

Neurologic Deficit Score at 4–5 Days Post-eCPR Predicts Long-Term Brain Dysfunction in Rats Following Cardiac Arrest

 , , ,
, , ,  ,
,  , and
, and 
Abstract
:
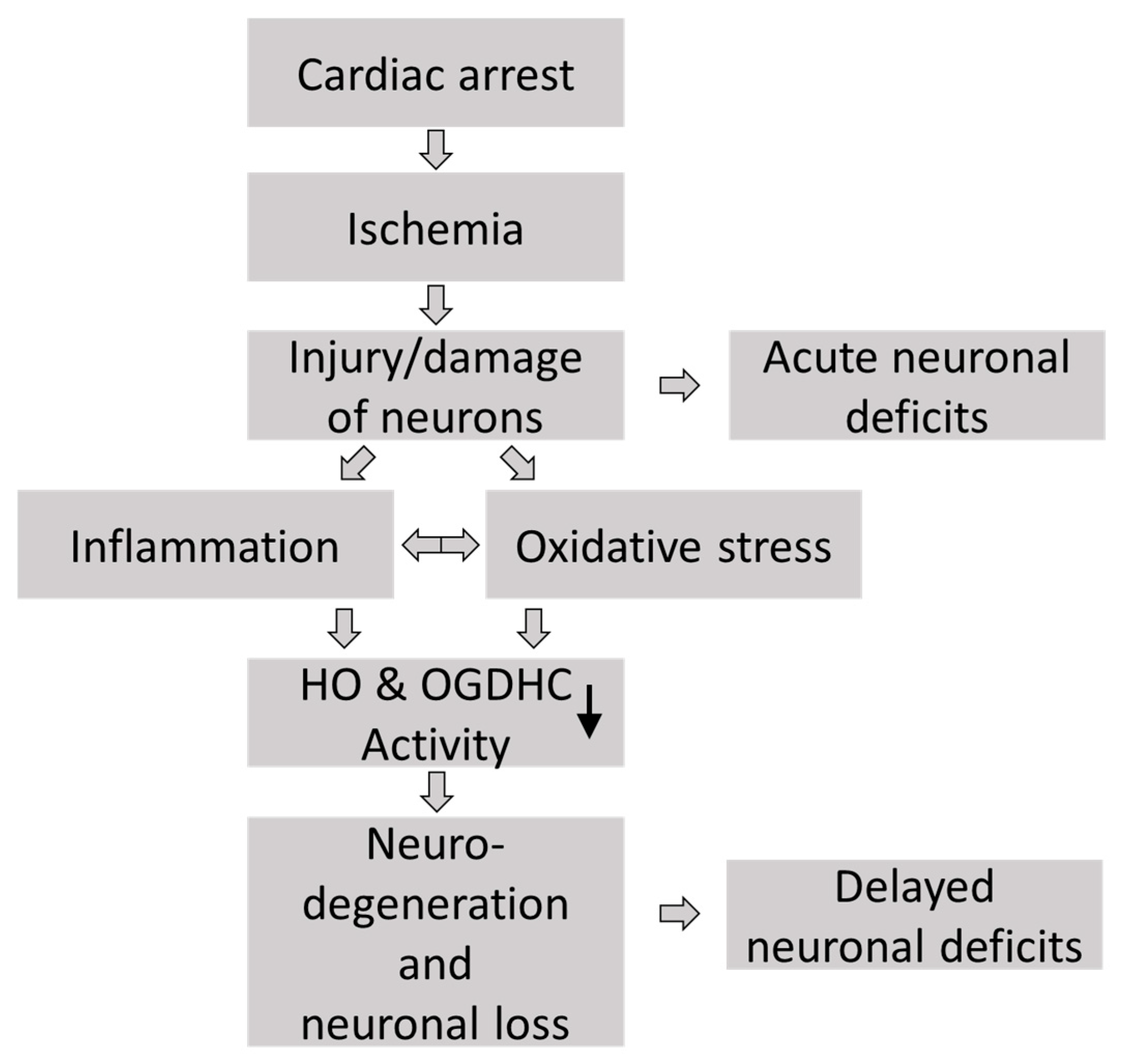
1. Introduction
2. Materials and Methods
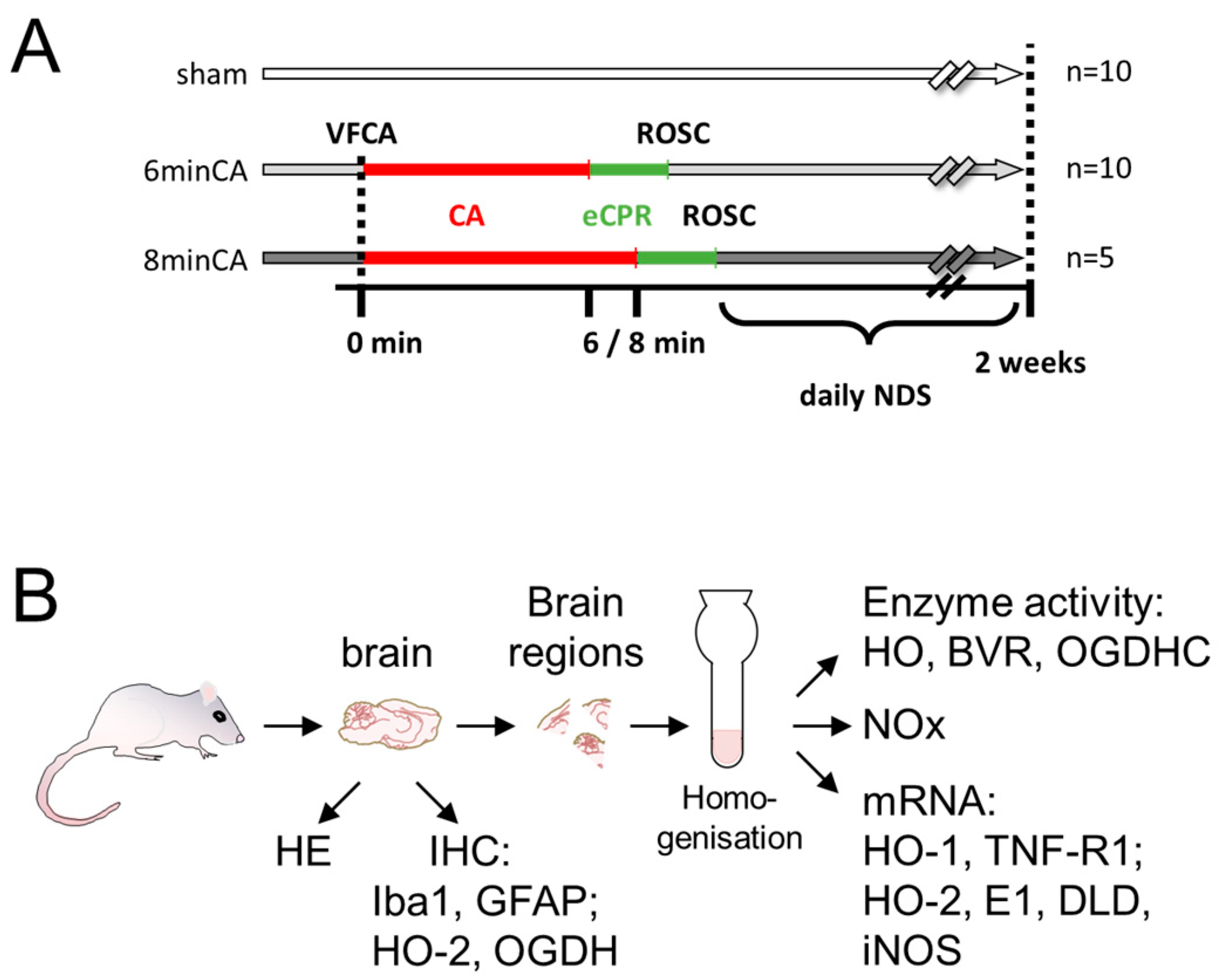
2.1. Animal Model of VFCA and eCPR
2.2. Neurologic Deficit Scoring
2.3. Sampling
2.4. Histological Analysis of Brain Regions
2.5. Preparation of Tissue Homogenates
2.6. Determination of Heme Oxygenase Activity in Brain Regions
2.7. Determination of Oxoglutarate Dehydrogenase Complex Activity in Brain Regions
2.8. Determination of Nitric Oxides (NOx)
2.9. Analysis of Gene Expression
2.10. Data Analysis and Statistics
3. Results
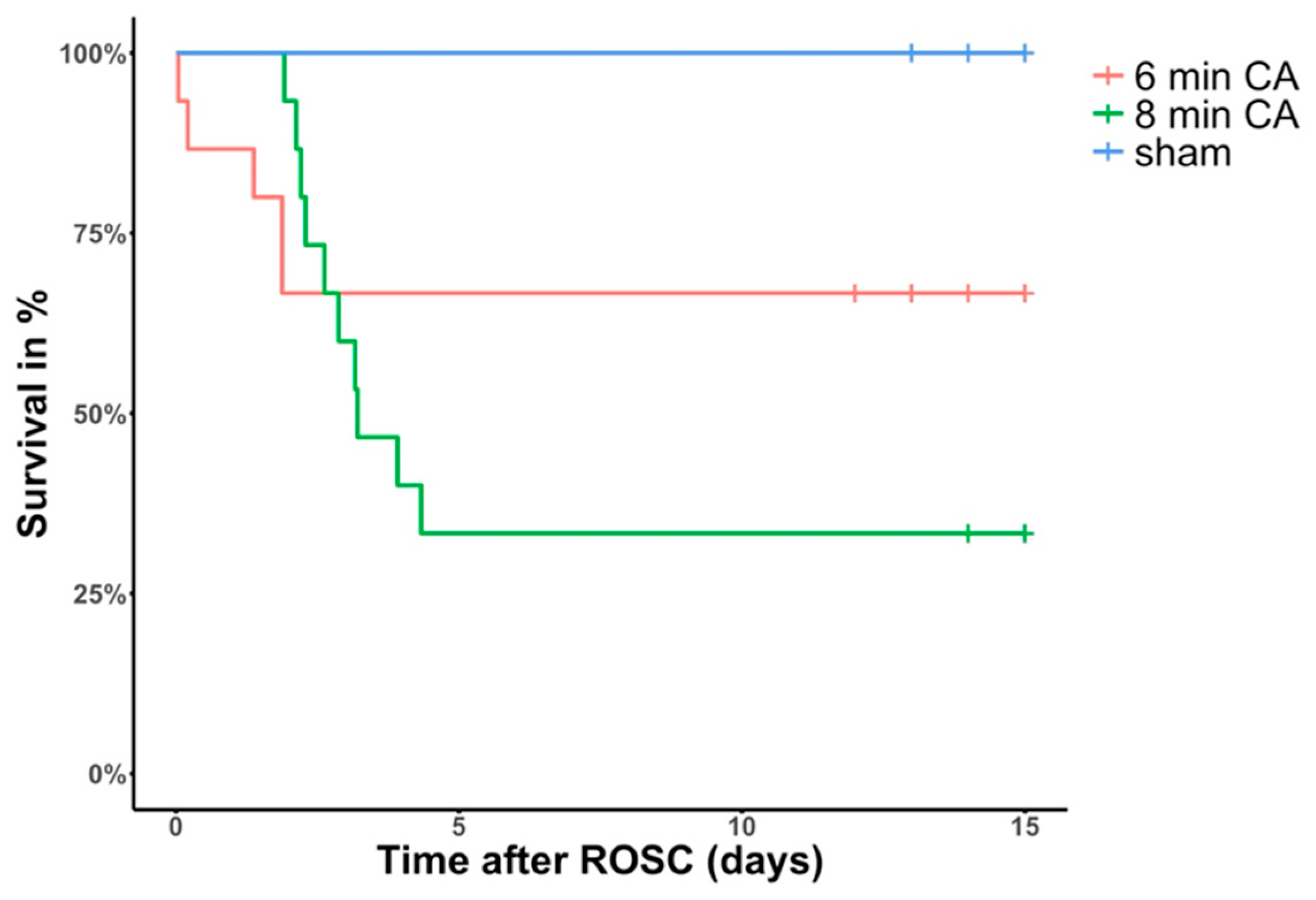
3.1. Animal Survival and Outcome
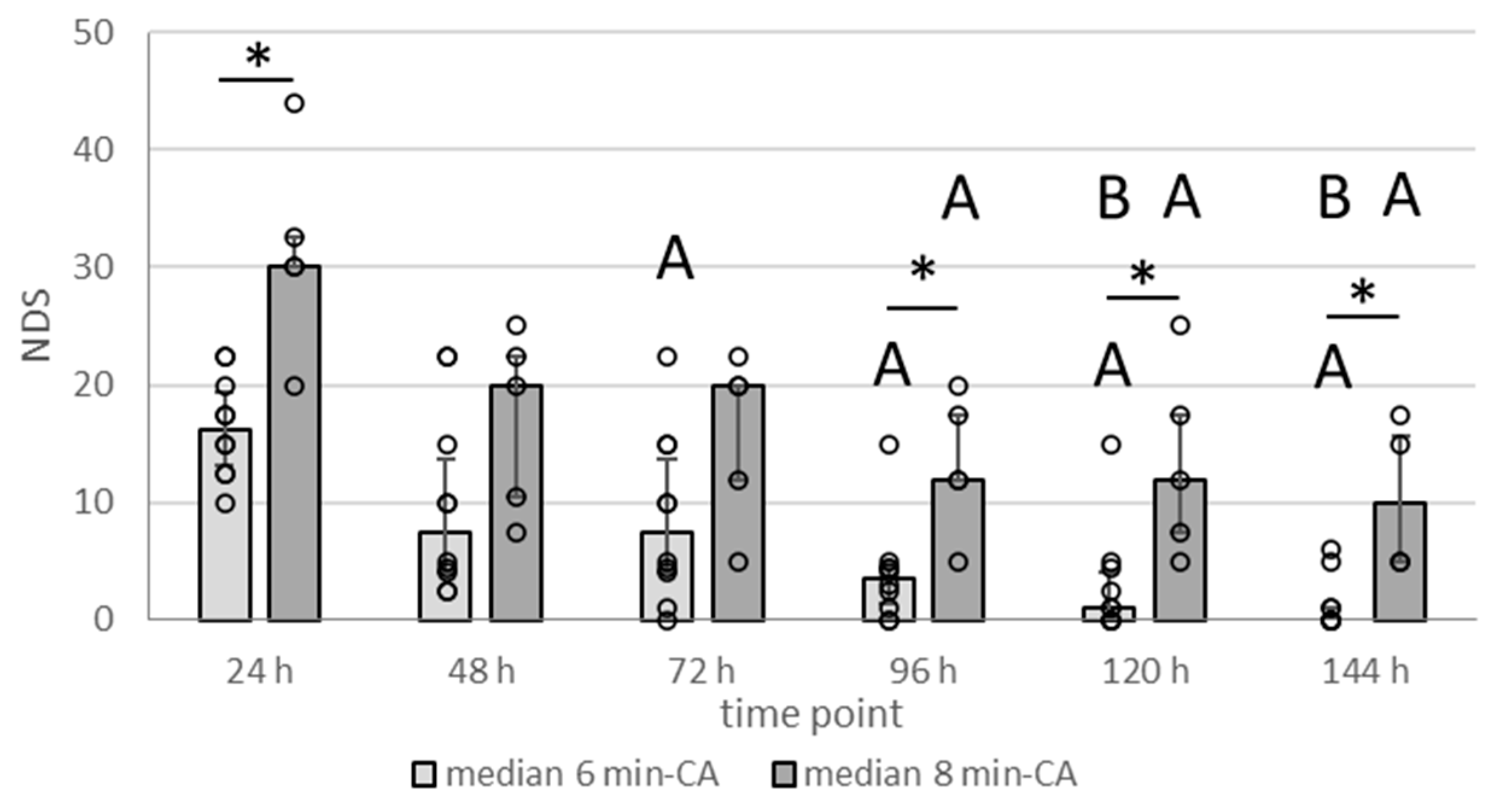
3.2. Neurologic Deficit Scores Within the First Week After CA
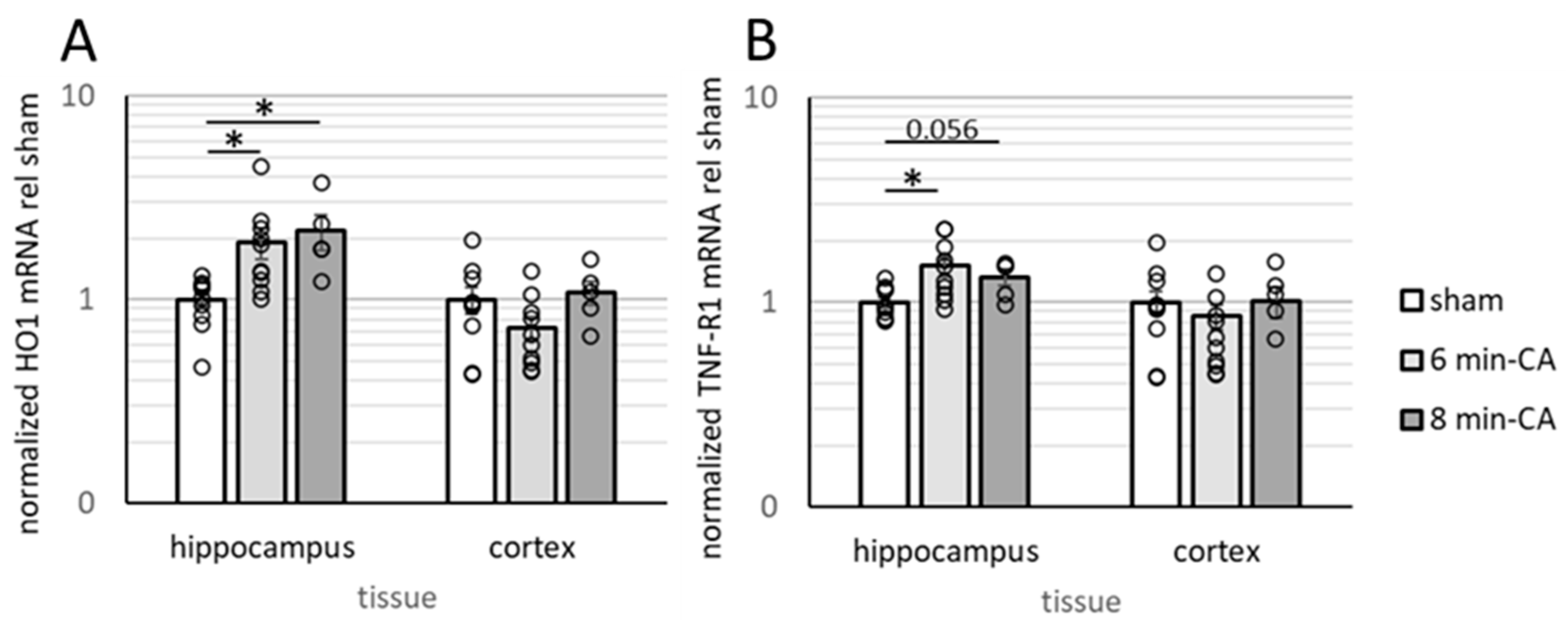
3.3. Determination of Neuroinflammation and Neurodegeneration in the Hippocampus, but Not in Cortex Tissue, 14 Days After CA
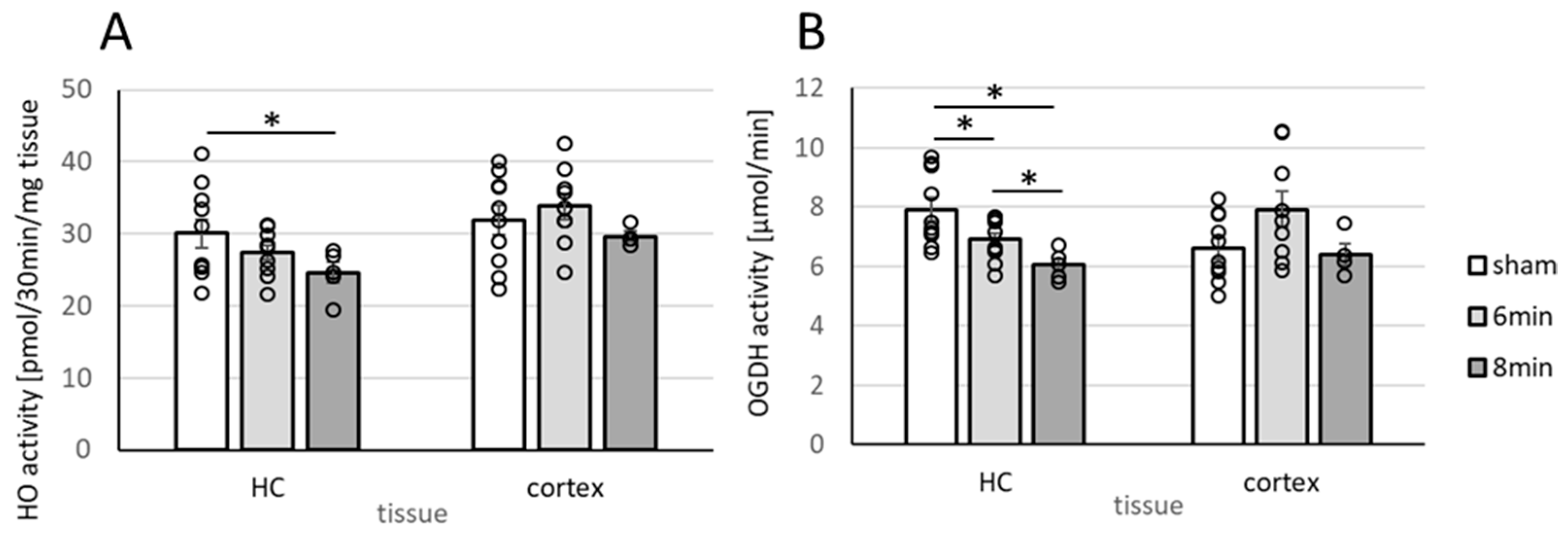
3.4. Determination of Enzyme Activities Relevant to Neuronal Function (HO and OGDHC) in Homogenates of the Hippocampus and Cortex 14 Days After ROSC
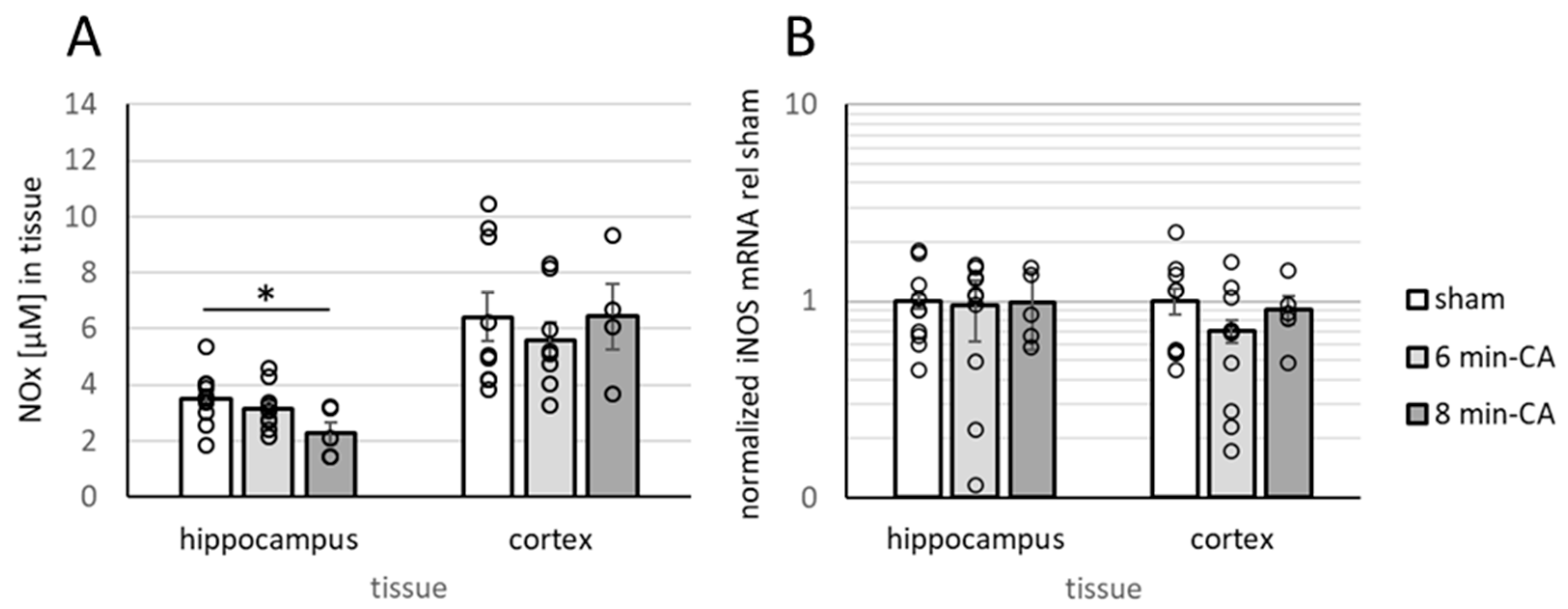
3.5. Determination of Oxidation Products of NO and Expression of iNOS
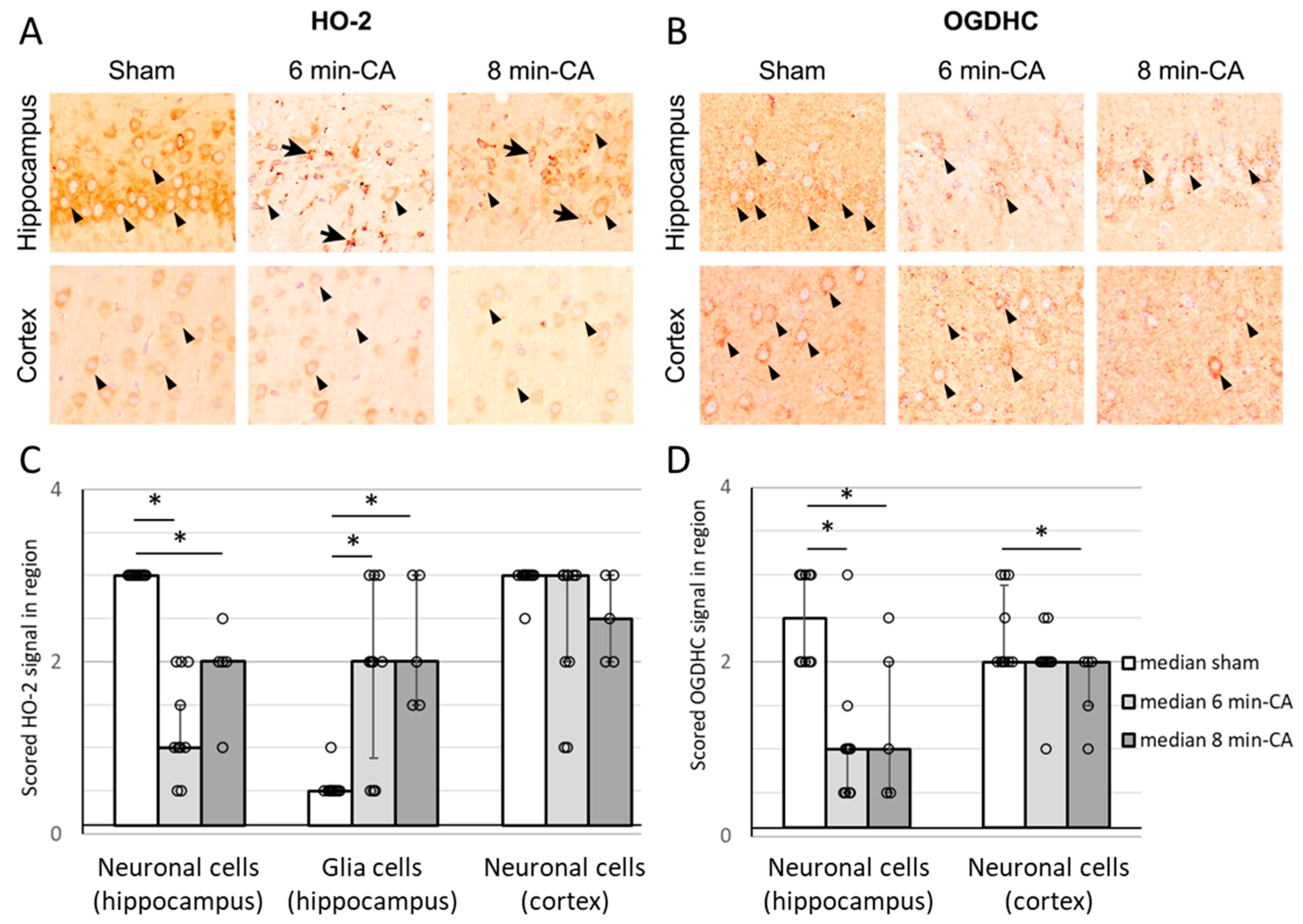
3.6. Semi-Quantitative Determination of HO-2- and OGDH-Positive Cells in Hippocampus and Cortex Tissue by Immunohistochemistry 14 Days After ROSC
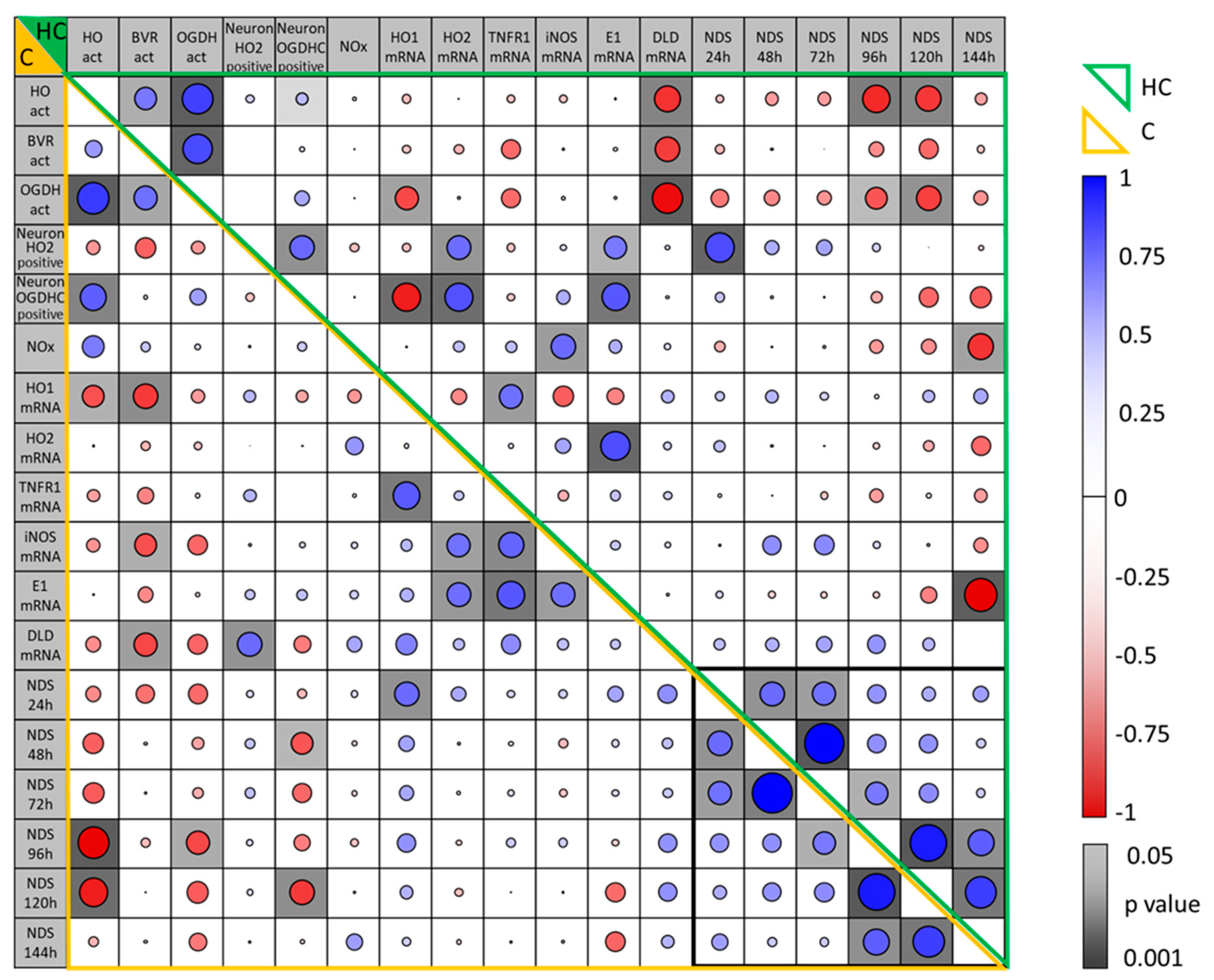
3.7. Correlation Between Parameters Determined in the Brain Regions, Hippocampus and Motor Cortex, of Rats Two Weeks After CA and Their Association with Early NDS
3.8. Association of Early Neurologic Deficit Scoring with the Manifestation of Delayed Changes in Enzyme Activities in Hippocampus and Cortex Tissue 14 Days After CA
4. Discussion
4.1. Animal Model
4.2. Increased Neuroinflammation and Gliosis in the Hippocampus Two Weeks After VFCA and eCPR
4.3. Neuronal Outcome Biomarkers Indicate Dysfunction and Degeneration of Neurons in the Hippocampus and Cortex Two Weeks After VFCA and eCPR
4.4. Scoring Neurological Deficits at 4 to 5 Days After CA Predicts Long-Term Neurological Outcome Following VFCA and Resuscitation by eCPR
4.5. Clinical Implications
4.6. Limitations of the Study
4.7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CA | Cardiac arrest |
| CA-1 | Cornu ammonis-1 |
| CPR | Cardiopulmonary resuscitation |
| ECMO | Extracorporeal membrane oxygenation |
| eCPR | Extracorporeal cardiopulmonary resuscitation |
| Hc | Hippocampus |
| HE | Hematoxylin and eosin |
| HO | Heme oxygenase |
| IHC | Immunohistochemistry |
| iNOS | Inducible NO synthase |
| mC | Motor cortex |
| NOx | Nitric oxide species (NO, NO2− + NO3− + SNO−) |
