathway was further validated when they treated the knockout mice with
S-nitroso-
N-acetylpenicillamine (SNAP), which acts as an NO donor and nitrosylation agent, and observed normalization of cardiomyocyte function. nNOS has been shown to be possibly anti-arrhythmogenic through its inhibition of L-type Ca
2+ channels by
S-nitrosylation when it relocates to the sarcolemma during heart failure, as well as regulating β-adrenergic (β-AR) responsiveness and Ca
2+ entering the cell from the extracellular environment [
2]. Arrhythmic heart contractions seem to be intimately related to nNOS expression, as mice that lacked nNOS presented diastolic Ca
2+ leaking, leading to contractile dysfunction. Regulation of heart action potentials is accomplished by several ion channels that are themselves regulated by S-nitrosylation; an important example of one of these is RyR2 S-nitrosylation by nNOS, which helps regulate intracellular Ca
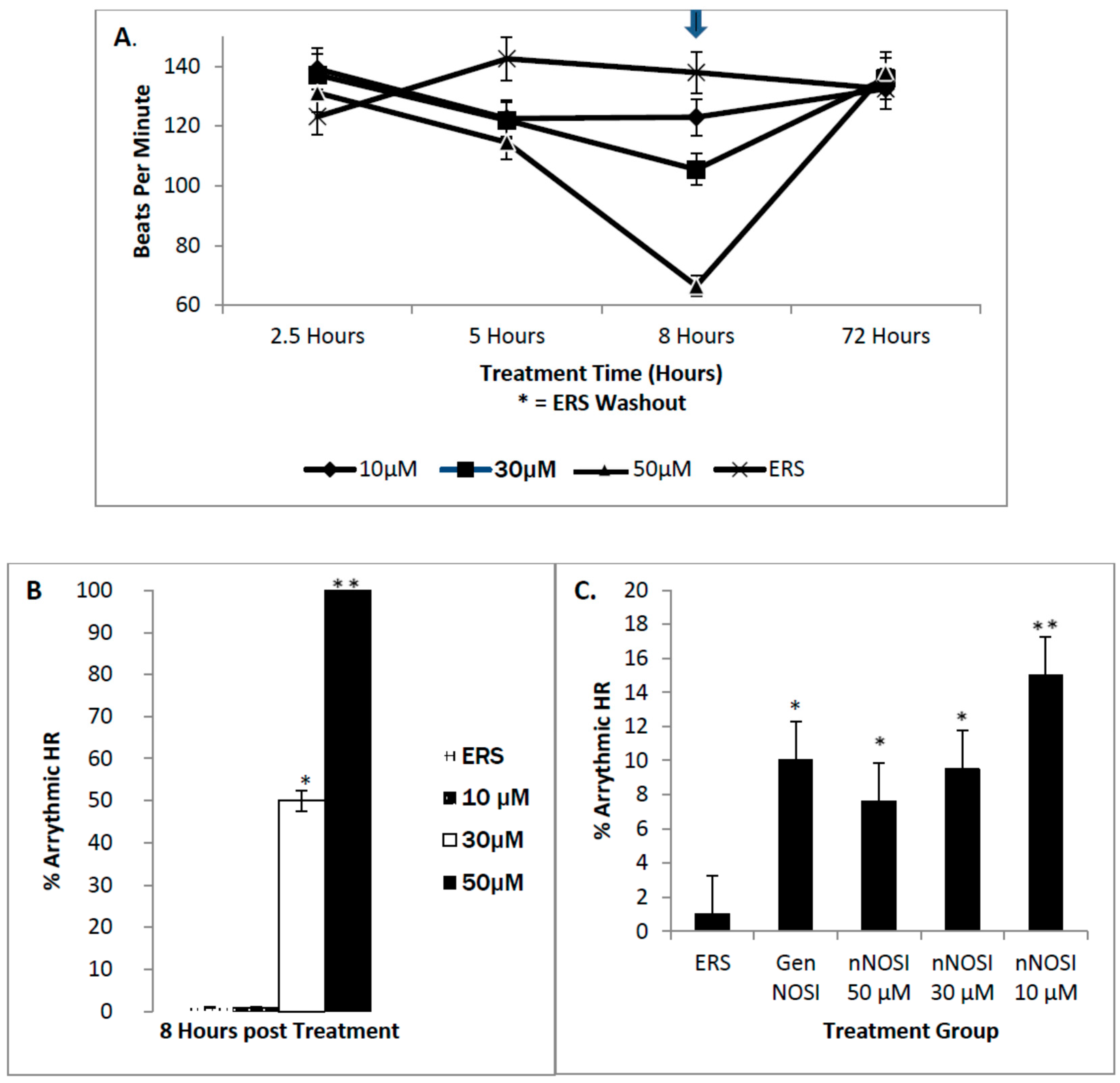
2+ concentration [
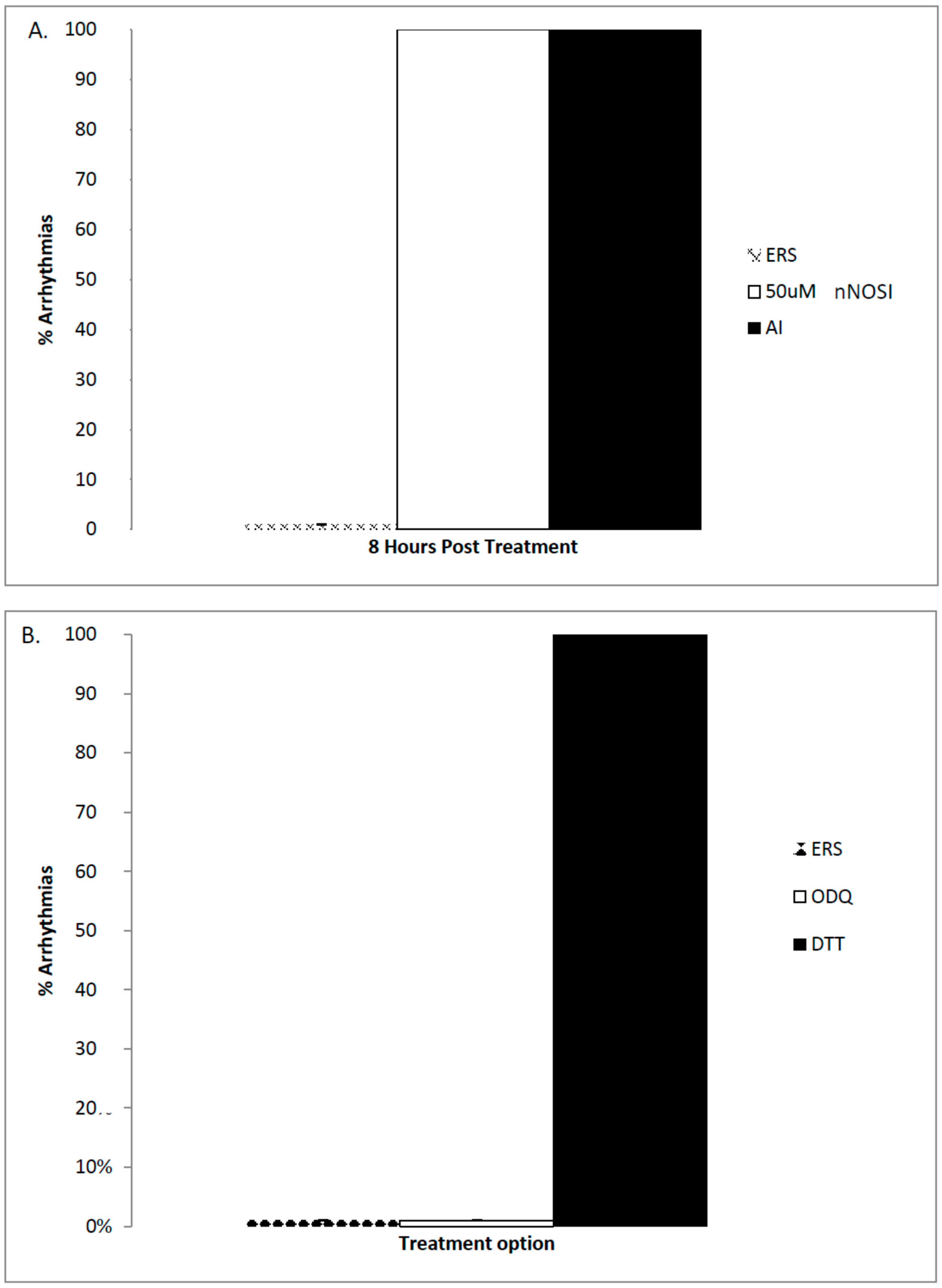
2]. Results from the current study again confirm this hypothesis in that DTT, an S-nitrosylation pathway inhibitor but not ODQ, an NO/sGC/cGMP pathway inhibitor, caused arrhythmias in 100% of the treated embryonic zebrafish population. eNOS is also active in cardiomyocytes using the
S-nitrosylation pathway to modify L-type Ca
2+ channels and thus contractility [
2]. eNOS may also act in an anti-atherosclerotic manner by preventing leucocytes from adhering to the walls of blood vessels, a key event in the beginning stages of atherosclerosis [
6]. eNOS has also been implicated in blood vessel angiogenesis by its mediation of signals that lie downstream of angiogenic factors [
6]. Although all isoforms of NOS bind to calmodulin, nNOS and eNOS are more heavily regulated by Ca
2+ release and calmodulin binding than iNOS [
6].
