Pathophysiological Mechanisms of Cognitive Impairment and Neurodegeneration by Toxoplasma gondii Infection

 , , and
, , and
Abstract
1. Introduction
2. Toxoplasma Gondii Effects in the CNS
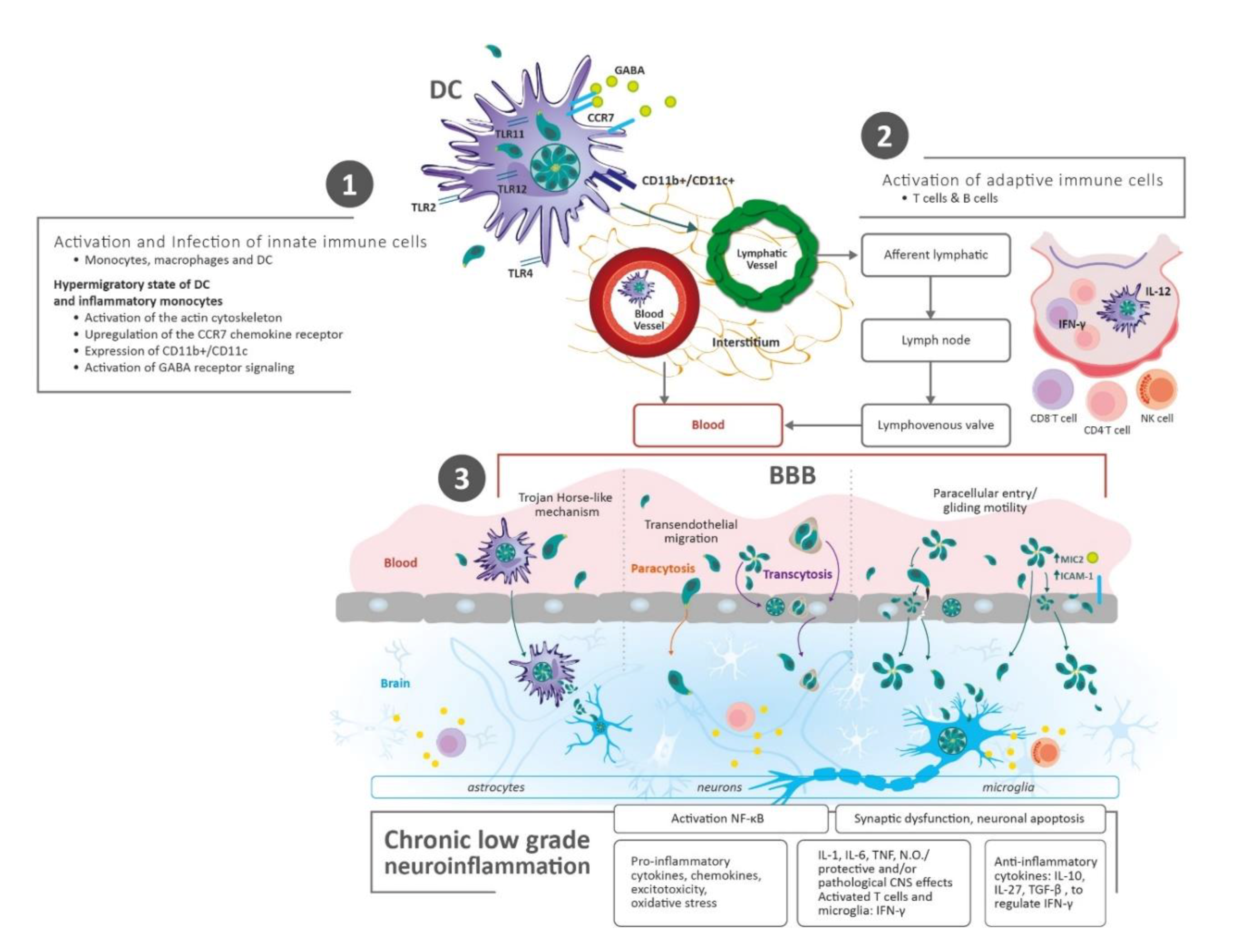
2.1. Parasite Transmission and Dissemination to the Brain
2.2. Chronic Immune Response in the Brain
3. Amyloid Beta Plaques Accumulation and Tau Pathology
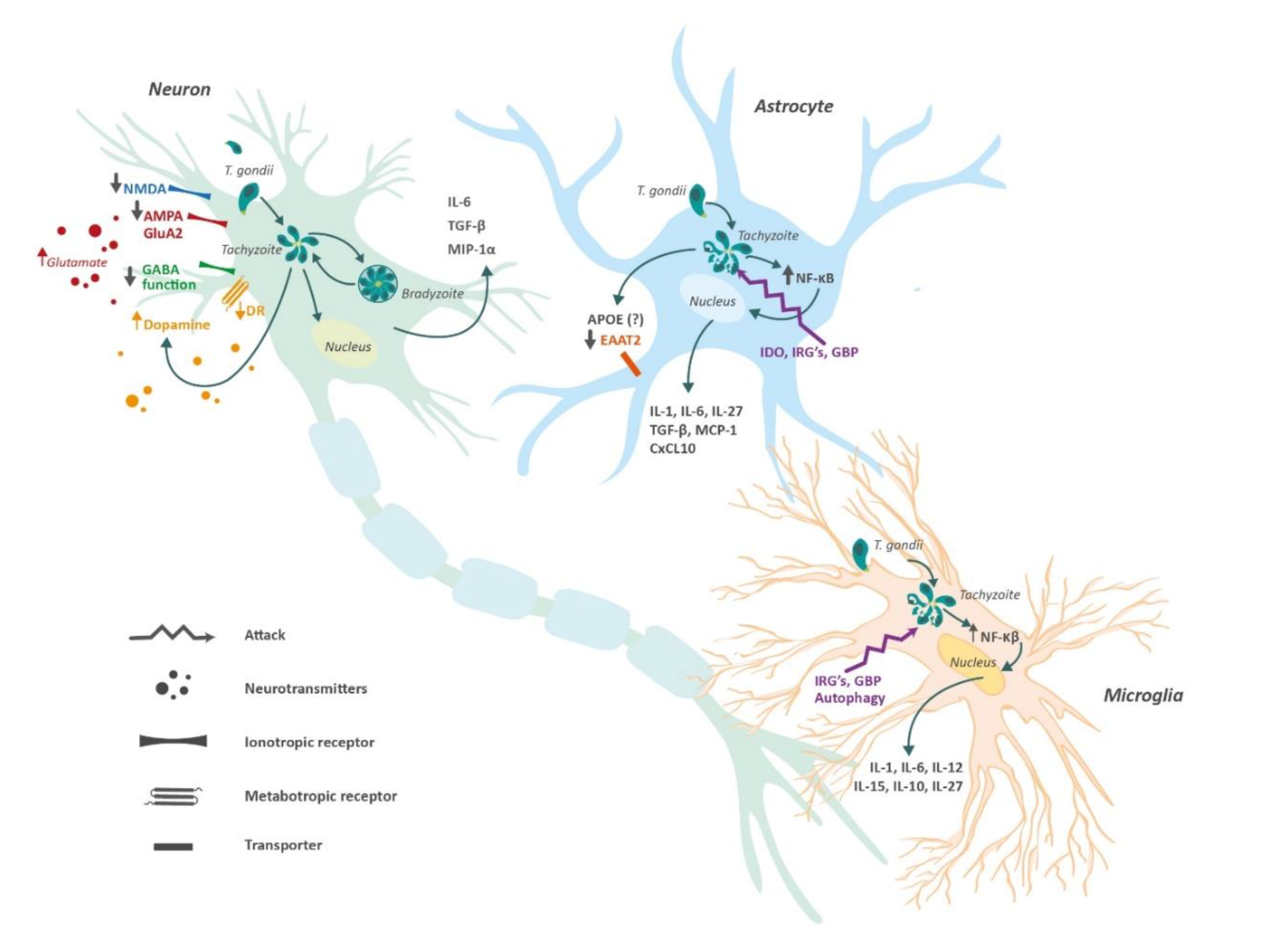
4. Neurotransmitter Imbalance Induced by Toxoplasma
4.1. Glutamate
4.2. GABA
4.3. Dopamine
5. Toxoplasmosis and ApoE
6. Final Considerations and Conclusions
Author Contributions
Funding
Acknowledgments
Ethical Statement
Conflicts of Interest
References
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of alzheimer’s disease: Report of the NINCDS-ADRDA work group* under the auspices of department of health and human services task force on alzheimer’s disease. Neurology 1984, 34, 939. [Google Scholar] [CrossRef]
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s disease and parkinson’s disease. N. Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Jack, C.R.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J. On the path to 2025: Understanding the alzheimer’s disease continuum. Alzheimer Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Hersi, M.; Irvine, B.; Gupta, P.; Gomes, J.; Birkett, N.; Krewski, D. Risk factors associated with the onset and progression of alzheimer’s disease: A systematic review of the evidence. Neurotoxicology 2017, 61, 143–187. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Guerrero, G.; Amador-Muñoz, D.; Calderón-Ospina, C.A.; López-Fuentes, D.; Nava-Mesa, M.O. Proton pump inhibitors and dementia: Physiopathological mechanisms and clinical consequences. Neural Plast. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Torres-Berrio, A.; Nava-Mesa, M.O. The opioid system in stress-induced memory disorders: From basic mechanisms to clinical implications in post-traumatic stress disorder and alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 88, 327–338. [Google Scholar] [CrossRef]
- Bu, X.; Yao, X.; Jiao, S.; Zeng, F.; Liu, Y.; Xiang, Y.; Liang, C.; Wang, Q.; Wang, X.; Cao, H. A study on the association between infectious burden and a lzheimer’s disease. Eur. J. Neurol. 2015, 22, 1519–1525. [Google Scholar] [CrossRef]
- Shima, K.; Kuhlenbäumer, G.; Rupp, J. Chlamydia pneumoniae infection and Alzheimer’s disease: A connection to remember? Med. Microbiol. Immunol. 2010, 199, 283–289. [Google Scholar] [CrossRef]
- Cetinkaya, Z.; Yazar, S.; Gecici, O.; Namli, M.N. Anti-Toxoplasma gondii antibodies in patients with schizophrenia—preliminary findings in a Turkish sample. Schizophr. Bull. 2007, 33, 789–791. [Google Scholar] [CrossRef]
- Pearce, B.D.; Kruszon-Moran, D.; Jones, J.L. The relationship between Toxoplasma gondii infection and mood disorders in the third National Health and Nutrition Survey. Biol. Psychiatry 2012, 72, 290–295. [Google Scholar] [CrossRef]
- Koseoglu, E.; Koc, I.; Yazar, S. Is Toxoplasma gondii a causal agent in migraine? Am. J. Med. Sci. 2009, 338, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Miman, O.; Mutlu, E.A.; Ozcan, O.; Atambay, M.; Karlidag, R.; Unal, S. Is there any role of toxoplasma gondii in the etiology of obsessive–compulsive disorder? Psychiatry Res. 2010, 177, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Kusbeci, O.Y.; Miman, O.; Yaman, M.; Aktepe, O.C.; Yazar, S. Could toxoplasma gondii have any role in alzheimer disease? Alzheimer Dis. Assoc. Disord. 2011, 25, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.; Bynoe, M.S. Toxoplasma gondii alters NMDAR signaling and induces signs of Alzheimer’s disease in wild-type, C57BL/6 mice. FASEB J. 2019, 33, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudvand, H.; Sheibani, V.; Shojaee, S.; Mirbadie, S.R.; Keshavarz, H.; Esmaeelpour, K.; Keyhani, A.R.; Ziaali, N. Toxoplasma gondii infection potentiates cognitive impairments of alzheimer’s disease in the BALB/c mice. J. Parasitol. 2016, 102, 629–635. [Google Scholar] [CrossRef]