| OGDH | Oxoglutarate dehydrogenase |
| OGDHC | Oxoglutarate dehydrogenase complex |
| qPCR | Quantitative real-time PCR |
| ROSC | Return of spontaneous circulation |
| RONS | Reactive oxygen and nitrogen species |
| ROS | Reactive oxygen species |
| VFCA | Ventricular fibrillation cardiac arrest |
| 6 min CA | 6 min of cardiac arrest |
| 8 min CA | 8 min of cardiac arrest |
References
- Nolan, J.P.; Sandroni, C.; Böttiger, B.W.; Cariou, A.; Cronberg, T.; Friberg, H.; Genbrugge, C.; Haywood, K.; Lilja, G.; Moulaert, V.R.M.; et al. European Resuscitation Council and European Society of Intensive Care Medicine Guidelines 2021: Post-resuscitation care. Resuscitation 2021, 161, 220–269. [Google Scholar] [CrossRef]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity from the Perspective of Glial Cells. Front. Cell. Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed]
- Anacker, C.; Hen, R. Adult hippocampal neurogenesis and cognitive flexibility—Linking memory and mood. Nat. Rev. Neurosci. 2017, 18, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Shimoda, T.; Uno, K.; Tateishi, N.; Furuya, S.; Tsuchihashi, Y.; Kawai, Y.; Naruse, S.; Fujita, S. Temporal and sequential changes of glial cells and cytokine expression during neuronal degeneration after transient global ischemia in rats. J. Neuroinflamm. 2011, 8, 70. [Google Scholar] [CrossRef]
- Smith, M.L.; Auer, R.N.; Siesjö, B.K. The density and distribution of ischemic brain injury in the rat following 2-10 min of forebrain ischemia. Acta Neuropathol. 1984, 64, 319–332. [Google Scholar] [CrossRef]
- Nakatomi, H.; Kuriu, T.; Okabe, S.; Yamamoto, S.; Hatano, O.; Kawahara, N.; Tamura, A.; Kirino, T.; Nakafuku, M. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 2002, 110, 429–441. [Google Scholar] [CrossRef]
- Janata, A.; Drabek, T.; Magnet, I.A.; Stezoski, J.P.; Janesko-Feldman, K.; Popp, E.; Garman, R.H.; Tisherman, S.A.; Kochanek, P.M. Extracorporeal versus conventional cardiopulmonary resuscitation after ventricular fibrillation cardiac arrest in rats: A feasibility trial. Crit. Care Med. 2013, 41, e211–e222. [Google Scholar] [CrossRef]
- Warenits, A.M.; Hatami, J.; Müllebner, A.; Ettl, F.; Teubenbacher, U.; Magnet, I.A.M.; Bauder, B.; Janata, A.; Miller, I.; Moldzio, R.; et al. Motor Cortex and Hippocampus Display Decreased Heme Oxygenase Activity 2 Weeks After Ventricular Fibrillation Cardiac Arrest in Rats. Front. Med. 2020, 7, 513. [Google Scholar] [CrossRef]
- Guo, Y.; Cho, S.M.; Wei, Z.; Wang, Q.; Modi, H.R.; Gharibani, P.; Lu, H.; Thakor, N.V.; Geocadin, R.G. Early Thalamocortical Reperfusion Leads to Neurologic Recovery in a Rodent Cardiac Arrest Model. Neurocrit. Care 2022, 37, 60–72. [Google Scholar] [CrossRef]
