1. Introduction
It has been shown that the length of a thin filament in the sarcomere of cardiomyocytes significantly affects the mechanics of the left ventricular myocardium [
1]. Dysregulation of thin filament length caused by dysfunction of proteins capping the actin filament contributes to heart failure [
1,
2,
3]. In muscle sarcomere, the length of a thin filament is determined by the dynamics of attachment and detachment of actin monomers at the slow-growing (pointed, minus) end, which are regulated by tropomodulin (Tmod) and leiomodin (Lmod) [
4,
5,
6,
7,
8]. Tmod binds to the minus end of the actin filament and prevents its disassembly and attachment of new actin monomers [
9], while Lmod displaces Tmod and promotes attachment of new actin monomers and elongation of the actin filament [
5,
10]. In striated muscle sarcomere, two Tmod isoforms are present. Tmod1, encoded by the
TMOD1 gene, is expressed in both cardiac and skeletal muscles, while Tmod4 (
TMOD4 gene) is found only in skeletal muscles [
11,
12]. Also, in the actin cytoskeleton that is not part of striated muscle sarcomere, Tmod3 (
TMOD3 gene) is expressed [
12].
Tmod1 is important for maintaining the length of the thin filament and the stability of the cardiomyocyte sarcomere structure. Tmod1 knockout disrupts the development of the heart chambers, with subsequent death of the embryo [
13]. Using a culture of cardiomyocytes, it was shown that overexpression of Tmod1 leads to the formation of short thin filaments, while its absence disrupts myofibrillogenesis [
14,
15].
Experiments on Xenopus showed that Tmod4 is required in myofibrillogenesis in skeletal muscles [
16]. In
Danio rerio with a knockout of the
TMOD4 gene, a significant decrease in the number of myofibrils and impaired muscle force generation were observed [
17]. Using a mouse model with
TMOD1 gene knockout, it was shown [
18,
19] that Tmod isoforms directly affect the formation of myosin cross-bridges and skeletal muscle force. Knockout of the
TMOD1 gene resulted in the replacement of Tmod1 by Tmod3/4 and a decrease in the mobility of the Tpm strand on the actin filament, reducing the number of cross-bridges in a strongly bound state and the force developed by the fiber [
18,
19]. It should be noted that in skeletal muscle, unlike the heart,
TMOD1 knockout does not affect the length of the thin filament, which means that the effect of Tmod on the regulation of actin–myosin interactions occurs through a change in the mobility of the Tpm strand [
19].
The Tmod1 molecule (~40 kDa) can be divided into two parts [
20,
21,
22]. The N-terminal domain of the Tmod1 molecule is predominantly unstructured. According to NMR data, only amino acid (a.a.) residues 24–35 form an α-helix. The N-terminal domain contains three functional sites: two tropomyosin (Tpm)-binding sites (a.a. residues 1–38, TMBS1, and 109–144, TMBS2) and one Tpm-dependent actin-capping site (a.a. residues 48–92, ABS1) [
22,
23,
24,
25,
26,
27,
28,
29]. The C-terminal part of the molecule is packed into a leucine-rich repeat (LRR) domain-containing second actin-binding site (ABS2) [
30,
31,
32,
33]. Using cultures of cardiomyocytes, it has been shown that the LRR domain is required for Tmod1 to bind to the actin filament’s minus end. Deletion of the LRR domain and changes in its structure due to point mutations lead to a complete loss of Tmod1 at the pointed end of the actin filament in the sarcomere of cardiomyocytes [
21,
34].
Due to the presence of two Tpm-binding sites, the Tmod1 molecule binds two Tpm dimers at the pointed end of the actin filament [
27]. The N-terminal part of the Tpm molecule plays an important role in the interactions of Tmods with the minus end of the actin filament [
22]. Tropomyosin enhances the binding of Tmod1 to the actin filament by three to four orders of magnitude [
9,
25,
27,
35,
36].
The question of the influence of Tmod1 on the regulation of actin–myosin interactions in the myocardium remains open. The importance of Tmod for myocardial contraction was demonstrated by the discovery of a mutation that causes dilated and restrictive cardiomyopathy [
37]. Here, we studied the effects of Tmod on the actin–myosin interaction in the myocardium using an in vitro motility assay. For the functioning of Tmod in striated muscles, the interaction of Tmod with Tpm is important. It was previously shown that the Tmod1 and Tmod3 isoforms bind Tpm isoforms with different affinities [
18,
31,
38]. In the myocardium, Tpm1.1 (αTpm) and Tpm1.2 (κTpm) are expressed. Tpm 1.2 is the result of alternative splicing of the TPM1 gene [
39]. In Tpm1.2 mRNA, exon 2b is replaced by exon 2a, which corresponds to ~ 40 amino acid residues (from a.a. 39 to a.a. 77) in the Tpm molecule [
39]. Activation of Tpm1.2-containing thin filaments has been shown to depend on myosin [
40].
It can be assumed that the binding of Tmod with the Tpm isoforms in the myocardium specifically modulates the actin–myosin interaction. To test this assumption, we studied the effects of Tmod on the interactions of cardiac myosin with actin using an in vitro motility assay. We assessed the interactions of Tmod with cardiac Tpm isoforms using cross-linking with glutaraldehyde, a pull-down assay, and size-exclusion chromatography. It can be assumed that the regulatory effect of Tmod on actin–myosin interactions depends on Tpm isoforms. We compared the effects of Tmod1 on the interactions of myosin with thin filaments containing Tpm1.1 and Tpm1.2. In addition, we tested whether the effect of Tmod on actin–myosin interactions in the myocardium depends on Tmod isoforms. For this, we used Tmod4, which is normally expressed in skeletal muscles.
2. Materials and Methods
2.1. Experimental Design, Animal Handling, and Ethics Requirements
A public corporation in Kamensk-Uralsky provided a sheep heart. Directive 2010/63/EU of the European Parliament was followed in the treatment of the rats used in the present study. The experimental protocol was approved by The Animal Care and Use Committee of the Institute of Immunology and Physiology. Unless otherwise indicated, Merck & Co. Inc. (Rahway, NJ, USA) was the supplier of all chemicals and reagents.
The institutional vivarium was occupied by 10-week-old male Wistar rats (250–300 g) that were provided free access to food (Delta Feeds LbK 120 S-19, BioPro, Novosibirsk, Russian) and water. An intramuscular injection of 2% Xylazine (1 mL/kg body weight, Alfasan, Woerden, The Netherlands) and Zoletil-100 (0.3 mL/kg body weight, Virbac, Carros, France) was used to anesthetize the rats. Then, heparin (5000 IU/kg, Ellara, Pokrov, Russian) was added, and the rats were euthanized through exsanguination. After removing the hearts, the left ventricles were frozen in portions for myosin isolation and stored at −86 °C.
