Probing the Mechanism of Action of Cry41Aa on HepG2 through the Establishment of a Resistant Subline

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
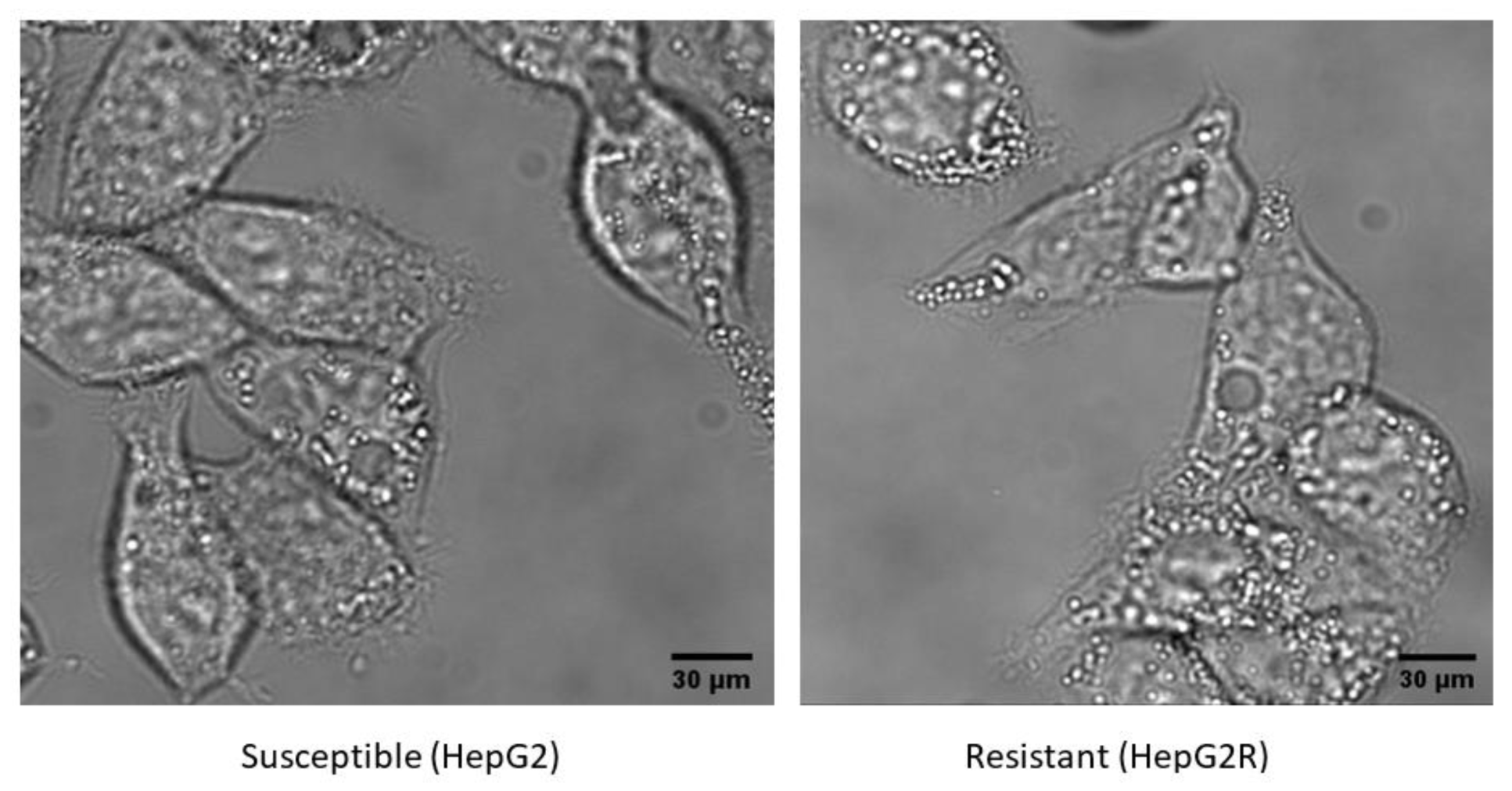
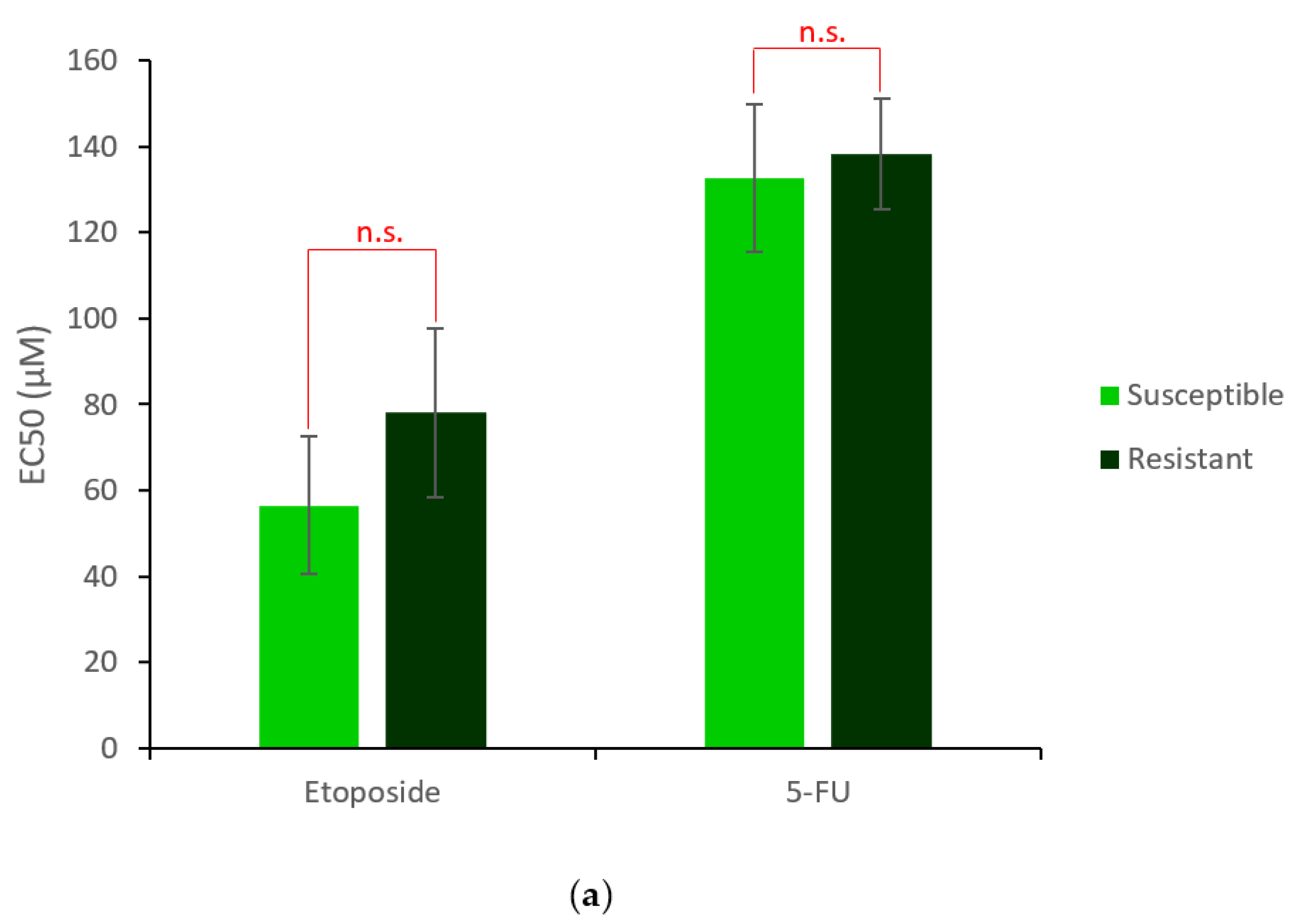