- McGovern, K.E.; Cabral, C.M.; Morrison, H.W.; Koshy, A.A. Aging with toxoplasma gondii results in pathogen clearance, resolution of inflammation, and minimal consequences to learning and memory. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.-K.; Pyo, K.-H.; Shin, K.Y.; Hwang, Y.S.; Lim, H.; Lee, S.J.; Moon, J.-H.; Lee, S.H.; Suh, Y.-H.; Chai, J.-Y. Toxoplasma gondii infection in the brain inhibits neuronal degeneration and learning and memory impairments in a murine model of alzheimer’s disease. PLoS ONE 2012, 7, e33312. [Google Scholar] [CrossRef]
- Perry, C.E.; Gale, S.D.; Erickson, L.; Wilson, E.; Nielsen, B.; Kauwe, J.; Hedges, D.W. Seroprevalence and serointensity of latent toxoplasma gondii in a sample of elderly adults with and without alzheimer disease. Alzheimer Dis. Assoc. Disord. 2016, 30, 123–126. [Google Scholar] [CrossRef]
- Alvarado-Esquivel, C.; Rico-Almochantaf, Y.D.R.; Hernández-Tinoco, J.; Quiñones-Canales, G.; Sánchez-Anguiano, L.F.; Torres-González, J.; Schott, B.; Liesenfeld, O.; Dunay, I.R. Toxoplasma gondii exposure and neurological disorders: An age-and gender-matched case-control pilot study. Eur. J. Microbiol. Immunol. 2017, 7, 303–309. [Google Scholar] [CrossRef]
- Bouscaren, N.; Pilleron, S.; Mbelesso, P.; Ndamba-Bandzouzi, B.; Dartigues, J.; Clément, J.; Preux, P.; Dardé, M.; Guerchet, M. EPIDEMCA group prevalence of toxoplasmosis and its association with dementia in older adults in central Africa: A result from the EPIDEMCA programme. Trop. Med. Int. Health 2018, 23, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Bayani, M.; Riahi, S.M.; Bazrafshan, N.; Ray Gamble, H.; Rostami, A. Toxoplasma gondii infection and risk of parkinson and alzheimer diseases: A systematic review and meta-analysis on observational studies. Acta Trop. 2019, 196, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Nayeri Chegeni, T.; Sarvi, S.; Moosazadeh, M.; Sharif, M.; Aghayan, S.A.; Amouei, A.; Hosseininejad, Z.; Daryani, A. Is toxoplasma gondii a potential risk factor for alzheimer’s disease? A systematic review and meta-analysis. Microb. Pathog. 2019, 137, 103751. [Google Scholar] [CrossRef] [PubMed]
- Berenreiterová, M.; Flegr, J.; Kuběna, A.A.; Němec, P. The distribution of toxoplasma gondii cysts in the brain of a mouse with latent toxoplasmosis: Implications for the behavioral manipulation hypothesis. PLoS ONE 2011, 6, e28925. [Google Scholar] [CrossRef]
- McConkey, G.A.; Martin, H.L.; Bristow, G.C.; Webster, J.P. Toxoplasma gondii infection and behavior—location, location, location? J. Exp. Biol. 2013, 216, 113–119. [Google Scholar] [CrossRef]
- Lang, D.; Schott, B.H.; van Ham, M.; Morton, L.; Kulikovskaja, L.; Herrera-Molina, R.; Pielot, R.; Klawonn, F.; Montag, D.; Jänsch, L.; et al. Chronic Toxoplasma infection is associated with distinct alterations in the synaptic protein composition. J. Neuroinflamm. 2018, 15, 216. [Google Scholar] [CrossRef]
- Parlog, A.; Harsan, L.-A.; Zagrebelsky, M.; Weller, M.; von Elverfeldt, D.; Mawrin, C.; Korte, M.; Dunay, I.R. Chronic murine toxoplasmosis is defined by subtle changes in neuronal connectivity. Dis. Model Mech. 2014, 7, 459–469. [Google Scholar] [CrossRef]
- Sanecka, A.; Frickel, E.-M. Use and abuse of dendritic cells by Toxoplasma gondii. Virulence 2012, 3, 678–689. [Google Scholar] [CrossRef]
- Wohlfert, E.A.; Blader, I.J.; Wilson, E.H. Brains and brawn: Toxoplasma infections of the central nervous system and skeletal muscle. Trends Parasitol. 2017, 33, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Dubey, J. Advances in the life cycle of toxoplasma gondii. Int. J. Parasitol. 1998, 28, 1019–1024. [Google Scholar] [CrossRef]
- Elmore, S.A.; Jones, J.L.; Conrad, P.A.; Patton, S.; Lindsay, D.S.; Dubey, J. Toxoplasma gondii: Epidemiology, feline clinical aspects, and prevention. Trends Parasitol. 2010, 26, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.B. Everyday and exotic foodborne parasites. Can. J. Infect. Dis. Med. Microbiol. 2000, 11, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Dubey, J. Waterborne toxoplasmosis–recent developments. Exp. Parasitol. 2010, 124, 10–25. [Google Scholar] [CrossRef]