2.2. Protein Extraction and Purification
Human Tpm1.1 and Tpm1.2 were expressed in
E. coli C41(DE3) as previously described [
40]. The Tpm1.1 and Tpm1.2 had Ala-Ser N-terminal extensions to mimic the naturally occurring acetylation of native Tpm [
41]. A recombinant human cardiac Tn complex composed of TnI, TnT, and TnC was provided by HyTest (Turku, Finland) (Cat.# 8ITCR). Rat Tmod1 was obtained as described by Kostyukova and her co-authors [
42,
43]. CDSs of human Tmod1 and Tmod4 were obtained from Cloning Facility (Moscow, Russia) in an EV expression vector. All constructs had a 6X N-terminal His-tag. The Tmods were expressed in
E. coli C41 (DE3) cells. A night culture was inoculated in 1 L of LB medium in 30 mM HEPES buffer (pH 7.3) with 100 mM NaCl and grew up to an optical density of 0.6 at 37 °C. Then, expression was induced by 0.2 mM IPTG and continued for 4 h at 30 °C. The Tmods were purified on a HisTrap HP column (GE Healthcare, Chicago, IL, USA) using a linear imidazole gradient from 15 to 500 mM. The samples were analyzed using SDS-PAGE electrophoresis. The best fractions were collected, combined, and loaded on a HiLoad 16/600 Superdex 200 pg column for additional purification. Finally, the Tmods were dialyzed against 30 mM HEPES-Na and 100 mM NaCl (pH 7.3) and stored at −80 °C.
The left ventricles and atria of the sheep and rat hearts were subjected to myosin extraction [
44]. The atrial myosin from the sheep heart was composed of atrial light chain (LC) and 80% α-myosin heavy chain (MHC) with 20% β-MHC (
Figure S1 in the
Supplementary Materials). The sheep ventricular myosin entirely comprised β-MHC and ventricular light chain (VC). The rat ventricular myosin contained 90% α-MHC, 10% β-MHC, and ventricular LC. The rat atrial myosin contained 90% α-MHC, 10% β-MHC, and atrial LC. Native thin filaments (NTFs) were extracted from the sheep left ventricles [
45]. Standard procedures [
46] were utilized to prepare rabbit skeletal muscle actin, which was then polymerized and labeled with TRITC-phalloidin at a 2-fold molecular excess.
2.3. Chemical Cross-Linking Between Tmod and Tropomyosin
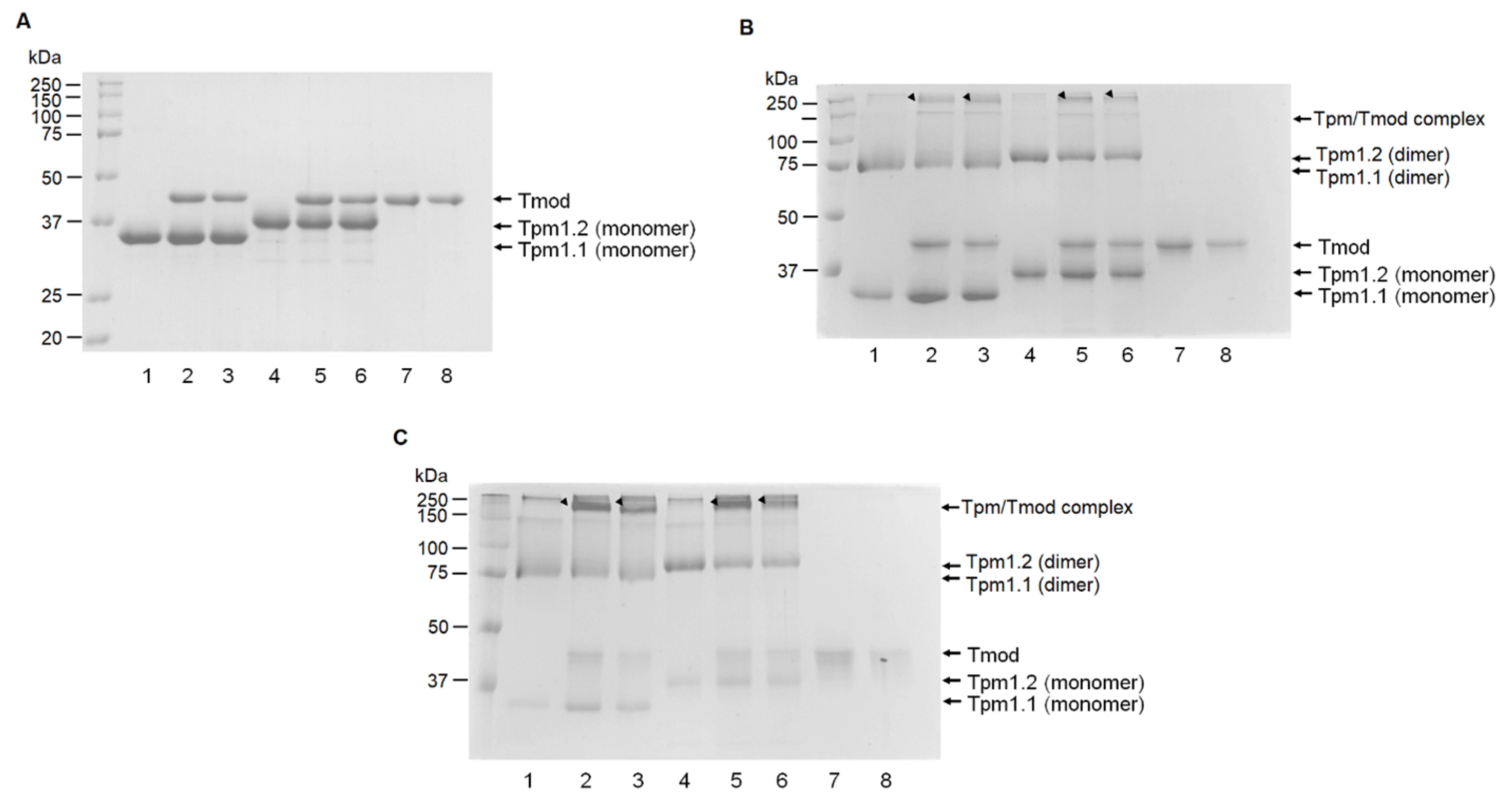
Tpm and Tmod samples were chemically cross-linked using an aqueous solution of glutaraldehyde (TED PELLA, Redding, CA, USA) in a 30 mM HEPES buffer, pH 7.3, with 200 mM NaCl. The concentrations of glutaraldehyde were 0.008% and 0.002%. The Tpm1.1 and Tpm1.2 concentrations were 0.2 mg/mL, and the Tmod1 and Tmod4 concentrations were 0.12 mg/mL. The cross-linking was performed for 15 and 60 min at 30 °C. After that, the samples were analyzed by SDS-PAGE in 12.5% gel.