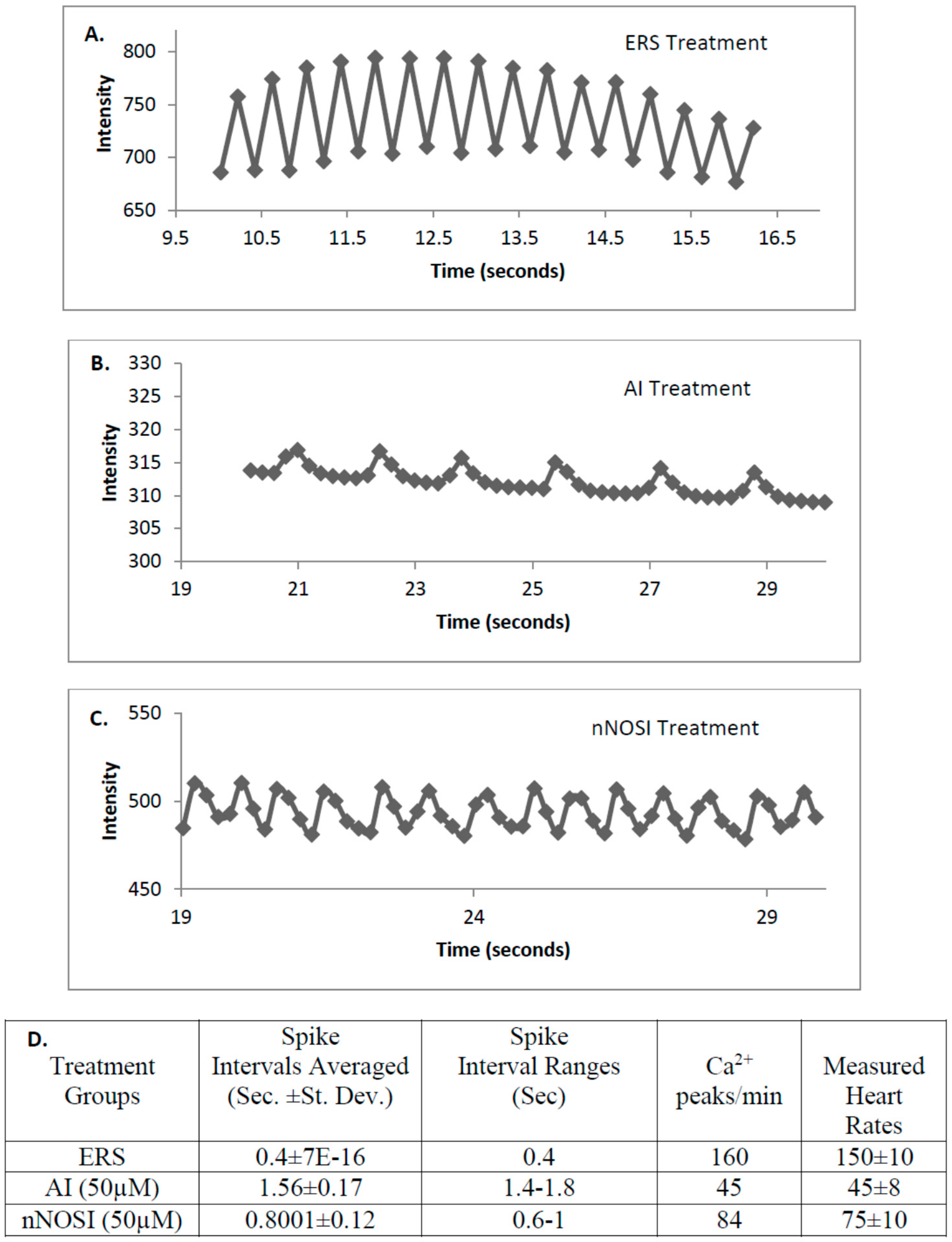
Mechanisms of cardiomyocyte Ca
2+ handling in zebrafish have not been completely explored; however, a recent study suggests that Ca
2+ handling in zebrafish ventricular cardiomyocytes may be initiated mainly via either the L-type Ca
2+ and/or the SR ryanodine channels [
32]. Specifically,
S-nitrosylation has also been shown to act on ryanodine receptors of the sarcoplasmic reticulum of cardiomyocytes to regulate intracellular Ca
2+ and thus heart contractility [
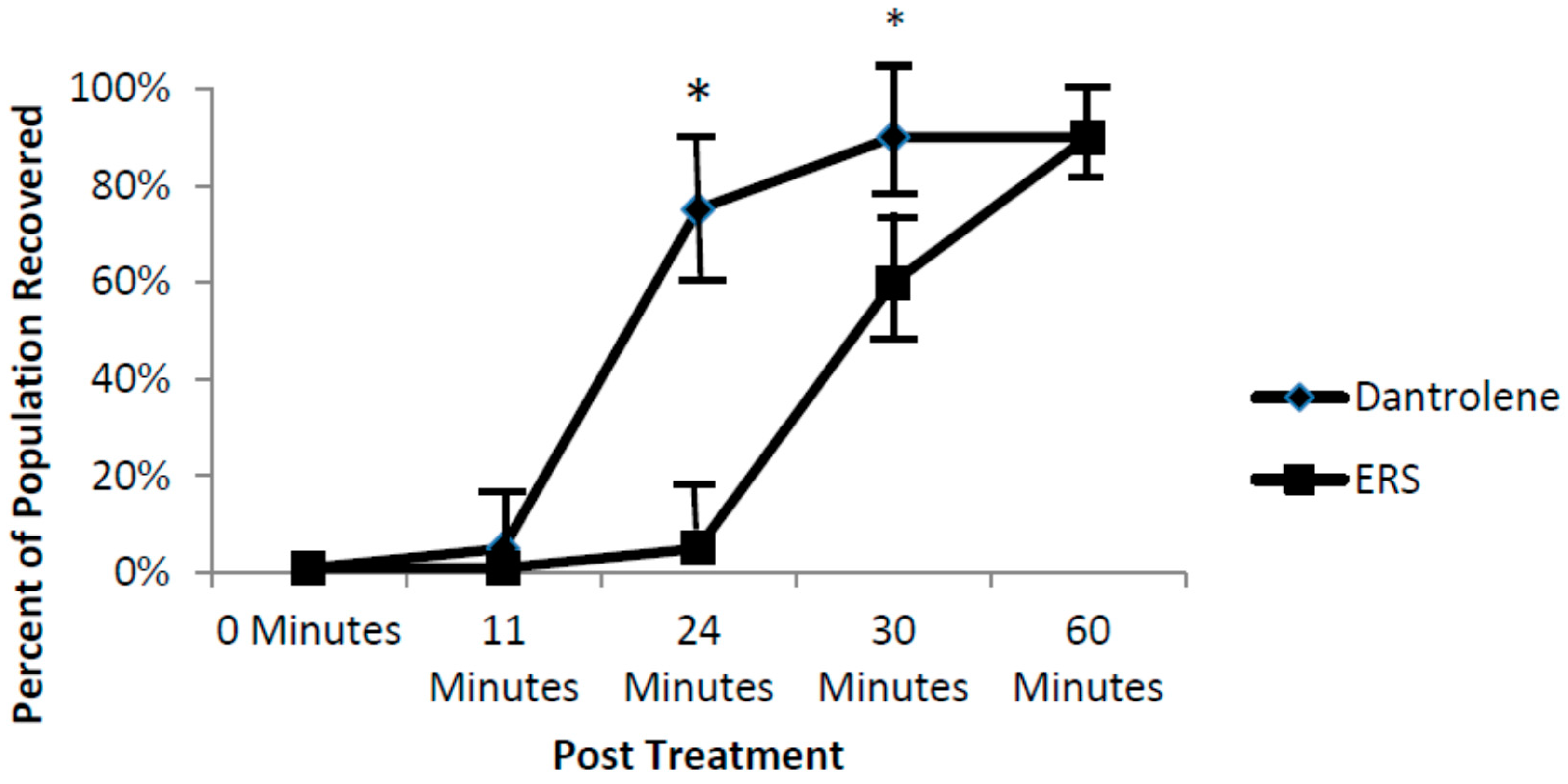
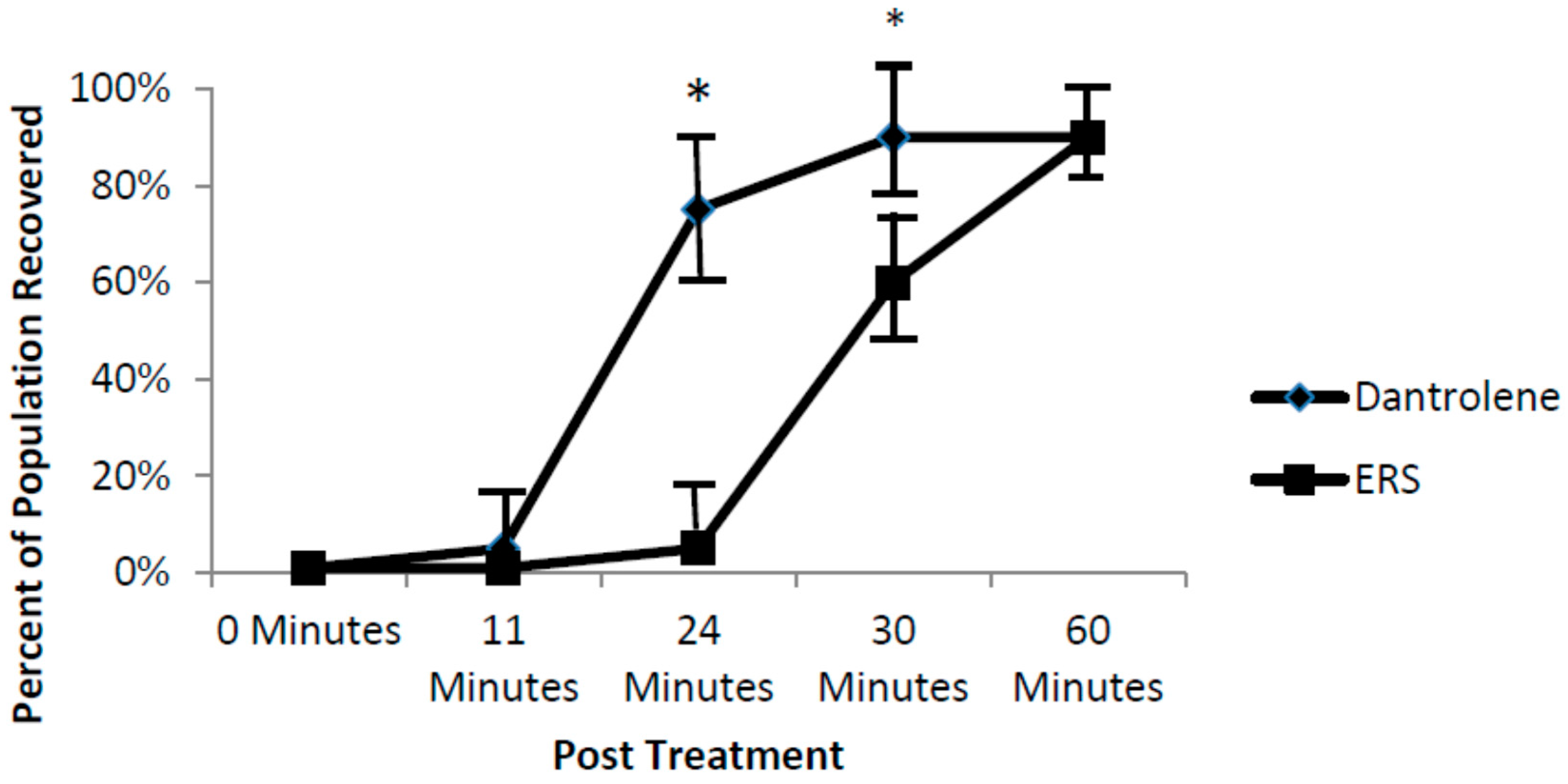
2]. Data from the current study points to the importance of SR ryanodine channels in arrhythmic zebrafish. Specifically, our findings show that washout with a ryanodine receptor blocker, dantrolene, accelerates recovery of fish from the nNOSI induced arrhythmias and cardiac death. The hypothesis here is that dantrolene washout serves to shut off the Ca