- Khurana, S.; Batra, N. Toxoplasmosis in organ transplant recipients: Evaluation, implication, and prevention. Trop. Parasitol. 2016, 6, 123. [Google Scholar]
- Cohen, S.; Denkers, E. The gut mucosal immune response to toxoplasma gondii. Parasite Immunol. 2015, 37, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Fuks, J.M.; Arrighi, R.B.G.; Weidner, J.M.; Kumar Mendu, S.; Jin, Z.; Wallin, R.P.A.; Rethi, B.; Birnir, B.; Barragan, A. GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog. 2012, 8, e1003051. [Google Scholar] [CrossRef]
- Weidner, J.M.; Kanatani, S.; Hernández-Castañeda, M.A.; Fuks, J.M.; Rethi, B.; Wallin, R.P.A.; Barragan, A. Rapid cytoskeleton remodelling in dendritic cells following invasion by toxoplasma gondii coincides with the onset of a hypermigratory phenotype. Cell. Microbiol. 2013, 15, 1735–1752. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef]
- Courret, N.; Darche, S.; Sonigo, P.; Milon, G.; Buzoni-Gâtel, D.; Tardieux, I. CD11c-and CD11b-expressing mouse leukocytes transport single toxoplasma gondii tachyzoites to the brain. Blood 2006, 107, 309–316. [Google Scholar] [CrossRef]
- Ueno, N.; Harker, K.S.; Clarke, E.V.; McWhorter, F.Y.; Liu, W.F.; Tenner, A.J.; Lodoen, M.B. Real-time imaging of toxoplasma-infected human monocytes under fluidic shear stress reveals rapid translocation of intracellular parasites across endothelial barriers. Cell. Microbiol. 2014, 16, 580–595. [Google Scholar] [CrossRef]
- Barragan, A.; Brossier, F.; Sibley, L.D. Transepithelial migration of toxoplasma gondii involves an interaction of intercellular adhesion molecule 1 (ICAM-1) with the parasite adhesin MIC2. Cell. Microbiol. 2005, 7, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Harker, K.S.; Jivan, E.; McWhorter, F.Y.; Liu, W.F.; Lodoen, M.B. Shear forces enhance toxoplasma gondii tachyzoite motility on vascular endothelium. MBio 2014, 5, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Lambert, H.; Hitziger, N.; Dellacasa, I.; Svensson, M.; Barragan, A. Induction of dendritic cell migration upon toxoplasma gondii infection potentiates parasite dissemination. Cell. Microbiol. 2006, 8, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Lachenmaier, S.M.; Deli, M.A.; Meissner, M.; Liesenfeld, O. Intracellular transport of Toxoplasma gondii through the blood-brain barrier. J. Neuroimmunol. 2011, 232, 119–130. [Google Scholar] [CrossRef]
- Furtado, J.M.; Bharadwaj, A.S.; Chipps, T.J.; Pan, Y.; Ashander, L.M.; Smith, J.R. Toxoplasma gondii tachyzoites cross retinal endothelium assisted by intercellular adhesion molecule-1 in vitro. Immunol. Cell Biol. 2012, 90, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Konradt, C.; Ueno, N.; Christian, D.A.; Delong, J.H.; Pritchard, G.H.; Herz, J.; Bzik, D.J.; Koshy, A.A.; McGavern, D.B.; Lodoen, M.B. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat. Microbiol. 2016, 1, 16001. [Google Scholar] [CrossRef]
- Gambuzza, M.E.; Sofo, V.; Salmeri, F.M.; Soraci, L.; Marino, S.; Bramanti, P. Toll-like receptors in Alzheimer’s disease: A therapeutic perspective. CNS Neurol. Disord. Drug Targets 2014, 13, 1542–1558. [Google Scholar] [CrossRef] [PubMed]
- Ravari, A.; Mirzaei, T.; Kennedy, D.; Kazemi Arababadi, M. Chronoinflammaging in alzheimer: A systematic review on the roles of toll like receptor 2. Life Sci. 2017, 171, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Atmaca, H.T.; Kul, O.; Karakuş, E.; Terzi, O.S.; Canpolat, S.; Anteplioğlu, T. Astrocytes, microglia/macrophages, and neurons expressing toll-like receptor 11 contribute to innate immunity against encephalitic toxoplasma gondii infection. Neuroscience 2014, 269, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, X.; Qin, W.; Zhang, X.; Wu, L.; Li, Y.; Zhou, C.; Zhou, H.; He, S.; Cong, H. From inflammatory reactions to neurotransmitter changes: Implications