- Soar, J.; Böttiger, B.W.; Carli, P.; Couper, K.; Deakin, C.D.; Djärv, T.; Lott, C.; Olasveengen, T.; Paal, P.; Pellis, T.; et al. European Resuscitation Council Guidelines 2021: Adult advanced life support. Resuscitation 2021, 161, 115–151. [Google Scholar] [CrossRef]
- Bartos, J.A.; Carlson, K.; Carlson, C.; Raveendran, G.; John, R.; Aufderheide, T.P.; Yannopoulos, D. Surviving refractory out-of-hospital ventricular fibrillation cardiac arrest: Critical care and extracorporeal membrane oxygenation management. Resuscitation 2018, 132, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Magnet, I.A.M.; Ettl, F.; Schober, A.; Warenits, A.M.; Grassmann, D.; Wagner, M.; Schriefl, C.; Clodi, C.; Teubenbacher, U.; Hogler, S.; et al. Extracorporeal Life Support Increases Survival After Prolonged Ventricular Fibrillation Cardiac Arrest in the Rat. Shock 2017, 48, 674–680. [Google Scholar] [CrossRef]
- Magnet, I.; Stommel, A.M.; Schriefl, C.; Mueller, M.; Poppe, M.; Grafeneder, J.; Testori, C.; Janata, A.; Schober, A.; Grassmann, D.; et al. Neuroprotection with hypothermic reperfusion and extracorporeal cardiopulmonary resuscitation—A randomized controlled animal trial of prolonged ventricular fibrillation cardiac arrest in rats. J. Cereb. Blood Flow Metab. 2024, 45, 476–485. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 2003, 34, 325–337. [Google Scholar] [CrossRef]
- Verma, A.; Hirsch, D.J.; Glatt, C.E.; Ronnett, G.V.; Snyder, S.H. Carbon monoxide: A putative neural messenger. Science 1993, 259, 381–384. [Google Scholar] [CrossRef]
- Kirkby, K.A.; Adin, C.A. Products of heme oxygenase and their potential therapeutic applications. Am. J. Physiol. Renal Physiol. 2006, 290, F563–F571. [Google Scholar] [CrossRef]
- Li, J.J.; Zou, Z.Y.; Liu, J.; Xiong, L.L.; Jiang, H.Y.; Wang, T.H.; Shao, J.L. Biliverdin administration ameliorates cerebral ischemia reperfusion injury in rats and is associated with proinflammatory factor downregulation. Exp. Ther. Med. 2017, 14, 671–679. [Google Scholar] [CrossRef]
- Wollborn, J.; Steiger, C.; Doostkam, S.; Schallner, N.; Schroeter, N.; Kari, F.A.; Meinel, L.; Buerkle, H.; Schick, M.A.; Goebel, U. Carbon Monoxide Exerts Functional Neuroprotection After Cardiac Arrest Using Extracorporeal Resuscitation in Pigs. Crit. Care Med. 2020, 48, e299–e307. [Google Scholar] [CrossRef]
- Mancuso, C.; Pistritto, G.; Tringali, G.; Grossman, A.B.; Preziosi, P.; Navarra, P. Evidence that carbon monoxide stimulates prostaglandin endoperoxide synthase activity in rat hypothalamic explants and in primary cultures of rat hypothalamic astrocytes. Mol. Brain Res. 1997, 45, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.S.; Zhuang, H.; Doré, S. Heme oxygenase-1 protects brain from acute excitotoxicity. Neuroscience 2006, 141, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P.; Kark, R.A.; Menon, N.K. Low activities of the pyruvate and oxoglutarate dehydrogenase complexes in five patients with Friedreich’s ataxia. N. Engl. J. Med. 1976, 295, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [CrossRef]