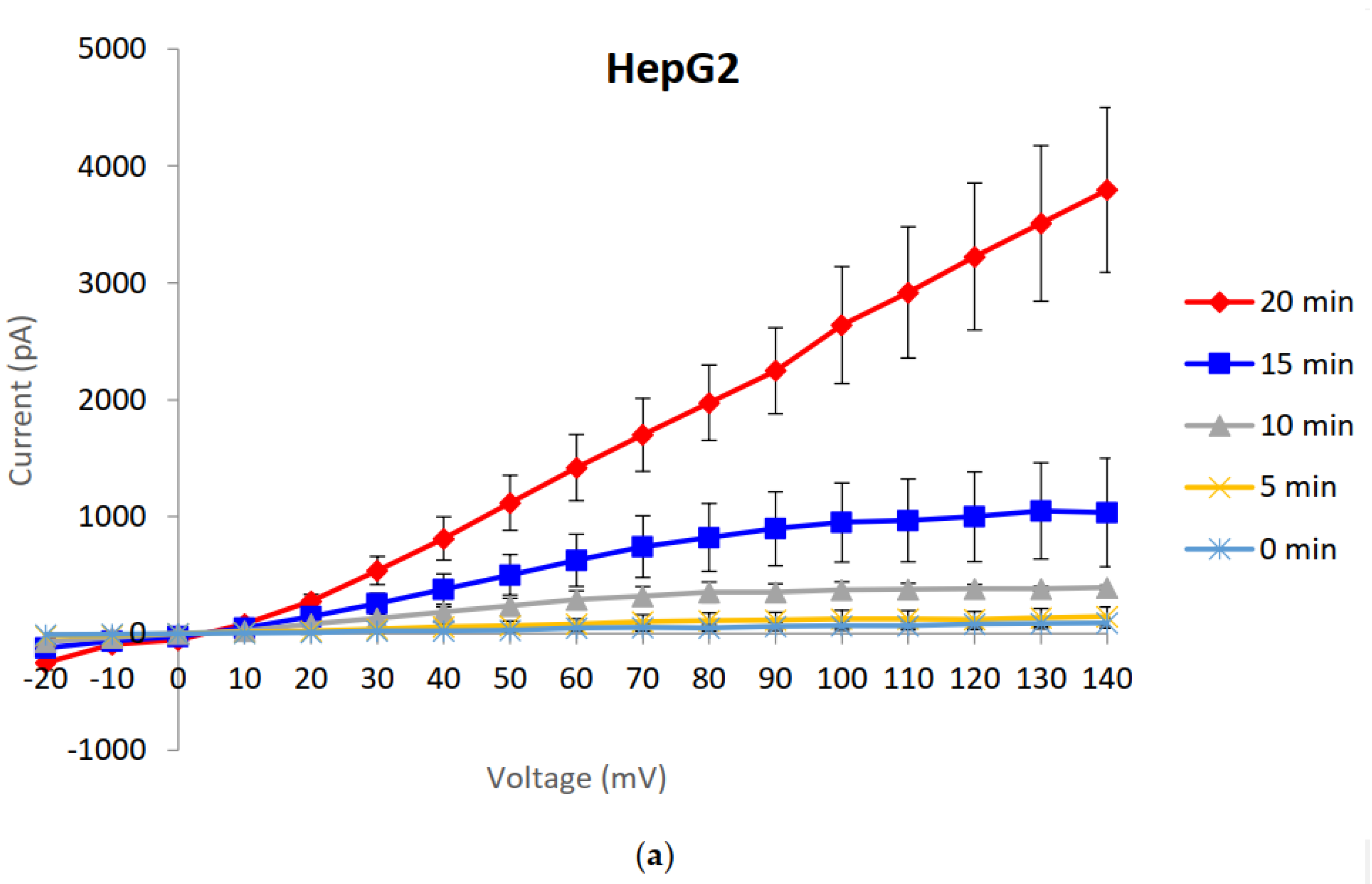
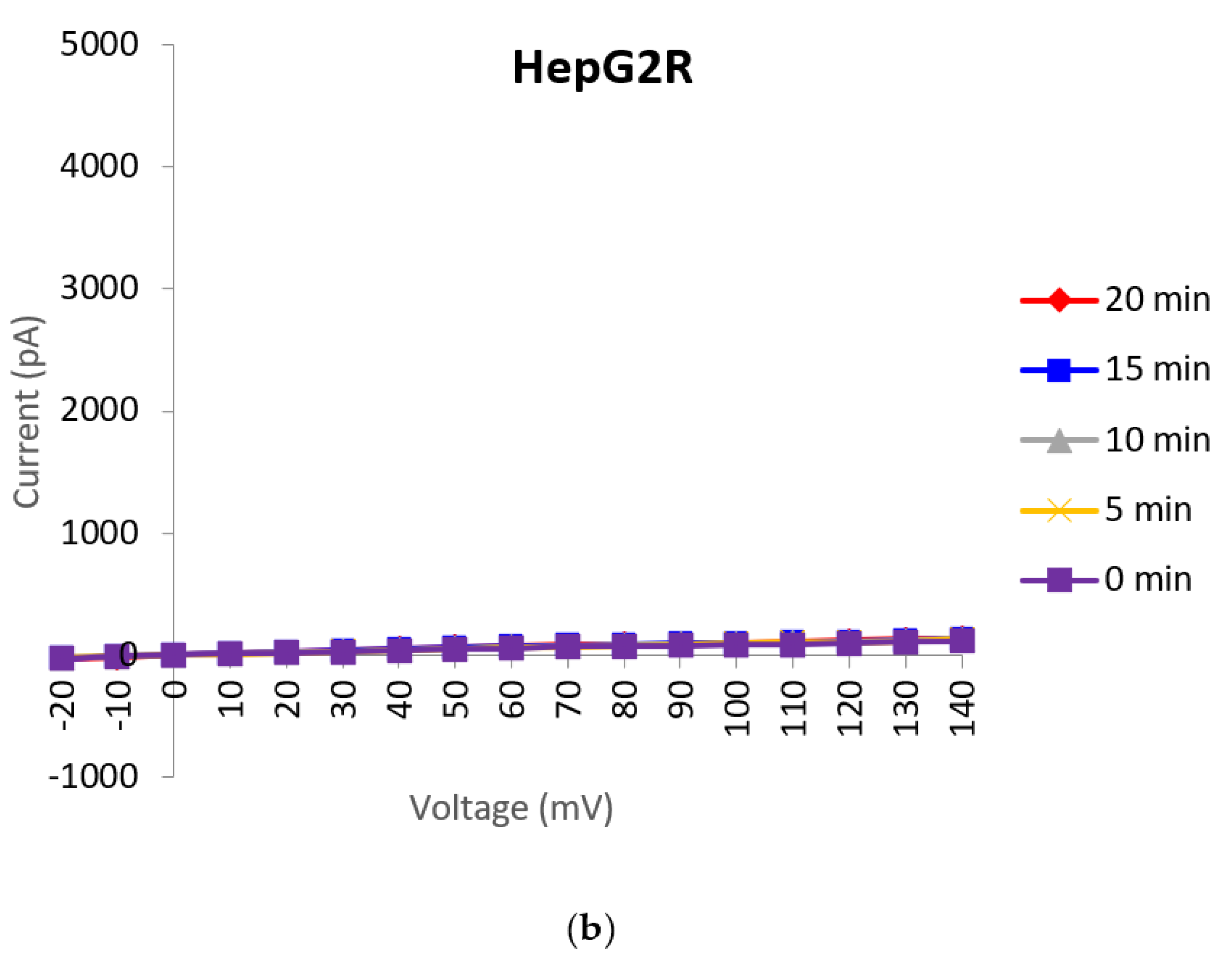
2.1. Generation and