for understanding the neurobehavioral changes in mice chronically infected with toxoplasma gondii. Behav. Brain Res. 2019, 359, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Casal, C.; Serratosa, J.; Tusell, J.M. Effects of beta-AP peptides on activation of the transcription factor NF-kappaB and in cell proliferation in glial cell cultures. Neurosci. Res. 2004, 48, 315–323. [Google Scholar] [CrossRef] [PubMed]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Liesenfeld, O.; Parvanova, I.; Zerrahn, J.; Han, S.; Heinrich, F.; Muñoz, M.; Kaiser, F.; Aebischer, T.; Buch, T.; Waisman, A. The IFN-?-inducible GTPase, Irga6, protects mice against toxoplasma gondii but not against plasmodium berghei and some other intracellular pathogens. PLoS ONE 2011, 6, e20568. [Google Scholar] [CrossRef]
- Munoz, M.; Liesenfeld, O.; Heimesaat, M.M. Immunology of Toxoplasma gondii. Immunol. Rev. 2011, 240, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Sturge, C.R.; Yarovinsky, F. Complex immune cell interplay in the gamma interferon response during toxoplasma gondii infection. Infect. Immun. 2014, 82, 3090–3097. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, D.; Kullberg, M.C.; Feng, C.G.; Goldszmid, R.S.; Collazo, C.M.; Wilson, M.; Wynn, T.A.; Kamanaka, M.; Flavell, R.A.; Sher, A. Conventional T-bet+ Foxp3− Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J. Exp. Med. 2007, 204, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Koshy, A.A.; Fouts, A.E.; Lodoen, M.B.; Alkan, O.; Blau, H.M.; Boothroyd, J.C. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nat. Methods 2010, 7, 307. [Google Scholar] [CrossRef]
- Schlüter, D.; Deckert, M.; Hof, H.; Frei, K. Toxoplasma gondii infection of neurons induces neuronal cytokine and chemokine production, but gamma interferon-and tumor necrosis factor-stimulated neurons fail to inhibit the invasion and growth of T. gondii. Infect. Immun. 2001, 69, 7889–7893. [Google Scholar] [CrossRef]
- Chao, C.C.; Anderson, W.R.; Hu, S.; Gekker, G.; Martella, A.; Peterson, P.K. Activated microgila inhibit multiplication of toxoplasma gondii via a nitric oxide mechanism. Clin. Immunol. Immunopathol. 1993, 67, 178–183. [Google Scholar] [CrossRef]
- Tomita, T.; Sugi, T.; Yakubu, R.; Tu, V.; Ma, Y.; Weiss, L.M. Making home sweet and sturdy: Toxoplasma gondii ppGalNAc-Ts glycosylate in hierarchical order and confer cyst wall rigidity. MBio 2017, 8, e2048-16. [Google Scholar] [CrossRef]
- Blanchard, N.; Dunay, I.R.; Schlüter, D. Persistence of Toxoplasma gondii in the central nervous system: A fine-tuned balance between the parasite, the brain and the immune system. Parasite Immunol. 2015, 37, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Dietrich, H.K.; Axtell, R.C.; Williams, A.M.; Egusquiza, R.; Wai, K.M.; Koshy, A.A.; Buckwalter, M.S. Astrocytic TGF-β signaling limits inflammation and reduces neuronal damage during central nervous system Toxoplasma infection. J. Immunol. 2014, 193, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Younkin, S.G. The role of Aβ42 in alzheimer’s disease. J. Physiol.-Paris 1998, 92, 289–292. [Google Scholar] [CrossRef]
- Möhle, L.; Israel, N.; Paarmann, K.; Krohn, M.; Pietkiewicz, S.; Müller, A.; Lavrik, I.N.; Buguliskis, J.S.; Schott, B.H.; Schlüter, D. Chronic Toxoplasma gondii infection enhances β-amyloid phagocytosis and clearance by recruited monocytes. Acta Neuropathol. Commun. 2016, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Cabral, C.M.; McGovern, K.E.; MacDonald, W.R.; Franco, J.; Koshy, A.A. Dissecting amyloid beta deposition using distinct strains of the neurotropic parasite Toxoplasma gondii as a novel tool. ASN Neuro 2017, 9, 1759091417724915. [Google Scholar] [CrossRef]
- Syn, G.; Anderson, D.; Blackwell, J.M.; Jamieson, S.E. Epigenetic dysregulation of host gene expression in toxoplasma infection with specific reference to dopamine and amyloid pathways. Infect. Genet. Evol. 2018, 65, 159–162. [Google Scholar] [CrossRef]