2.4. Analytical Size-Exclusion Chromatography
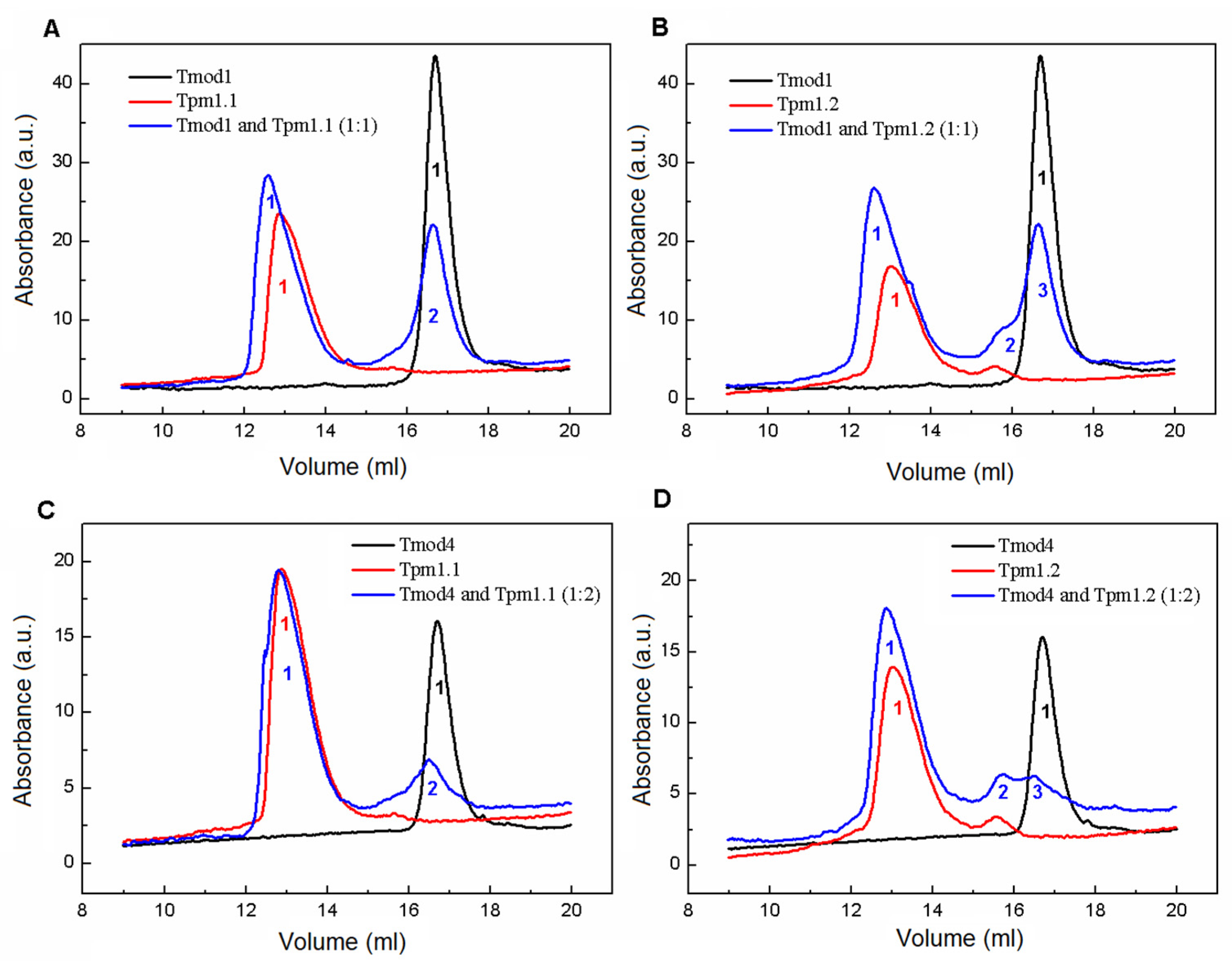
Size-exclusion chromatography was performed using a Varian ProStar 3250 system (Varian, Belrose, Australia) on a Superose 6 Increase 10/300 GL column (GE Healthcare, Stockholm, Sweden) equilibrated by a 50 mM HEPES-Na buffer (pH 7.3) with 150 mM NaCl and 1 mM DTT. The sample volume was 100 µL with 30 µM Tpm and Tmod1 in all elution profiles with Tmod1 and 26 µM Tpm and 13 µM Tmod4 in all elution profiles with Tmod4. The elution profiles were recorded at 280 nm with a 0.5 mL/min flow rate for all experiments. The following protein standards were used: thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), ovalbumin (43 kDa), and carboanhydrase (29 kDa), with RNAse (13.7 kDa) used for column calibration.
2.5. Pull-Down Assay