- Humphries, K.M.; Szweda, L.I. Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: Reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry 1998, 37, 15835–15841. [Google Scholar] [CrossRef]
- Trofimova, L.K.; Araujo, W.L.; Strokina, A.A.; Fernie, A.R.; Bettendorff, L.; Bunik, V.I. Consequences of the alpha-ketoglutarate dehydrogenase inhibition for neuronal metabolism and survival: Implications for neurodegenerative diseases. Curr. Med. Chem. 2012, 19, 5895–5906. [Google Scholar] [CrossRef]
- Ferris, S.H.; de Leon, M.J.; Wolf, A.P.; Farkas, T.; Christman, D.R.; Reisberg, B.; Fowler, J.S.; Macgregor, R.; Goldman, A.; George, A.E.; et al. Positron emission tomography in the study of aging and senile dementia. Neurobiol. Aging 1980, 1, 127–131. [Google Scholar] [CrossRef]
- Weidinger, A.; Milivojev, N.; Hosmann, A.; Duvigneau, J.C.; Szabo, C.; Toro, G.; Rauter, L.; Vaglio-Garro, A.; Mkrtchyan, G.V.; Trofimova, L.; et al. Oxoglutarate dehydrogenase complex controls glutamate-mediated neuronal death. Redox Biol. 2023, 62, 102669. [Google Scholar] [CrossRef]
- Mkrtchyan, G.V.; Üçal, M.; Müllebner, A.; Dumitrescu, S.; Kames, M.; Moldzio, R.; Molcanyi, M.; Schaefer, S.; Weidinger, A.; Schaefer, U.; et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 925–931. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Stommel, A.M.; Högler, S.; Mueller, M.; Magnet, I.A.M.; Kodajova, P.; Ullram, B.; Szinovatz, A.; Panzer, F.P.; Engenhart-Seyrl, A.; Kaschmekat, J.; et al. A ventricular fibrillation cardiac arrest model with extracorporeal cardiopulmonary resuscitation in rats: 8 minutes arrest time leads to increased myocardial damage but does not increase neuronal damage compared to 6 minutes. Front. Vet. Sci. 2023, 10, 1276588. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.; Ebmeyer, U.; Safar, P.; Radovsky, A.; Neumar, R. Outcome model of asphyxial cardiac arrest in rats. J. Cereb. Blood Flow Metab. 1995, 15, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 7th ed.; Academic Press: San Diego, CA, USA, 2013. [Google Scholar]
- Kozlov, A.V.; Duvigneau, J.C.; Miller, I.; Nurnberger, S.; Gesslbauer, B.; Kungl, A.; Ohlinger, W.; Hartl, R.T.; Gille, L.; Staniek, K.; et al. Endotoxin causes functional endoplasmic reticulum failure, possibly mediated by mitochondria. Biochim. Biophys. Acta 2009, 1792, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Soler, L.; Gutierrez, A.; Mullebner, A.; Ceron, J.J.; Duvigneau, J.C. Towards a better understanding of salivary and meat juice acute phase proteins determination in pigs: An expression study. Vet. Immunol. Immunopathol. 2013, 156, 91–98. [Google Scholar] [CrossRef]