- Benveniste, E.N. Cytokine actions in the central nervous system. Cytokine Growth Factor Rev. 1998, 9, 259–275. [Google Scholar] [CrossRef]
- Mastrangelo, M.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Interferon-γ differentially affects alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am. J. Pathol. 2009, 175, 2076–2088. [Google Scholar] [CrossRef] [PubMed]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L., Jr.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 1995, 374, 647. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiyota, T.; Horiba, M.; Buescher, J.L.; Walsh, S.M.; Gendelman, H.E.; Ikezu, T. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am. J. Pathol. 2007, 170, 680–692. [Google Scholar] [CrossRef]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-γ and interleukin-1β or tumor necrosis factor α on the synthesis of Aβ1-40 and Aβ1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.-A.; Mills, K.H.; Lynch, M.A. IFN-γ production by amyloid β–Specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of alzheimer’s disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Deczkowska, A.; Rosenzweig, N.; Tsitsou-Kampeli, A.; Sharif, A.M.; Matcovitch-Natan, O.; Kertser, A.; David, E.; Amit, I.; Schwartz, M. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of alzheimer’s disease. Nat. Med. 2016, 22, 135. [Google Scholar] [CrossRef] [PubMed]
- Lucchese, G. From toxoplasmosis to schizophrenia via NMDA dysfunction: Peptide overlap between toxoplasma gondii and N-Methyl-d-Aspartate receptors as a potential mechanistic link. Front. Psychiatry 2017, 8, 37. [Google Scholar] [CrossRef]
- Cai, X.; Zhou, H.; Xie, Y.; Yu, D.; Wang, Z.; Ren, H. Anti-N-methyl-D-aspartate receptor encephalitis associated with acute toxoplasma gondii infection: A case report. Medicine (Baltimore) 2018, 97, 9924. [Google Scholar] [CrossRef]
- David, C.N.; Frias, E.S.; Szu, J.I.; Vieira, P.A.; Hubbard, J.A.; Lovelace, J.; Michael, M.; Worth, D.; McGovern, K.E.; Ethell, I.M.; et al. GLT-1-Dependent disruption of CNS glutamate homeostasis and neuronal function by the protozoan parasite toxoplasma gondii. PLoS Pathog. 2016, 12, e1005643. [Google Scholar] [CrossRef]
- Kannan, G.; Crawford, J.A.; Yang, C.; Gressitt, K.L.; Ihenatu, C.; Krasnova, I.N.; Cadet, J.L.; Yolken, R.H.; Severance, E.G.; Pletnikov, M.V. Anti-NMDA receptor autoantibodies and associated neurobehavioral pathology in mice are dependent on age of first exposure to toxoplasma gondii. Neurobiol. Dis. 2016, 91, 307–314. [Google Scholar] [CrossRef]
- Bhandage, A.K.; Kanatani, S.; Barragan, A. Toxoplasma-Induced hypermigration of primary cortical microglia implicates GABAergic signaling. Front. Cell Infect. Microbiol. 2019, 9, 73. [Google Scholar] [CrossRef]
- Kanatani, S.; Fuks, J.M.; Olafsson, E.B.; Westermark, L.; Chambers, B.; Varas-Godoy, M.; Uhlén, P.; Barragan, A. Voltage-dependent calcium channel signaling mediates GABAA receptor-induced migratory activation of dendritic cells infected by toxoplasma gondii. PLoS Pathog. 2017, 13, e1006739. [Google Scholar] [CrossRef]
- Brooks, J.M.; Carrillo, G.L.; Su, J.; Lindsay, D.S.; Fox, M.A.; Blader, I.J. Toxoplasma gondii infections alter GABAergic synapses and signaling in the central nervous system. MBio 2015, 6, e01428-15. [Google Scholar] [CrossRef]
- McFarland, R.; Wang, Z.T.; Jouroukhin, Y.; Li, Y.; Mychko, O.; Coppens, I.; Xiao, J.; Jones-Brando, L.; Yolken, R.H.; Sibley, L.D. AAH2 gene is not required for dopamine-dependent neurochemical and behavioral abnormalities produced by Toxoplasma infection in mouse. Behav. Brain Res. 2018, 347, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.L.; Alsaady, I.; Howell, G.; Prandovszky, E.; Peers, C.; Robinson, P.; McConkey, G.A. Effect of parasitic infection on dopamine biosynthesis in dopaminergic cells. Neuroscience 2015, 306, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Prandovszky, E.; Gaskell, E.; Martin, H.; Dubey, J.; Webster, J.P.; McConkey, G.A. The neurotropic parasite toxoplasma gondii increases dopamine metabolism. PLoS ONE 2011, 6, e23866. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Li, Y.; Prandovszky, E.; Karuppagounder, S.S.; Talbot, C.C.; Dawson, V.L.; Dawson, T.M.; Yolken, R.H. MicroRNA-132 dysregulation in toxoplasma gondii infection has implications for dopamine signaling pathway. Neuroscience 2014, 268, 128–138. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Caraci, F.; Nicoletti, F.; Copani, A. Metabotropic glutamate receptors: The potential for therapeutic applications in alzheimer’s disease. Curr. Opin. Pharmacol. 2018, 38, 1–7. [Google Scholar] [CrossRef]