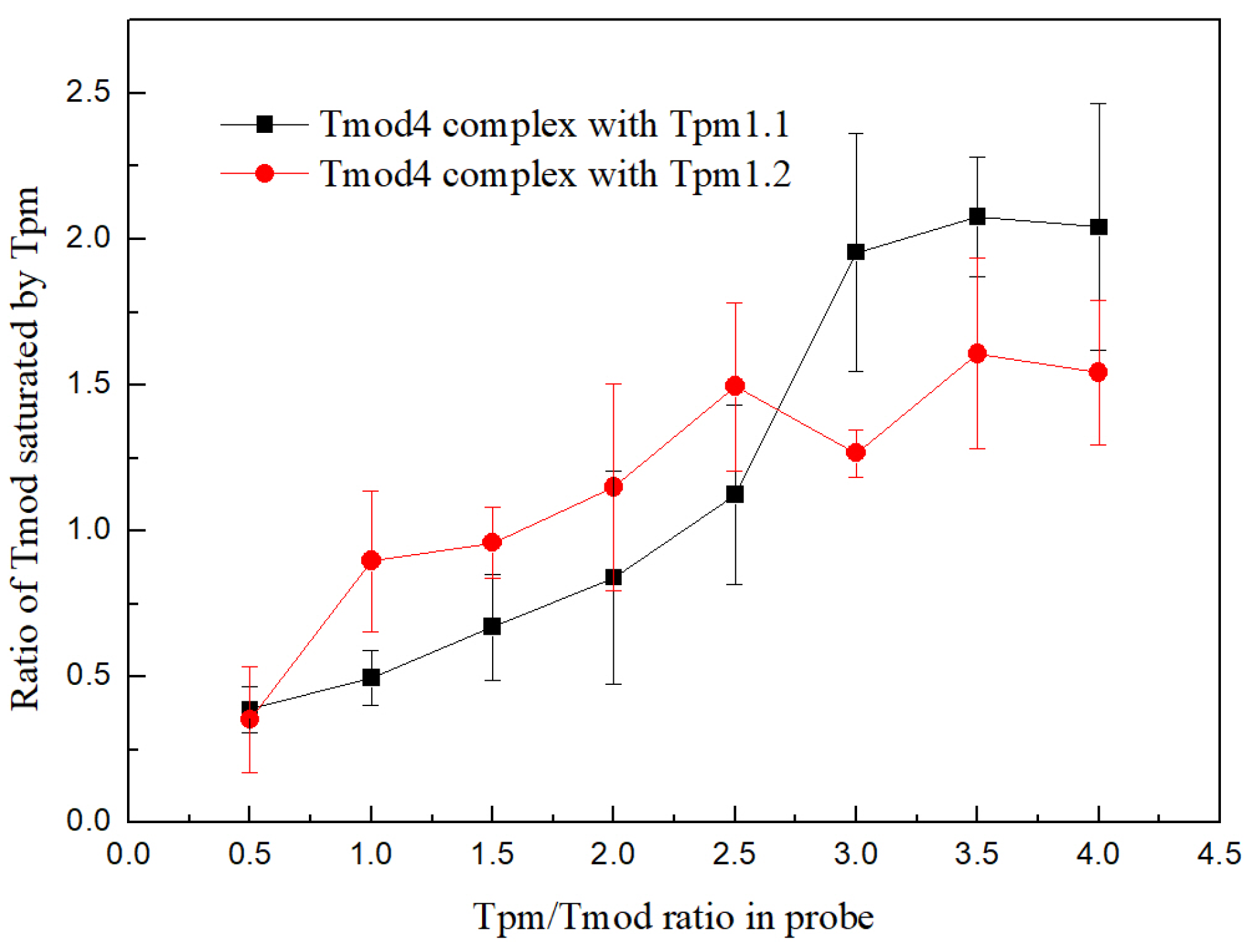
Experiments were performed by adding an affinity IMAC Ni-charged resin (Bio-Rad, Hercules, CA, USA) to Tpm/Tmod complexes. Before the experiments, the resin was washed three times by MQ water, then three times by a 30 mM HEPES-Na buffer, pH 7.3. Tmod1 and Tmod4 were incubated for 20 min in a 30 mM HEPES-Na buffer with 200 mM NaCl, pH 7.3, with increasing concentrations of Tpm1.1 and Tpm1.2 to the concentration ratio of 1:4 for Tmod/Tpm. After this, the resin was added to the complexes, and they were mixed and incubated for 5 min. The resin was pelleted by centrifugation and washed by a 30 mM HEPES-Na buffer with 200 mM NaCl, pH 7.3. Then, the resin was pelleted again and washed by 50 mM HEPES-Na, 300 mM NaCl, and 500 mM imidazole (pH 7.3). SDS-PAGE was used to analyze the supernatant