- Beschorner, R.; Adjodah, D.; Schwab, J.M.; Mittelbronn, M.; Pedal, I.; Mattern, R.; Schluesener, H.J.; Meyermann, R. Long-term expression of heme oxygenase-1 (HO-1, HSP-32) following focal cerebral infarctions and traumatic brain injury in humans. Acta Neuropathol. 2000, 100, 377–384. [Google Scholar] [CrossRef]
- Schober, A.; Warenits, A.M.; Testori, C.; Weihs, W.; Hosmann, A.; Högler, S.; Sterz, F.; Janata, A.; Scherer, T.; Magnet, I.A.; et al. Microdialysis Assessment of Cerebral Perfusion during Cardiac Arrest, Extracorporeal Life Support and Cardiopulmonary Resuscitation in Rats—A Pilot Trial. PLoS ONE 2016, 11, e0155303. [Google Scholar] [CrossRef]
- Kinobe, R.; Ji, Y.; Nakatsu, K. Peroxynitrite-mediated inactivation of heme oxygenases. BMC Pharmacol. 2004, 4, 26. [Google Scholar] [CrossRef]
- Park, L.C.; Zhang, H.; Sheu, K.F.; Calingasan, N.Y.; Kristal, B.S.; Lindsay, J.G.; Gibson, G.E. Metabolic impairment induces oxidative stress, compromises inflammatory responses, and inactivates a key mitochondrial enzyme in microglia. J. Neurochem. 1999, 72, 1948–1958. [Google Scholar] [CrossRef]
- Nulton-Persson, A.C.; Starke, D.W.; Mieyal, J.J.; Szweda, L.I. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry 2003, 42, 4235–4242. [Google Scholar] [CrossRef]
- Reynolds, J.C.; Grunau, B.E.; Rittenberger, J.C.; Sawyer, K.N.; Kurz, M.C.; Callaway, C.W. Association Between Duration of Resuscitation and Favorable Outcome After Out-of-Hospital Cardiac Arrest: Implications for Prolonging or Terminating Resuscitation. Circulation 2016, 134, 2084–2094. [Google Scholar] [CrossRef]
- Reddy, S.; Garcia, S.; Hostetter, L.J.; Finch, A.S.; Bellolio, F.; Guru, P.; Gerberi, D.J.; Smischney, N.J. Extracorporeal-CPR Versus Conventional-CPR for Adult Patients in Out of Hospital Cardiac Arrest- Systematic Review and Meta-Analysis. J. Intensive Care Med. 2025, 40, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Magnet, I.; Behringer, W.; Eibensteiner, F.; Ettl, F.; Grafeneder, J.; Heinz, G.; Holzer, M.; Krammel, M.; Lobmeyr, E.; Losert, H.; et al. Extracorporeal Cardiopulmonary Resuscitation: Outcomes Improve with Center Experience. Ann. Emerg. Med. 2025, 85, 421–427. [Google Scholar] [CrossRef]
- Ijuin, S.; Liu, K.; Gill, D.; Kyun Ro, S.; Vukovic, J.; Ishihara, S.; Belohlavek, J.; Li Bassi, G.; Suen, J.Y.; Fraser, J.F. Current animal models of extracorporeal cardiopulmonary resuscitation: A scoping review. Resusc. Plus 2023, 15, 100426. [Google Scholar] [CrossRef] [PubMed]
- Chalkias, A.; Xanthos, T. Post-cardiac arrest brain injury: Pathophysiology and treatment. J. Neurol. Sci. 2012, 315, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sandroni, C.; Cronberg, T.; Sekhon, M. Brain injury after cardiac arrest: Pathophysiology, treatment, and prognosis. Intensive Care Med. 2021, 47, 1393–1414. [Google Scholar] [CrossRef]
- Parfenova, H.; Leffler, C.W. Cerebroprotective functions of HO-2. Curr. Pharm. Des. 2008, 14, 443–453. [Google Scholar] [CrossRef]