- Cai, Z.; Wan, C.-Q.; Liu, Z. Astrocyte and alzheimer’s disease. J. Neurol. 2017, 264, 2068–2074. [Google Scholar] [CrossRef]
- Johnston, D.; Williams, S.; Jaffe, D.; Gray, R. NMDA-receptor-independent long-term potentiation. Annu. Rev. Physiol. 1992, 54, 489–505. [Google Scholar] [CrossRef]
- Widagdo, J.; Chai, Y.J.; Ridder, M.C.; Chau, Y.Q.; Johnson, R.C.; Sah, P.; Huganir, R.L.; Anggono, V. Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep. 2015, 10, 783–795. [Google Scholar] [CrossRef]
- Greger, I.H.; Khatri, L.; Ziff, E.B. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 2002, 34, 759–772. [Google Scholar] [CrossRef]
- Wenthold, R.J.; Petralia, R.S.; Blahos, J., II; Niedzielski, A.S. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 1996, 16, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Gaisler-Salomon, I.; Kravitz, E.; Feiler, Y.; Safran, M.; Biegon, A.; Amariglio, N.; Rechavi, G. Hippocampus-specific deficiency in RNA editing of GluA2 in alzheimer’s disease. Neurobiol. Aging 2014, 35, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Sans, N.; Vissel, B.; Petralia, R.S.; Wang, Y.-X.; Chang, K.; Royle, G.A.; Wang, C.-Y.; O’Gorman, S.; Heinemann, S.F.; Wenthold, R.J. Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J. Neurosci. 2003, 23, 9367–9373. [Google Scholar] [CrossRef]
- Wright, A.; Vissel, B. The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front. Mol. Neurosci. 2012, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Verpelli, C.; Dvoretskova, E.; Vicidomini, C.; Rossi, F.; Chiappalone, M.; Schoen, M.; Di Stefano, B.; Mantegazza, R.; Broccoli, V.; Böckers, T.M.; et al. Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J. Biol. Chem. 2011, 286, 34839–34850. [Google Scholar] [CrossRef]
- Roussignol, G.; Ango, F.; Romorini, S.; Tu, J.C.; Sala, C.; Worley, P.F.; Bockaert, J.; Fagni, L. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J. Neurosci. 2005, 25, 3560–3570. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.R.; LeBeauf, A.; Sharfman, N.M.; Lukiw, W.J. MicroRNA-34a (miRNA-34a) mediated down-regulation of the post-synaptic cytoskeletal element SHANK3 in sporadic alzheimer’s disease (AD). Front. Neurol. 2019, 10, 28. [Google Scholar] [CrossRef]
- Robinson, M.B.; Jackson, J.G. Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem. Int. 2016, 98, 56–71. [Google Scholar] [CrossRef]
- Ouyang, Y.-B.; Xu, L.; Liu, S.; Giffard, R.G. Role of astrocytes in delayed neuronal death: GLT-1 and its novel regulation by microRNAs. Adv. Neurobiol. 2014, 11, 171–188. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Hoshi, A.; Tsunoda, A.; Yamamoto, T.; Tada, M.; Kakita, A.; Ugawa, Y. Altered expression of glutamate transporter-1 and water channel protein aquaporin-4 in human temporal cortex with alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2018, 44, 628–638. [Google Scholar] [CrossRef]
- Swanson, A.; Wolf, T.; Sitzmann, A.; Willette, A.A. Neuroinflammation in alzheimer’s disease: Pleiotropic roles for cytokines and neuronal pentraxins. Behav. Brain Res. 2018, 347, 49–56. [Google Scholar] [CrossRef]
- Nava-Mesa, M.O.; Jiménez-Díaz, L.; Yajeya, J.; Navarro-Lopez, J.D. GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of alzheimer’s disease. Front. Cell. Neurosci. 2014, 8, 167. [Google Scholar] [CrossRef]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of functional GABA(A) receptors in the alzheimer diseased brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076. [Google Scholar] [CrossRef]