probes. Protein bands were analyzed by ImageJ2 software (Scion, Frederick, MD, USA). For each Tpm/Tmod complex, three independent experiments were conducted.
2.6. In Vitro Motility Assay
The in vitro motility assay was carried out in accordance with a previous description [
40,
47,
48]. Myosin (300 µg/mL) in an AB buffer (25 mM KCl, 25 mM imidazole, 4 mM MgCl
2, 1 mM EGTA, and 20 mM DTT, pH 7.5) with 0.5 M KCl was loaded into a flow cell. After 2 min, 0.5 mg/mL BSA was added for 1 min. F-actin without labeling was added to an AB buffer with 2 mM ATP and left for 5 min. In order to produce regulated thin filaments, TRITC-phalloidin-labeled F-actin was added to the cell for 5 min. AB buffer was used to wash out the thin filaments that were not bound. The Maxchelator program (
http://www.stanford.edu/~cpatton/webmaxc/webmaxcS.html (accessed on 1 November 2018)) was used to calculate the appropriate Ca
2+ concentration. Finally, the cell was washed with an AB buffer containing 0.5 mg/mL BSA, an oxygen scavenger system, 20 mM DTT, 2 mM ATP, 0.5% methylcellulose, 100 nM Tpm/Tn, and Ca
2+ ions. In each flow cell, ten 30 s image sequences were recorded at 30 °C from different fields containing ~30–50 thin filaments. GMimPro2023 software [
49] was used to measure the sliding velocities of the filaments. We measured the sliding velocities of filaments that were at least 2 μm long and moved for at least 10 frames. We found that adding Tmod did not affect the lengths of the F-actin and thin filaments (
Figure S2 and Table S1 in the Supplementary Materials).
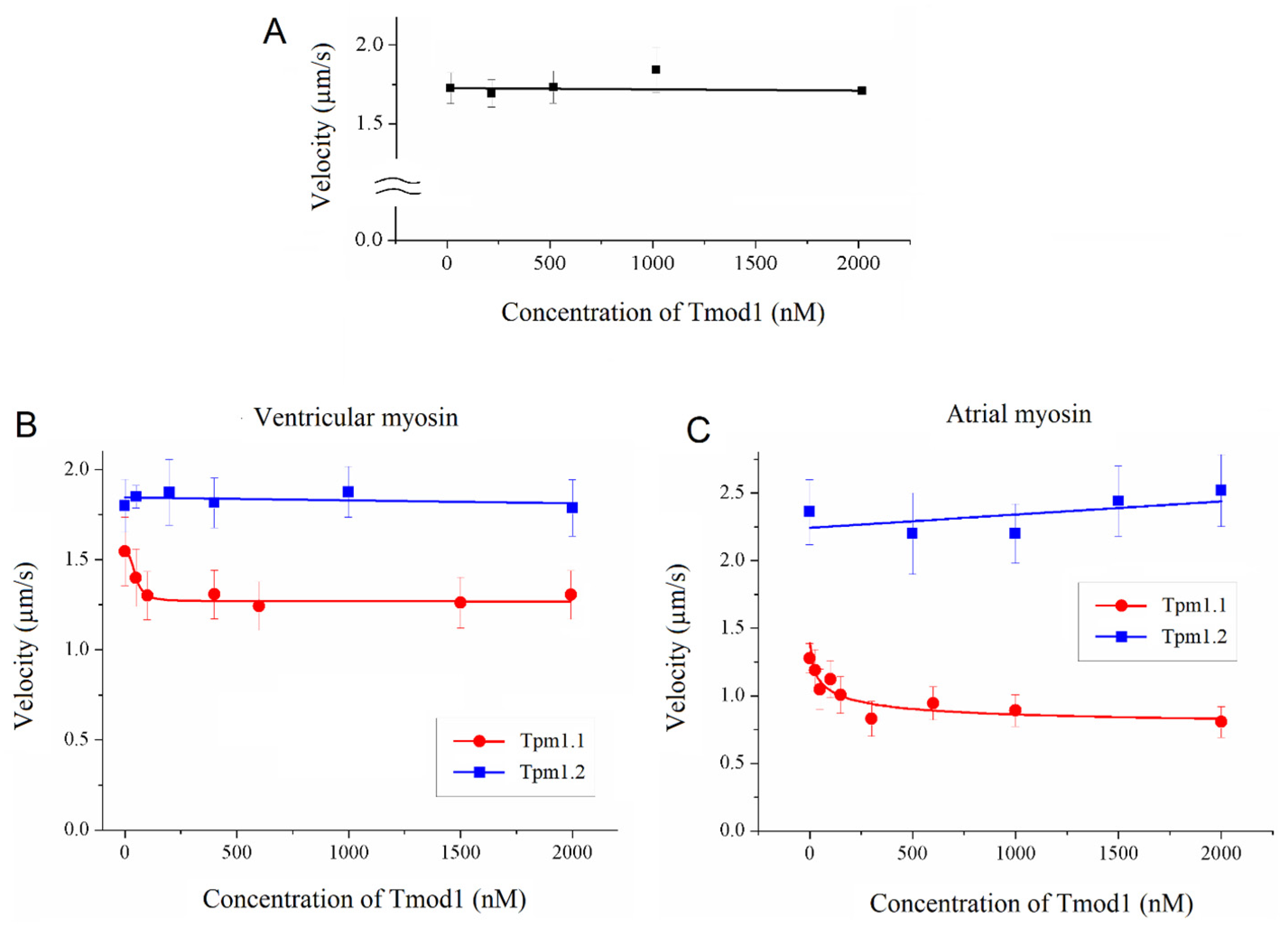
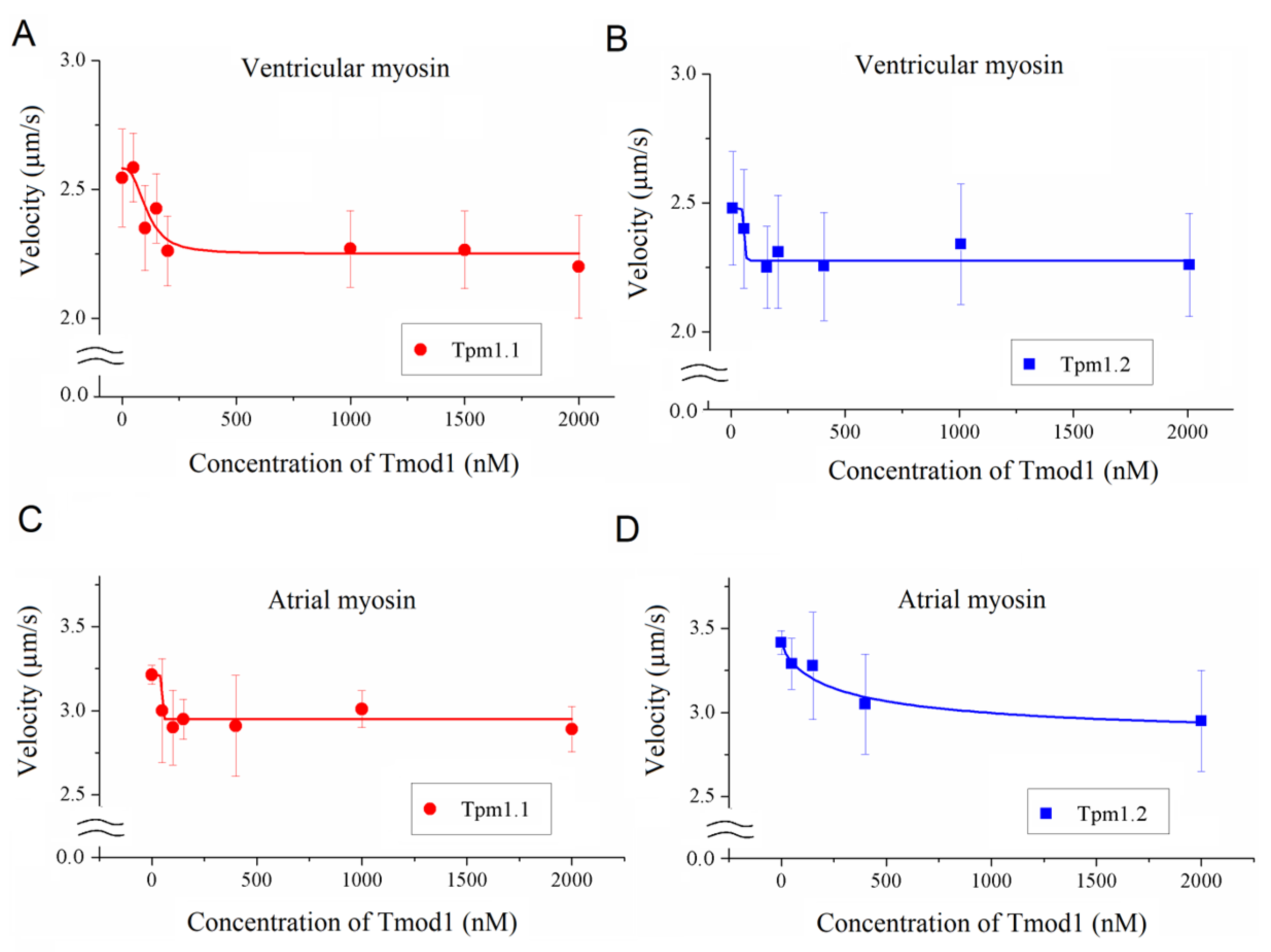