- Lee, T.K.; Kim, H.; Song, M.; Lee, J.C.; Park, J.H.; Ahn, J.H.; Yang, G.E.; Kim, H.; Ohk, T.G.; Shin, M.C.; et al. Time-course pattern of neuronal loss and gliosis in gerbil hippocampi following mild, severe, or lethal transient global cerebral ischemia. Neural Regen. Res. 2019, 14, 1394–1403. [Google Scholar] [CrossRef]
- Trakshel, G.M.; Kutty, R.K.; Maines, M.D. Resolution of the rat brain heme oxygenase activity: Absence of a detectable amount of the inducible form (HO-1). Arch. Biochem. Biophys. 1988, 260, 732–739. [Google Scholar] [CrossRef]
- Dore, S.; Sampei, K.; Goto, S.; Alkayed, N.J.; Guastella, D.; Blackshaw, S.; Gallagher, M.; Traystman, R.J.; Hurn, P.D.; Koehler, R.C.; et al. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol. Med. 1999, 5, 656–663. [Google Scholar] [CrossRef]
- Dore, S.; Goto, S.; Sampei, K.; Blackshaw, S.; Hester, L.D.; Ingi, T.; Sawa, A.; Traystman, R.J.; Koehler, R.C.; Snyder, S.H. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 2000, 99, 587–592. [Google Scholar] [CrossRef]
- Chang, E.F.; Wong, R.J.; Vreman, H.J.; Igarashi, T.; Galo, E.; Sharp, F.R.; Stevenson, D.K.; Noble-Haeusslein, L.J. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J. Neurosci. 2003, 23, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Ewing, J.F.; Maines, M.D. Histochemical localization of heme oxygenase-2 protein and mRNA expression in rat brain. Brain Res. Protoc. 1997, 1, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid. Redox Signal. 2006, 8, 929–947. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Sultana, R.; Coccia, R.; Mancuso, C.; Perluigi, M.; Butterfield, D.A. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2012, 52, 2292–2301. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Coccia, R.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J. Alzheimer’s Dis. 2011, 25, 623–633. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase—A protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta 2011, 1812, 480–487. [Google Scholar] [CrossRef]
- Iadecola, C.; Zhang, F.; Xu, S.; Casey, R.; Ross, M.E. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 378–384. [Google Scholar] [CrossRef]
- Fukuyama, N.; Takizawa, S.; Ishida, H.; Hoshiai, K.; Shinohara, Y.; Nakazawa, H. Peroxynitrite formation in focal cerebral ischemia-reperfusion in rats occurs predominantly in the peri-infarct region. J. Cereb. Blood Flow Metab. 1998, 18, 123–129. [Google Scholar] [CrossRef]
- Hosmann, A.; Milivojev, N.; Dumitrescu, S.; Reinprecht, A.; Weidinger, A.; Kozlov, A.V. Cerebral nitric oxide and mitochondrial function in patients suffering aneurysmal subarachnoid hemorrhage-a translational approach. Acta Neurochir. 2021, 163, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Jaffrey, S.R.; Snyder, S.H. Nitric oxide: A neural messenger. Annu. Rev. Cell Dev. Biol. 1995, 11, 417–440. [Google Scholar] [CrossRef]