2+ leak faster than the ERS control thus restoring HRs and arrhythmias to normal levels. Also, it would appear from the ODQ and findings that the preferred NO pathway in establishing developing zebrafish cardiac stability is the NO/cGMP independent-pathway. Specifically, the current data demonstrated that DTT treatment, which is an inhibitor of the NO-cGMP-independent pathway by preventing S-nitrosylation events, elicits the arrhythmic phenotype in 100% of the fish population. In contrast, no arrhythmic phenotypic expression is evident with ODQ treatment, which blocks the activity of sGC in the NO/sGC/cGMP-dependent pathway. Other reports have also shown that in cardiomyocytes,
S-nitrosylation seems to have protective functions against the development of arrhythmias by way of the nNOS derived NO pathway [
2].
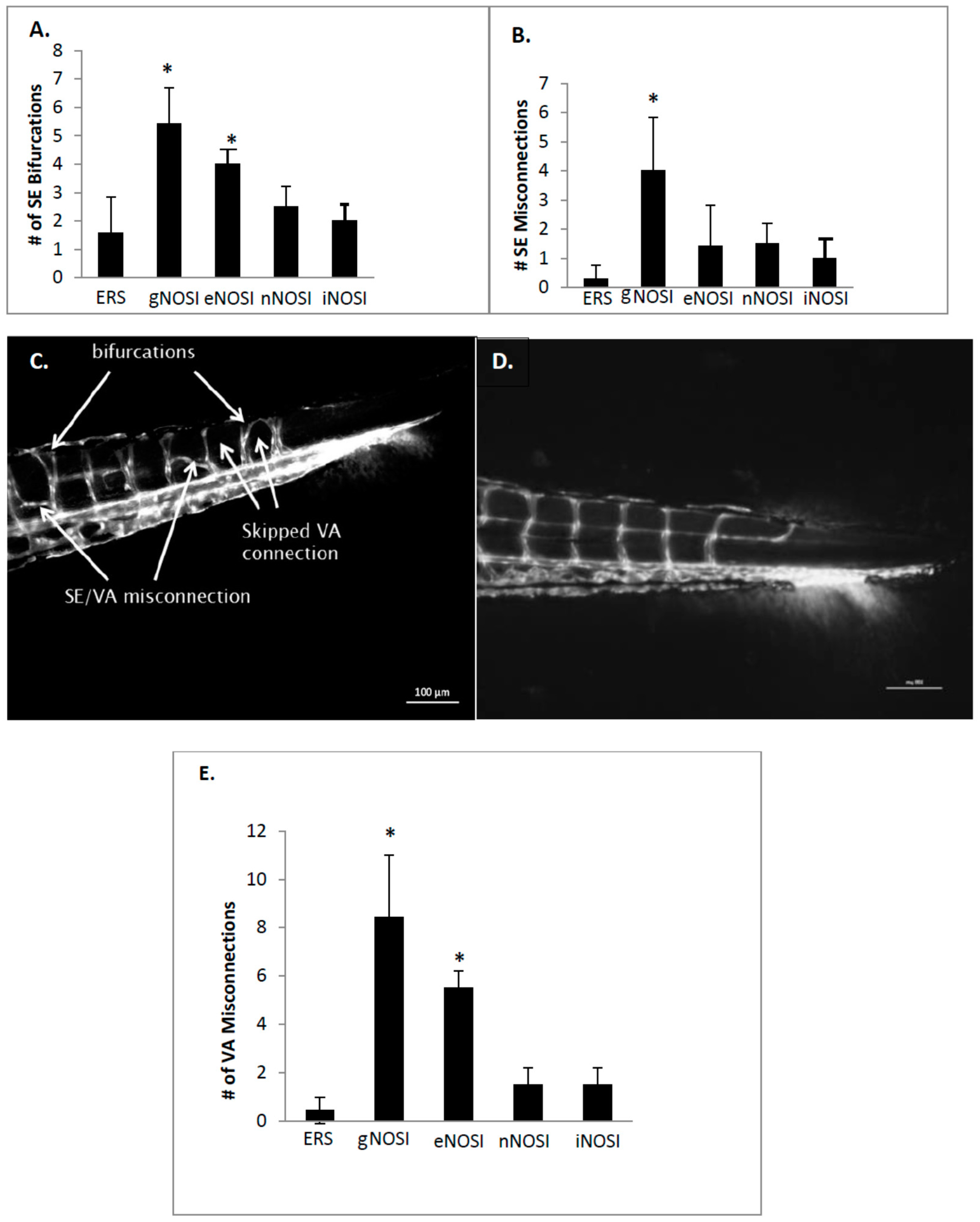
This study also tested the effects of the three NOS isoforms (eNOS, nNOS, and iNOS) on the integrity and maintenance of the developing zebrafish vascular bed. Angiogenesis is the process of new blood vessel development from the migration and restructuring of endothelial and other necessary cells from pre-existing cells of the vasculature [
33]. One of the primary signaling molecules associated with angiogenic stimulation is the vascular endothelial growth factor (VEGF), which, in part, acts through NO to affect vascular development [
22]. VEGF activity can be induced by E2, which also induces NO and fibroblast growth factor-2 and its isoforms, making E2 crucial for angiogenesis [
34]. The NO-sGC/cGMP dependent-pathway seems to act on mitosis and migratory behavior of endothelial cells, which is critical for the development of new blood vessels [
22]. It has been shown that sGC expression coincides with angiogenesis and inhibition coincides with significantly reduced angiogenesis [
33].