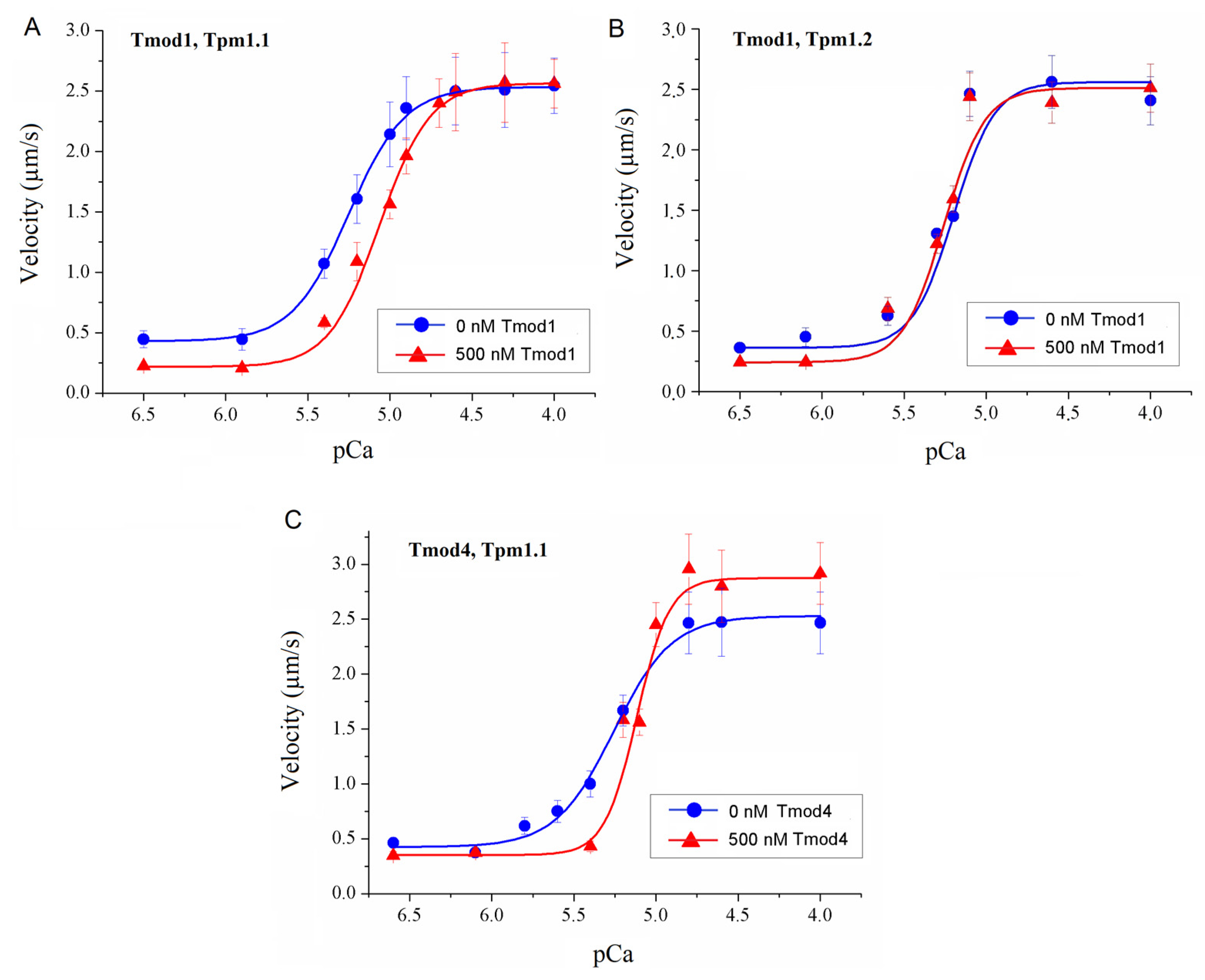
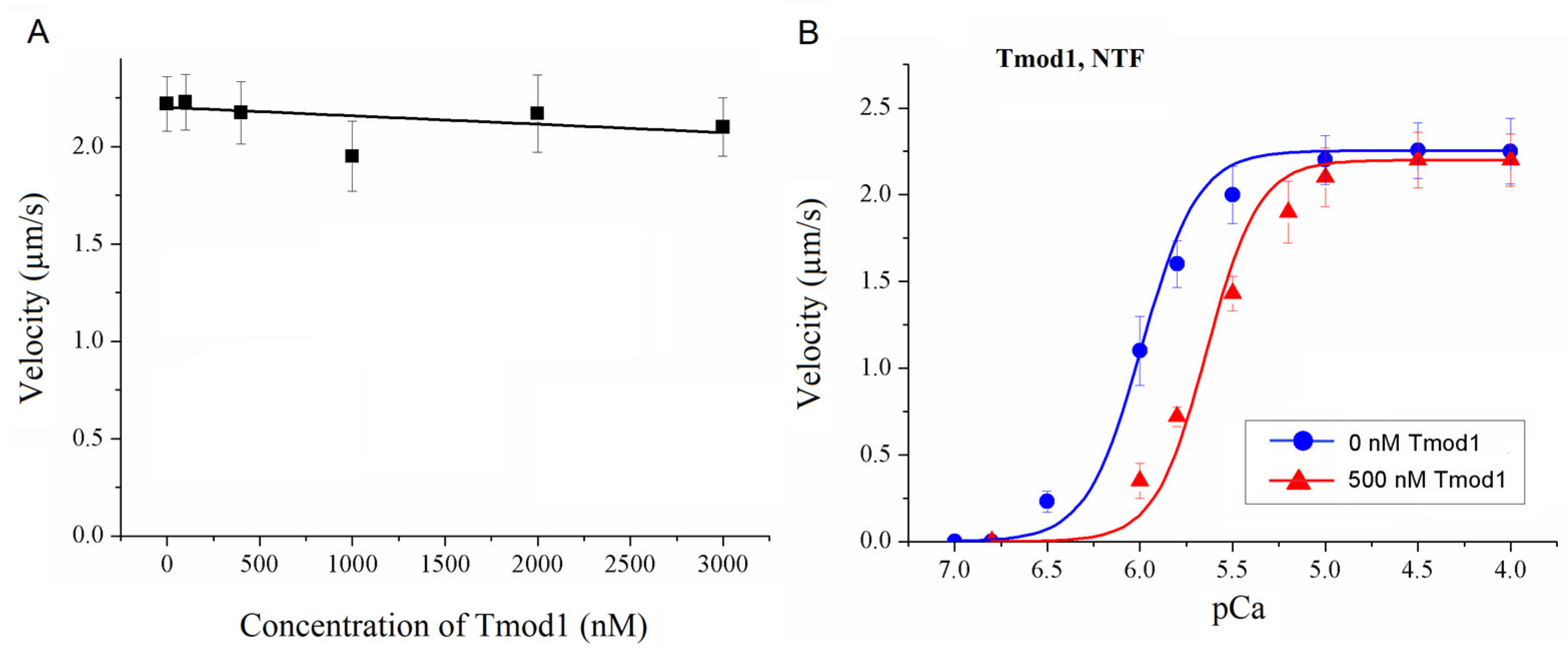
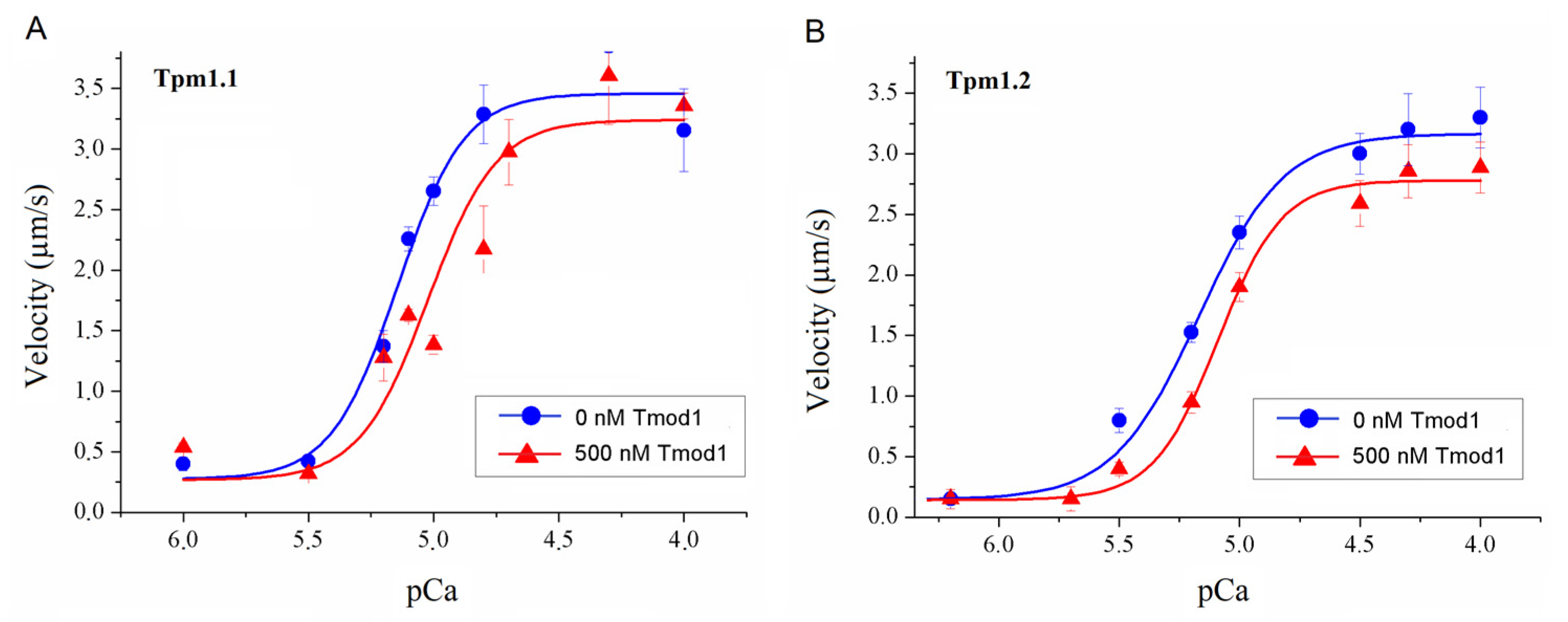
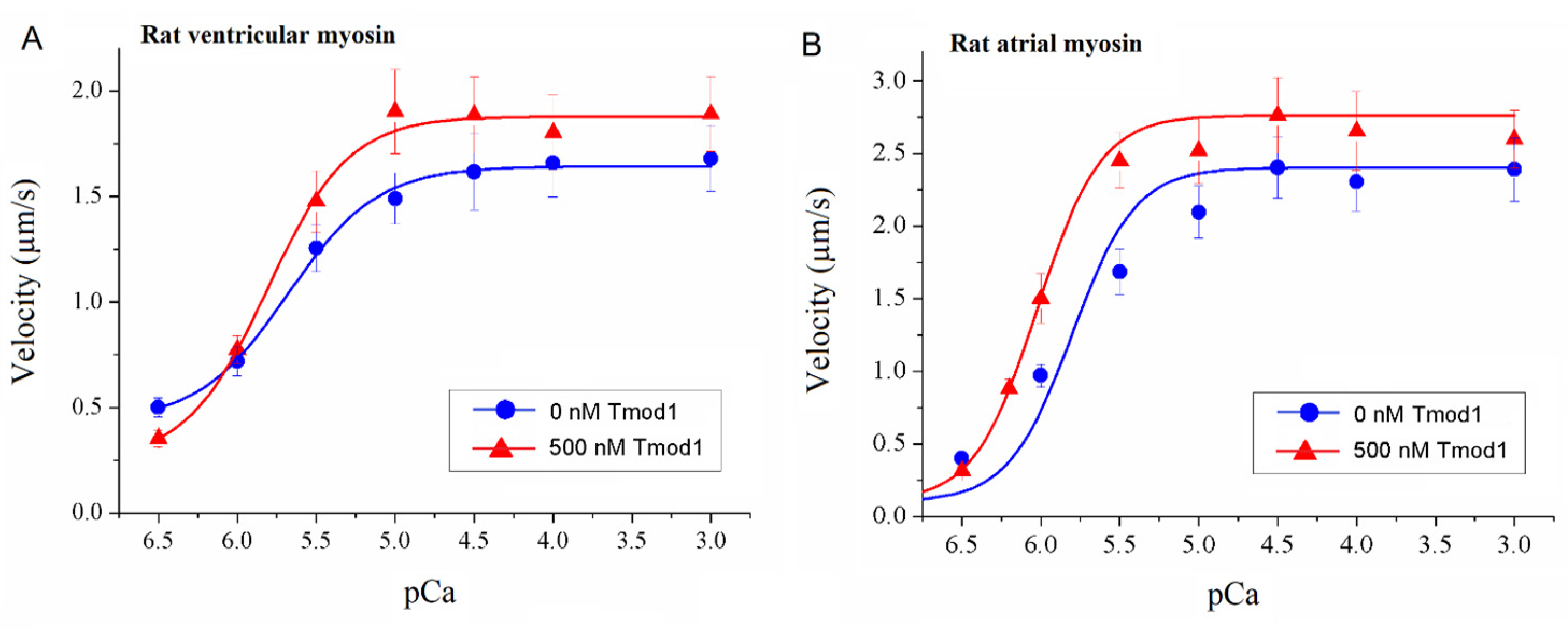
To study the effect of Tmod on the Ca2+ regulation of the actin–myosin interaction, we analyzed the dependence of the sliding velocity of the thin filaments over atrial and ventricular myosin on the Ca2+ concentration. Tmod was added to an AB buffer containing ATP. The means of individual experiments were fitted using the Hill equation: V = Vmax × (1 + 10h(pCa- pCa50))−1, where V and Vmax are the velocity and the maximal velocity at the saturating Ca2+ concentration, respectively; pCa50 (i.e., the Ca2+ sensitivity) is the pCa at which the velocity is half-maximal; and h is the Hill cooperativity coefficient. The parameters of individual experiments were averaged.