- Ulrich, D. Amyloid-β impairs synaptic inhibition via GABA(A) receptor endocytosis. J. Neurosci. 2015, 35, 9205–9210. [Google Scholar] [CrossRef]
- Azuma, H.; Inamoto, T.; Sakamoto, T.; Kiyama, S.; Ubai, T.; Shinohara, Y.; Maemura, K.; Tsuji, M.; Segawa, N.; Masuda, H.; et al. Gamma-aminobutyric acid as a promoting factor of cancer metastasis; induction of matrix metalloproteinase production is potentially its underlying mechanism. Cancer Res. 2003, 63, 8090–8096. [Google Scholar]
- Wheeler, D.W.; Thompson, A.J.; Corletto, F.; Reckless, J.; Loke, J.C.T.; Lapaque, N.; Grant, A.J.; Mastroeni, P.; Grainger, D.J.; Padgett, C.L.; et al. Anaesthetic impairment of immune function is mediated via GABA(A) receptors. PLoS ONE 2011, 6, e17152. [Google Scholar] [CrossRef]
- MacRae, J.I.; Sheiner, L.; Nahid, A.; Tonkin, C.; Striepen, B.; McConville, M.J. Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of toxoplasma gondii. Cell Host Microbe 2012, 12, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.-M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Kaminga, A.C.; Wen, S.W.; Wu, X.; Acheampong, K.; Liu, A. Dopamine and dopamine receptors in alzheimer’s disease: A systematic review and network meta-analysis. Front. Aging Neurosci. 2019, 11, 175. [Google Scholar] [CrossRef]
- Nobili, A.; Latagliata, E.C.; Viscomi, M.T.; Cavallucci, V.; Cutuli, D.; Giacovazzo, G.; Krashia, P.; Rizzo, F.R.; Marino, R.; Federici, M.; et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of alzheimer’s disease. Nat. Commun. 2017, 8, 14727. [Google Scholar] [CrossRef] [PubMed]
- Cordella, A.; Krashia, P.; Nobili, A.; Pignataro, A.; La Barbera, L.; Viscomi, M.T.; Valzania, A.; Keller, F.; Ammassari-Teule, M.; Mercuri, N.B.; et al. Dopamine loss alters the hippocampus-nucleus accumbens synaptic transmission in the Tg2576 mouse model of alzheimer’s disease. Neurobiol. Dis. 2018, 116, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.P.; McConkey, G.A. Toxoplasma gondii-altered host behaviour: Clues as to mechanism of action. Folia Parasitol. 2010, 57, 95. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.; Sun, P.; Guo, Y.; Zhang, Z.; Jin, G.; Zhen, X. Neuroprotective effects of atypical D1 receptor agonist SKF83959 are mediated via D1 receptor-dependent inhibition of glycogen synthase kinase-3β and a receptor-independent anti-oxidative action. J. Neurochem. 2008, 104, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Stibbs, H. Changes in brain concentrations of catecholamines and indoleamines in toxoplasma gondii infected mice. Ann. Trop. Med. Parasitol. 1985, 79, 153–157. [Google Scholar] [CrossRef]
- Ogawa, N.; Asanuma, M.; Miyazaki, I.; Diaz-Corrales, F.J.; Miyoshi, K. L-DOPA treatment from the viewpoint of neuroprotection. J. Neurol. 2005, 252, 23–31. [Google Scholar] [CrossRef]
- Goodwin, D.; Hrubec, T.C.; Klein, B.G.; Strobl, J.S.; Werre, S.R.; Han, Q.; Zajac, A.M.; Lindsay, D.S. Congenital infection of mice with toxoplasma gondii induces minimal change in behavior and no change in neurotransmitter concentrations. J. Parasitol. 2012, 98, 706–713. [Google Scholar] [CrossRef][Green Version]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nägler, K.; Schumacher, S.; Göritz, C.; Müller, E.-C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Brown, C.M.; Cook, D.; Needham, L.K.; Xu, Q.; Czapiga, M.; Saunders, A.M.; Schmechel, D.E.; Rasheed, K.; Vitek, M.P. APOE and the regulation of microglial nitric oxide production: A link between genetic risk and oxidative stress. Neurobiol. Aging 2002, 23, 777–785. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 causes widespread molecular and cellular alterations associated with alzheimer’s disease phenotypes in human iPSC-Derived brain cell types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant alzheimer’s disease in an APOE3 christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Yahya, R.S.; Awad, S.I.; El-Baz, H.A.; Saudy, N.; Abdelsalam, O.A.; Al-Din, M.S.S. Impact of ApoE genotypes variations on toxoplasma patients with dementia. J. Clin. Neurosci. 2017, 39, 184–188. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Neurotransmitter | Toxoplasma Effects | Study Type | References |