NO increases endothelial cell growth and migration even in the absence of sGC, but when NO is combined with increased levels of sGC, it has been observed that the angiogenic influence of NO is increased dramatically [
35]. However, VEGF has also been shown to act through
S-nitrosylation to stimulate angiogenesis through the activation of mitogen-activated protein kinase phosphatase 7 (MKP7), which activates c-Jun N-terminal kinase 3 (JNK3), which facilitates endothelial cell migration, a key event in angiogenesis [
2]. NO mediates both of these pathways since it activates both sGC and
S-nitrosylation once it has been stimulated by VEGF [
2,
16,
17]. eNOS has been shown to be active in many different functions of the vascular system, from angiogenesis to anti-atherosclerotic activities, but nNOS also is expressed in low levels in vascular smooth muscle to maintain some vasodilation when eNOS is not active or inhibited or otherwise incapacitated [
6].
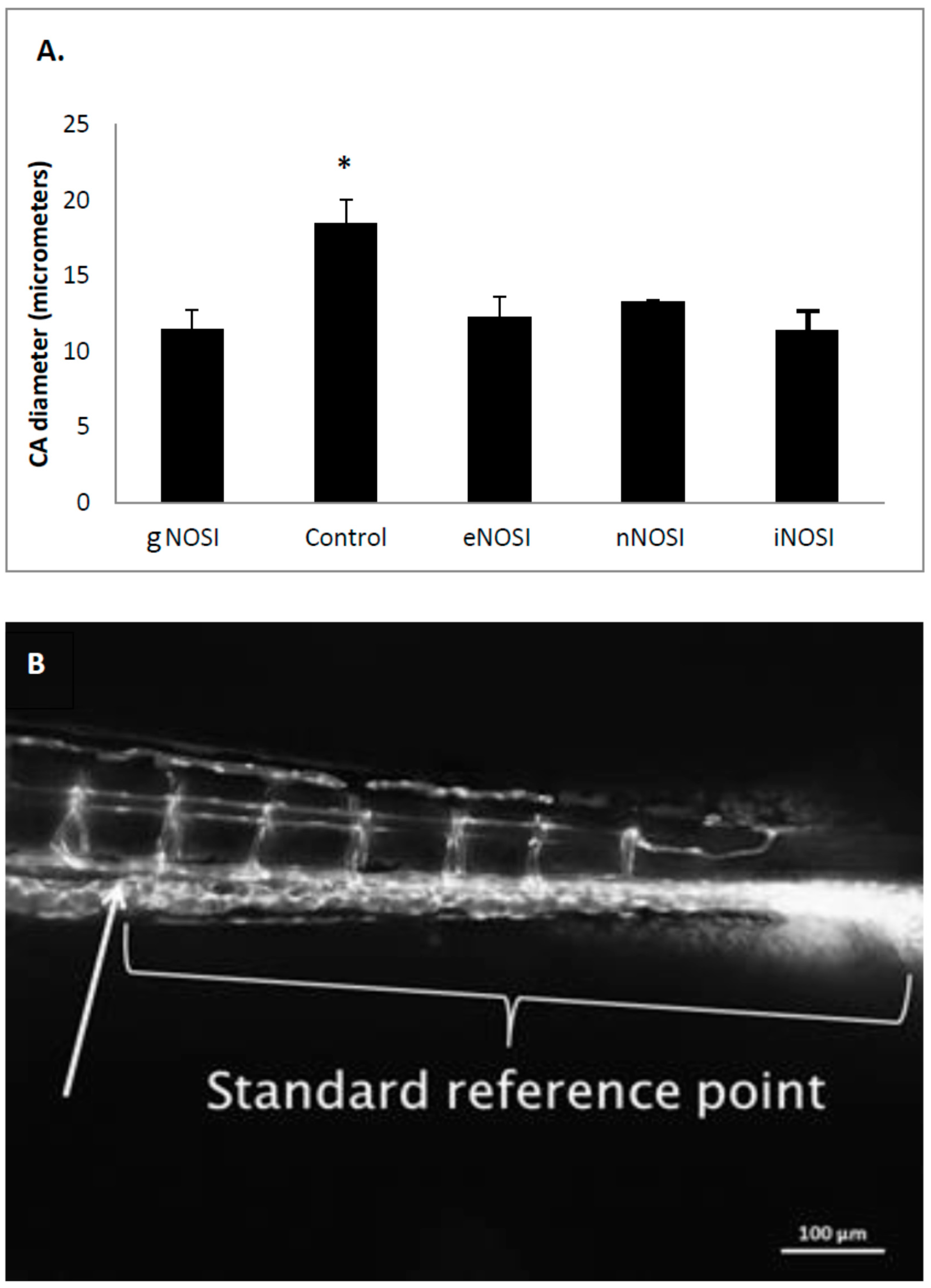
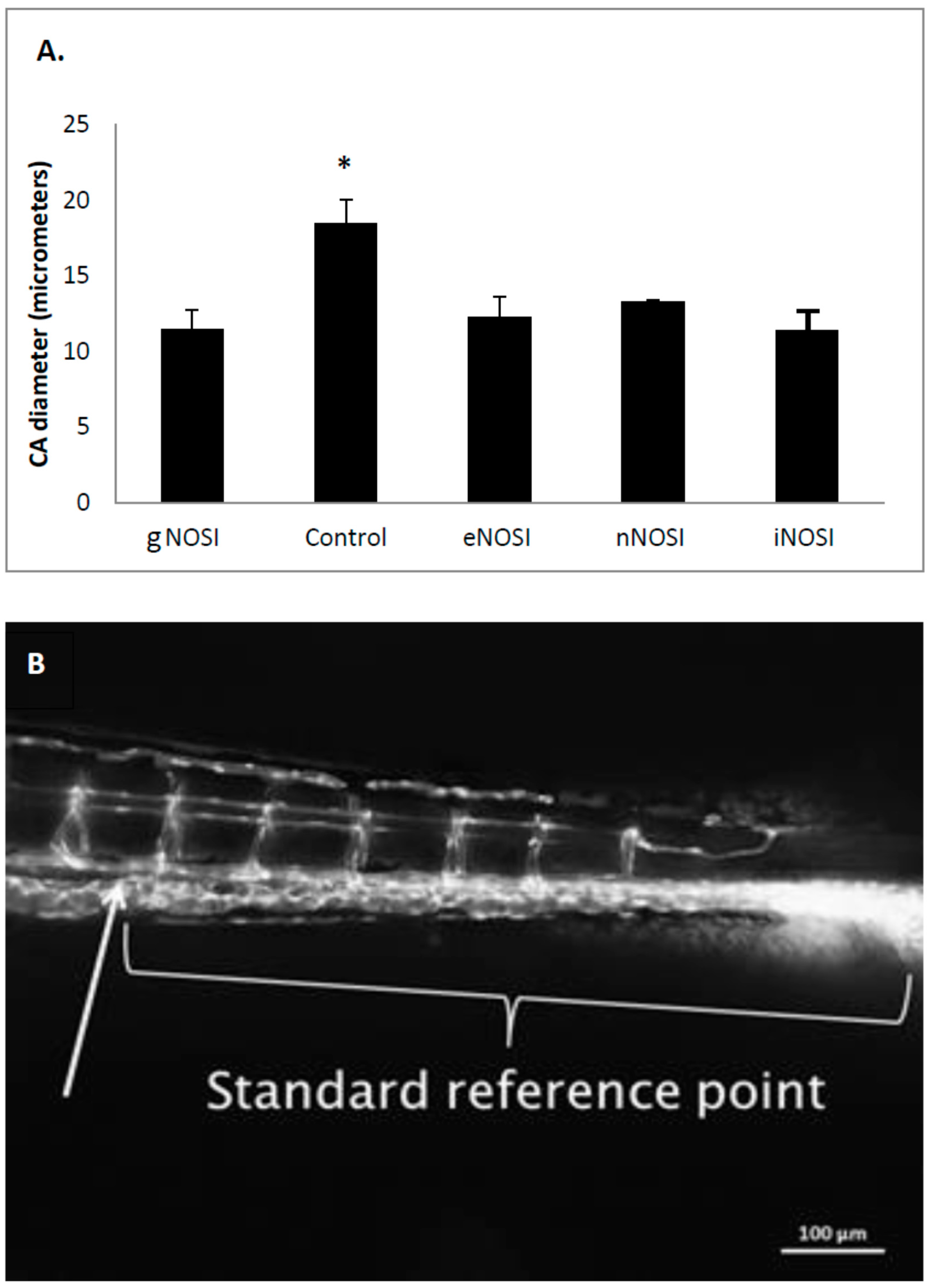
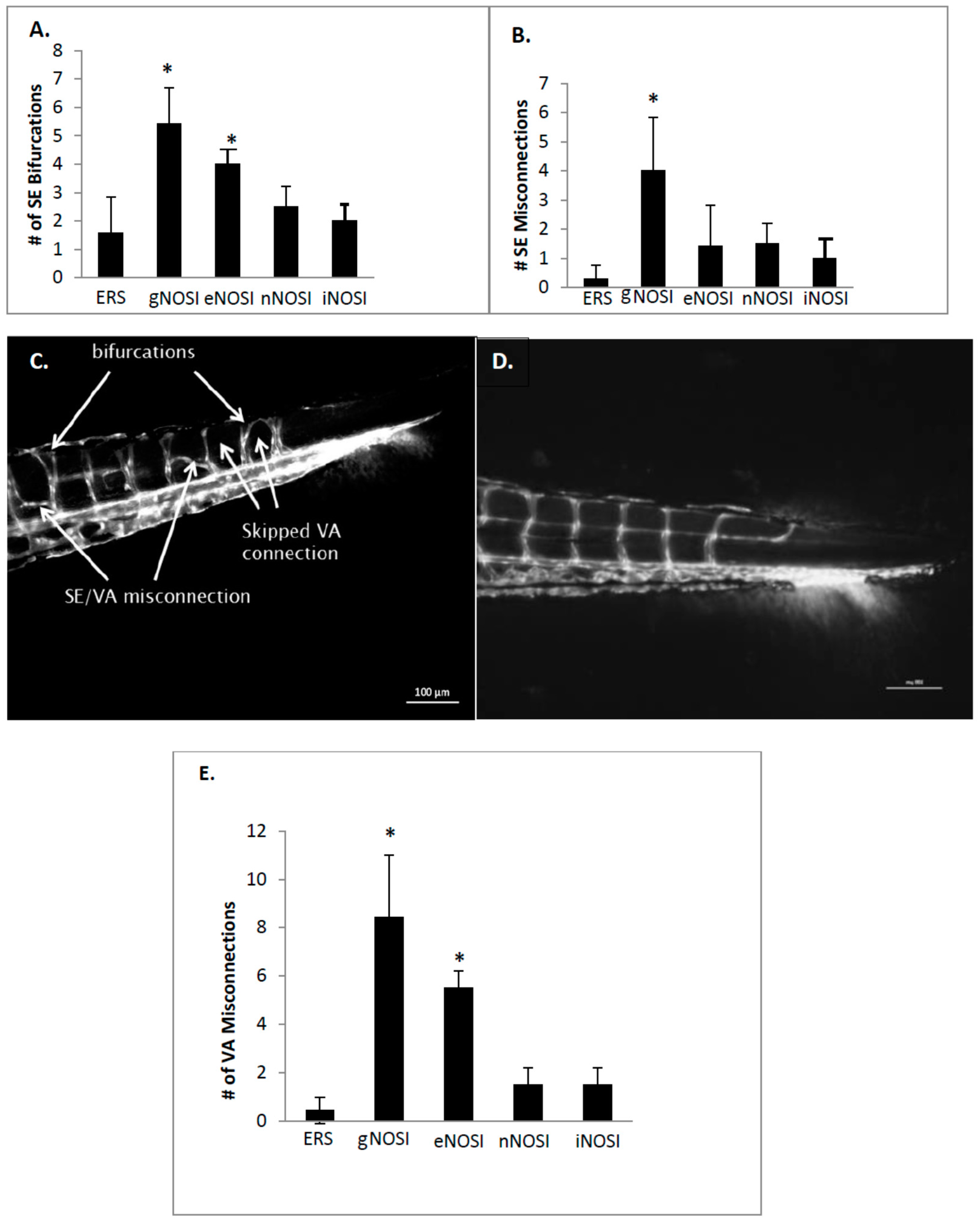