- Gibson, G.E.; Chen, H.L.; Xu, H.; Qiu, L.; Xu, Z.; Denton, T.T.; Shi, Q. Deficits in the mitochondrial enzyme alpha-ketoglutarate dehydrogenase lead to Alzheimer’s disease-like calcium dysregulation. Neurobiol. Aging 2012, 33, 1121.e13–1121.e24. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.; Walsh, J.M.; Dennis, S.C.; Clark, J.B. Synaptic and non-synaptic mitochondria from rat brain: Isolation and characterization. J. Neurochem. 1977, 28, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Vammen, L.; Munch Johannsen, C.; Magnussen, A.; Povlsen, A.; Riis Petersen, S.; Azizi, A.; Lofgren, B.; Andersen, L.W.; Granfeldt, A. Cardiac Arrest in Pigs with 48 hours of Post-Resuscitation Care Induced by 2 Methods of Myocardial Infarction: A Methodological Description. J. Am. Heart Assoc. 2021, 10, e022679. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wang, X.; Yu, Z. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem. Pharmacol. 2016, 5, 213. [Google Scholar] [CrossRef]
- Roof, R.L.; Schielke, G.P.; Ren, X.; Hall, E.D. A comparison of long-term functional outcome after 2 middle cerebral artery occlusion models in rats. Stroke 2001, 32, 2648–2657. [Google Scholar] [CrossRef]
- Gräsner, J.T.; Wnent, J.; Herlitz, J.; Perkins, G.D.; Lefering, R.; Tjelmeland, I.; Koster, R.W.; Masterson, S.; Rossell-Ortiz, F.; Maurer, H.; et al. Survival after out-of-hospital cardiac arrest in Europe—Results of the EuReCa TWO study. Resuscitation 2020, 148, 218–226. [Google Scholar] [CrossRef]
- Postl, A.; Zifko, C.; Hartl, R.T.; Ebel, T.; Miller, I.; Moldzio, R.; Redl, H.; Kozlov, A.V.; Bahrami, S.; Duvigneau, J.C. Transient increase of free iron in rat livers following hemorrhagic-traumatic shock and reperfusion is independent of heme oxygenase 1 upregulation. Shock 2011, 36, 501–509. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Score 1 | Characteristics | ||||
|---|---|---|---|---|---|---|
| Maximal points (worst condition) | Percentage (max 100%) | Description | ||||
| General deficit | Consciousness | 20 | 20 | 20 (comatose) | 10 (somnolent) | 0 (normal) |
| Respiration | 20 | 20 | 20 (abnormal breathing) | 0 (normal breathing) | ||
| Cranial nerve reflexes deficit | Olfactory (sniffing food) | 4 | 4 | 4 (no reaction) | 0 (normal) | |
| Vision (follows hand) | 4 | 4 | 4 (no reaction) | 0 (normal) | ||
| Corneal reflex | 4 | 4 | 4 (absent) | 0 (present) | ||
| Whisker Movement | 4 | 4 | 4 (no whisker movement) | 0 (normal) | ||
| Hearing (turns to clapped hands) | 4 | 4 | 4 (no reaction) | 0 (normal) | ||
| Motor deficit | Motor | 10 | 10 | 2.5 points for motoric problems for each affected extremity | 0 (normal) | |
| Sensory deficit | Sensory | 10 | 10 | 2.5 points for sensory loss for each affected extremity | 0 (normal) | |