|---|---|---|---|
| Glutamate | Cross-reactivity with NMDA-2D receptors. | In silico (UniProt database and Peptide Match program) | [74] |
| AD signs associated with loss of NMDAR expression and neuronal death. | In vivo and in vitro (C57BL/6 mice). | [15] | |
| Downregulation of synaptosomal EAAT2, AMPA receptor subunit GluA1, and the NMDA receptor subunit GluN1 | In vitro (Naval Medical Research Institute—NMRI-mice) | [26] | |
| Development of anti-NMDA encephalitis. | Case report | [75] | |
| Reduction in the astrocytic glutamate transporter, GLT-1 and increase in extracellular levels of glutamate. Abnormal EEG recordings. | In vivo and in vitro (C57BL/6 and BALB/c mice) | [76] | |
| Elevation of GLUN2 autoantibodies and reduction in Glun2A expression (NMDAR subunits). Reduction in the vesicular glutamate 1 transporter (VGLUT1) and post-synaptic density 95 (PSD-95). | In vitro (BALB/c mice) | [77] | |
| GABA | Dendritic cells hypermigration through GABAergic signaling which allows parasitic systemic dissemination. | In vivo and in vitro (C57BL/6 mice bone marrow-derived DC and human monocyte-derived DC) | [36] |
| Increased microglial cells hypermigration via GABAergic transmission. | In vitro (C57BL/6 mice astrocyte and microglia cell cultures) | [78] | |
| Activation of GABA-A receptors and L-type voltage-dependent calcium channels to modulate microglial activation and migration. | In vivo, ex vivo and in vitro (cell line NE-4C, mouse bone marrow-derived DCs and C57BL/6 mice) | [79] | |
| Diminished expression and altered cortical GAD67 distribution. Reduction in GABAergic transmission. | In vivo and in vitro (BALB/c and C57BL/6 mice) | [77,80] | |
| Dopamine | Reduction in DRD1, DRD2, DRD4, and GRK6 gene expression, reducing receptor availability and increasing dopamine concentration. | In vitro (BALB/c mice) | [50] |
| Decreased expression of Dopamine Transporter (DAT) and Vesicular Monoamine Transporter 2. Increasing locomotor activity to dopamine psychostimulants. | In vivo and in vitro (BALB/c mice) | [81] | |
| Increased dopamine synthesis and release. Increased DOPA decarboxylase (DDC) levels. | In vivo and in vitro (rat pheochromocytoma PC12 cells and Swiss Webster mouse) | [82,83] | |
| Decrease in D1-like receptors (DRD1, DRD5), MAO-A, and DARPP-32 gene expression, via MiR-132 RNA transcription. | In vitro (human neuroepithelioma cell line and CD-1 mice) | [84] | |
| Disruptions of dopamine-related pathways with DARPP-32 feedback and APP production. | In vitro (human WERI-Rb-1 eye cell line culture). Genome-wide analysis | [66] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Guerrero, G.; Gonzalez-Reyes, R.E.; de-la-Torre, A.; Medina-Rincón, G.; Nava-Mesa, M.O. Pathophysiological Mechanisms of Cognitive Impairment and Neurodegeneration by Toxoplasma gondii Infection. Brain Sci. 2020, 10, 369. https://doi.org/10.3390/brainsci10060369
Ortiz-Guerrero G, Gonzalez-Reyes RE, de-la-Torre A, Medina-Rincón G, Nava-Mesa MO. Pathophysiological Mechanisms of Cognitive Impairment and Neurodegeneration by Toxoplasma gondii Infection. Brain Sciences. 2020; 10(6):369. https://doi.org/10.3390/brainsci10060369
Chicago/Turabian StyleOrtiz-Guerrero, Gloria, Rodrigo E. Gonzalez-Reyes, Alejandra de-la-Torre, German Medina-Rincón, and Mauricio O. Nava-Mesa. 2020. "Pathophysiological Mechanisms of Cognitive Impairment and Neurodegeneration by Toxoplasma gondii Infection" Brain Sciences 10, no. 6: 369. https://doi.org/10.3390/brainsci10060369
APA StyleOrtiz-Guerrero, G., Gonzalez-Reyes, R. E., de-la-Torre, A., Medina-Rincón, G., & Nava-Mesa, M. O. (2020). Pathophysiological Mechanisms of Cognitive Impairment and Neurodegeneration by Toxoplasma gondii Infection. Brain Sciences, 10(6), 369. https://doi.org/10.3390/brainsci10060369