The current data indicated that eNOS was the isoform most implicated in the maintenance of an intact developing fish vascular system. Specifically, eNOS deprivation resulted in numerous intersegmental vessel misconnections, as well as patchy development or deterioration in the dorsal longitudinal anastomatic vessels and vertebral arteries. These vessels are some of the last to develop in the zebrafish and are thus most sensitive to the various eNOSI manipulations during our study period of 2–4 dpf [
13]. In a previous study, we reported similarities in vessel anomalies during development in E2 deprived AI-treated fish, which could be rescued with E2 co-treatment [
13]. These comparisons and similarities may be explained by the fact that E2 exerts control over the CVS, in part through the regulation of NOS activity [
2,
4] either directly or through its stimulatory effects on VEGF activity [
34]. Specifically, VEGF activity is induced by E2, which also induces NO and fibroblast growth factor-2 and its isoforms, making E2 crucial for angiogenesis [
34]. Specifically, NO has been shown to be one of the primary signaling molecules associated with angeogenic stimulation through VEGF, which, in part, acts through NO to affect vascular development [
22]. The NO-sGC/cGMP pathway seems to act on mitosis and migratory behavior of endothelial cells, which is critical for the development of new blood vessels [
22]. sGC expression coincides with angiogenesis and its inhibition coincides with inhibited angiogenesis [
33]. NO increases endothelial cell growth and migration even in the absence of sGC, but when NO is combined with increased levels of sGC the angiogenic influence of NO is increased dramatically [
33]. However, VEGF also acts through
S-nitrosylation to stimulate angiogenesis through the activation of mitogen-activated protein kinase phosphatase 7 (MKP7), which activates c-Jun N-terminal kinase 3 (JNK3) which facilitates endothelial cell migration, a key event in angiogenesis [
2]. NO mediates both of these pathways since it activates both sGC and
S-nitrosylation once it has been stimulated by VEGF [
2,
22].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}