| Coordination deficit | Travel ledge | 5 | 5 | 5 (no ability to walk on a beam) | 2.5 (ability to walk on a beam with some help or for shorter periods) | 0 (physiological behaviour) |
| Placing test (front paws reaching when lifted from ground by tail) | 5 | 5 | 5 (absent) | 0 (present) | ||
| Righting reflex (attempting to right self when placed on back) | 5 | 5 | 5 (absent) | 0 (present) | ||
| Stop at table edge | 5 | 5 | 5 (absent) | 2.5 (animal recognises table edge, but is too weak to prevent falling down) | 0 (present) | |
| Parameter 1 | Parameter 2 | Correlation Coefficient |
|---|---|---|
| HO activity in Hc | HO activity in mC | 0.895 ** |
| HO activity in Hc | OGDHC activity in Hc | 0.744 ** |
| HO activity in Hc | OGDHC activity in mC | 0.819 ** |
| HO activity in mC | OGDHC activity in Hc | 0.699 ** |
| HO activity in mC | OGDHC activity in mC | 0.762 ** |
| OGDHC activity in Hc | OGDHC activity in mC | 0.512 |
| Functional Outcome After 2 Weeks | NDS at 24 h | NDS at 96 h | NDS at 120 h |
|---|---|---|---|
| Cortex HO-1 mRNA | 0.59 * | 0.45 | 0.31 |
| Cortex HO activity [pmolBR/30 min/mg tissue] | −0.36 | −0.75 ** | −0.68 ** |
| Cortex OGDHC activity [µmol/min/mg tissue] | −0.46 | −0.56 * | −0.51 |
| Hippocampal HO activity [pmol/30 min/mg tissue] | −0.2 | −0.65 ** | −0.61 * |
| Hippocampal OGDHC activity [µmol/min/mg tissue] | −0.42 | −0.52 * | −0.59 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weihs, W.; Stommel, A.-M.; Müllebner, A.; Szinovatz, A.F.; Müller, M.; Magnet, I.; Holzer, M.; Kozlov, A.V.; Högler, S.; Duvigneau, J.C. Neurologic Deficit Score at 4–5 Days Post-eCPR Predicts Long-Term Brain Dysfunction in Rats Following Cardiac Arrest. Biomolecules 2025, 15, 732. https://doi.org/10.3390/biom15050732
Weihs W, Stommel A-M, Müllebner A, Szinovatz AF, Müller M, Magnet I, Holzer M, Kozlov AV, Högler S, Duvigneau JC. Neurologic Deficit Score at 4–5 Days Post-eCPR Predicts Long-Term Brain Dysfunction in Rats Following Cardiac Arrest. Biomolecules. 2025; 15(5):732. https://doi.org/10.3390/biom15050732
Chicago/Turabian StyleWeihs, Wolfgang, Alexandra-Maria Stommel, Andrea Müllebner, Alexander Franz Szinovatz, Matthias Müller, Ingrid Magnet, Michael Holzer, Andrey V. Kozlov, Sandra Högler, and J. Catharina Duvigneau. 2025. "Neurologic Deficit Score at 4–5 Days Post-eCPR Predicts Long-Term Brain Dysfunction in Rats Following Cardiac Arrest" Biomolecules 15, no. 5: 732. https://doi.org/10.3390/biom15050732
APA StyleWeihs, W., Stommel, A.-M., Müllebner, A., Szinovatz, A. F., Müller, M., Magnet, I., Holzer, M., Kozlov, A. V., Högler, S., & Duvigneau, J. C. (2025). Neurologic Deficit Score at 4–5 Days Post-eCPR Predicts Long-Term Brain Dysfunction in Rats Following Cardiac Arrest. Biomolecules, 15(5), 732. https://doi.org/10.3390/biom15050732