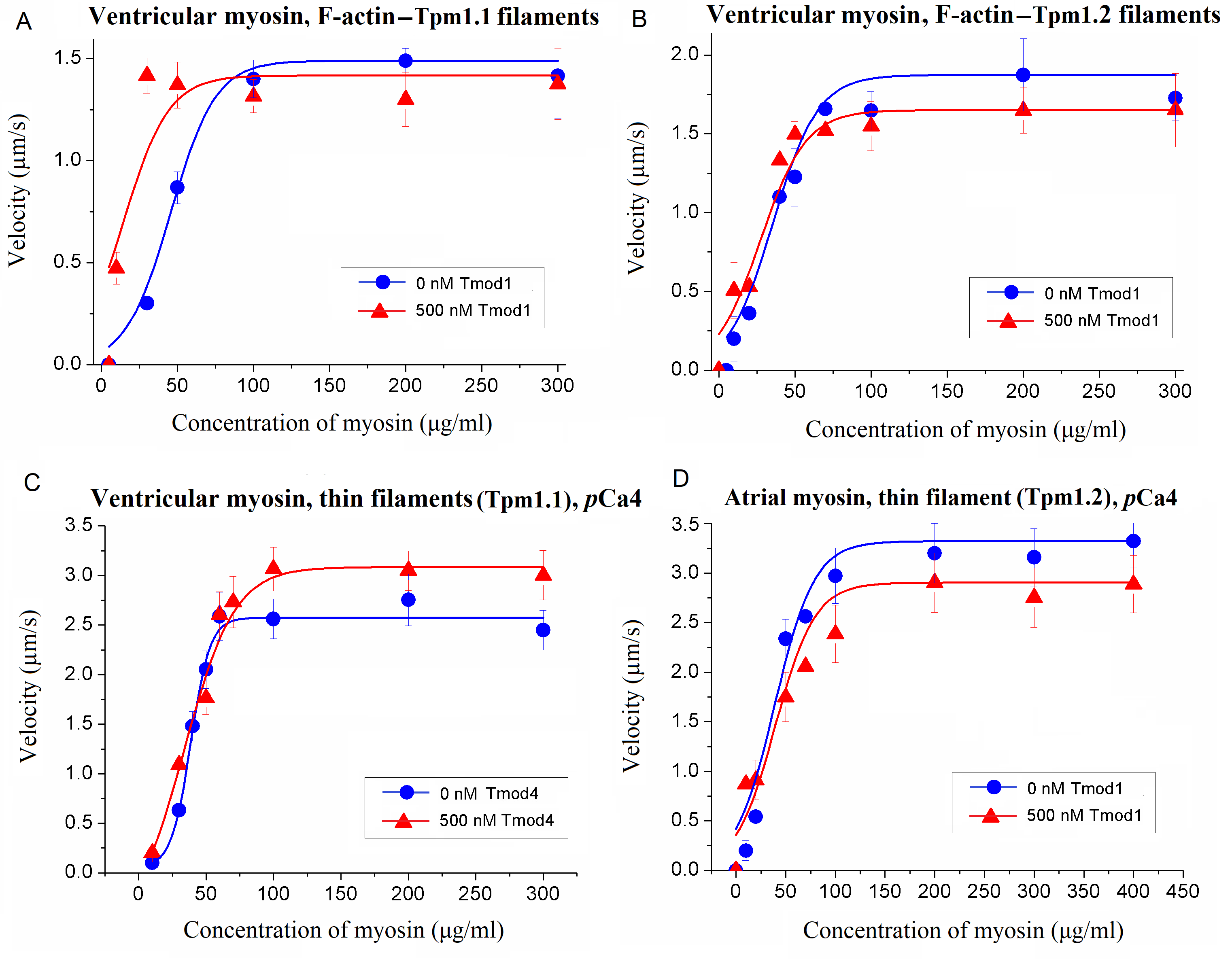
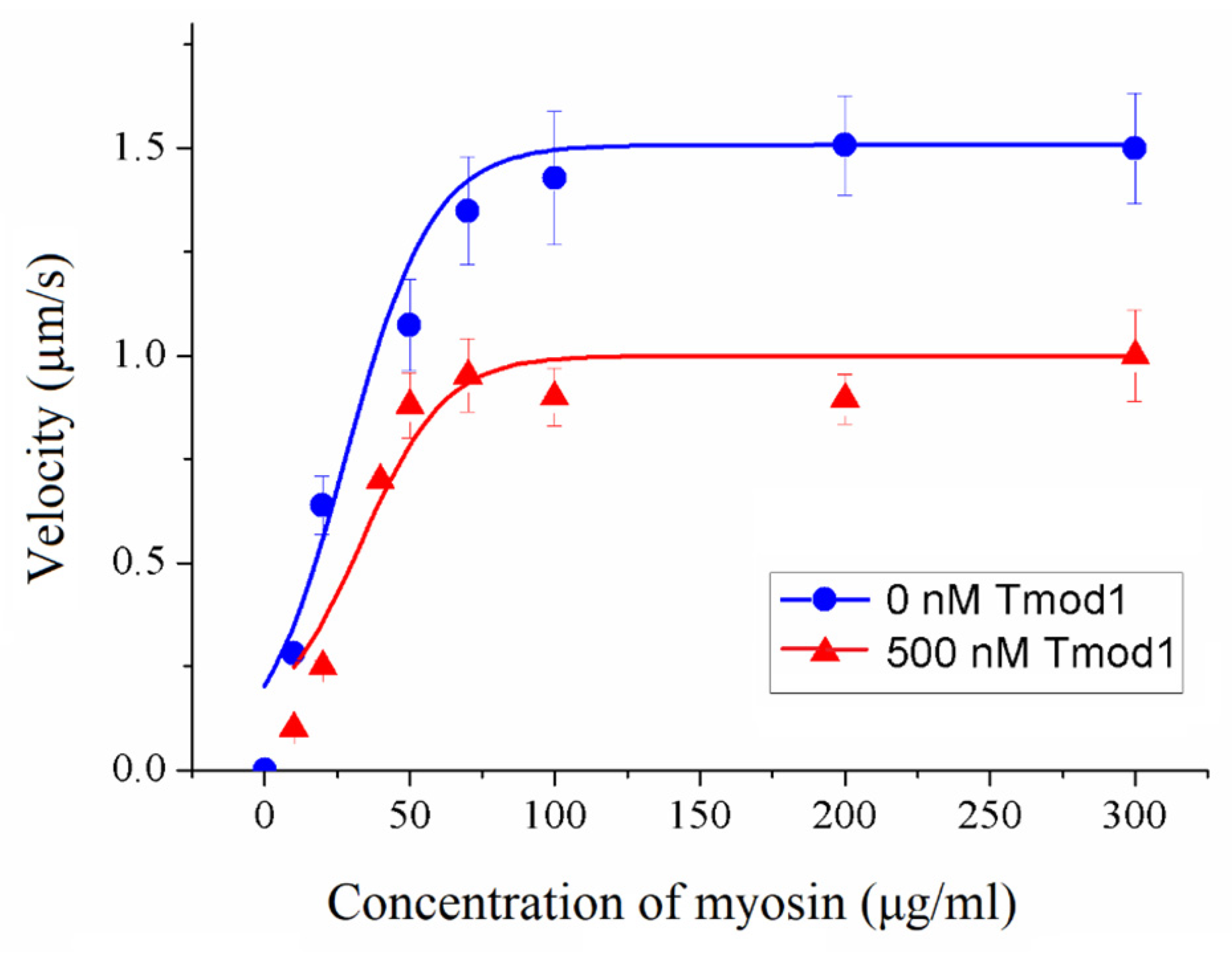
To assess the impact of Tmod on cross-bridge cooperation, the sliding velocities of thin filaments were evaluated in relation to the concentration of myosin added to the flow cell. This dependence was fitted with the modified Hill equation [
50] V = V
max ×
ch × (
c50h +
ch)
−1, where V
max is the maximal sliding velocity,
C is the myosin concentration,
C50 is the concentration required to achieve the half-maximal velocity, and h is the Hill coefficient.
Data analysis was performed using Excel 16 (Microsoft Corp., Redmond, WA, USA) and Origin 8.0 (Origin Lab, Northampton, MA, USA). All values were expressed as means ± SDs after three repetitions of the experiments. The Mann–Whitney U test was used to estimate the statistical significance of the characteristics of the Hill equation. In experiments where the effect of Tmod on the sliding velocity was studied, we measured the velocities of about 100 filaments, and comparisons of the filament sliding velocities at different Tmod concentrations were performed using Student’s t-test.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}