Characterisation of a Resistant HepG2 Cell Line
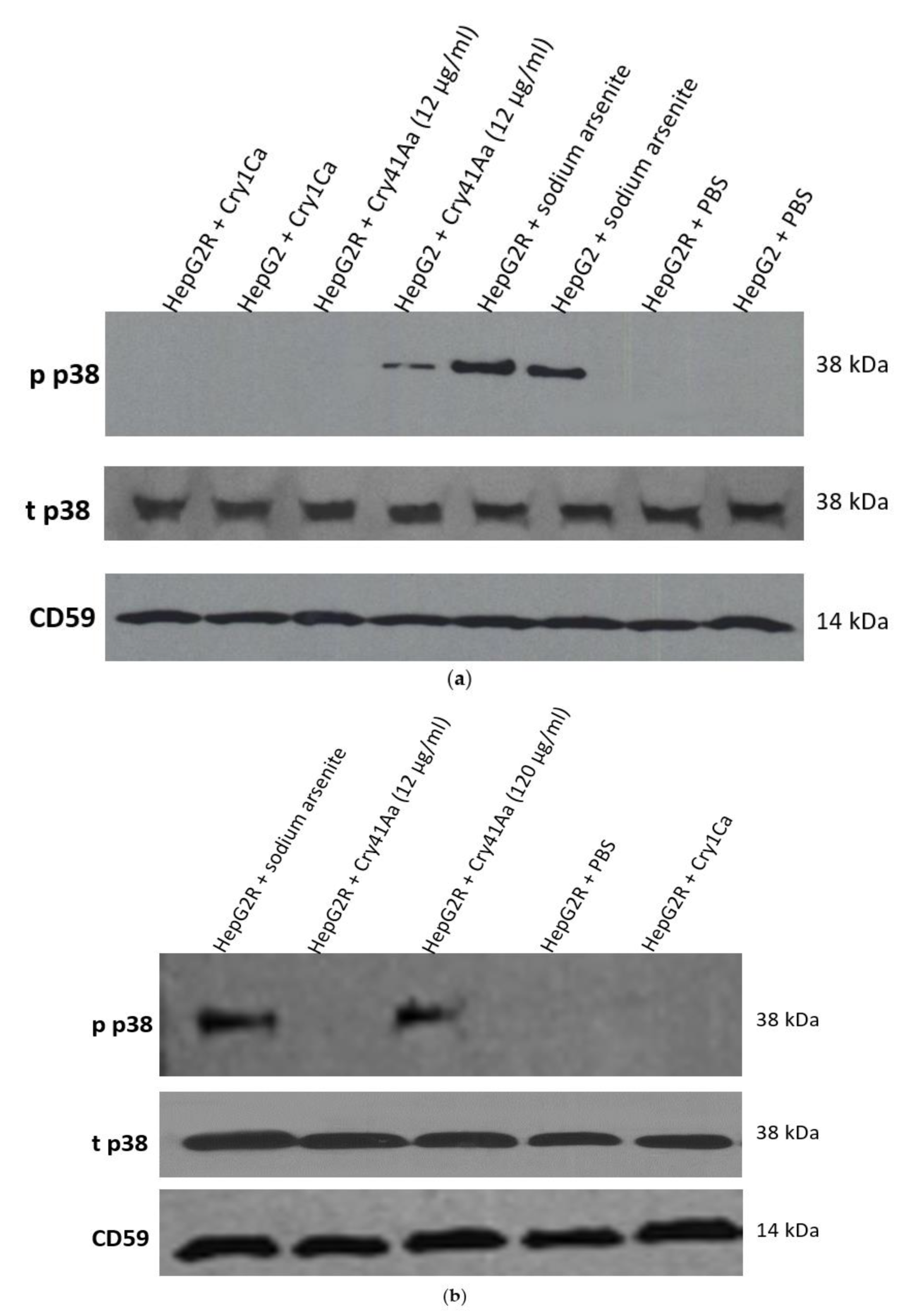
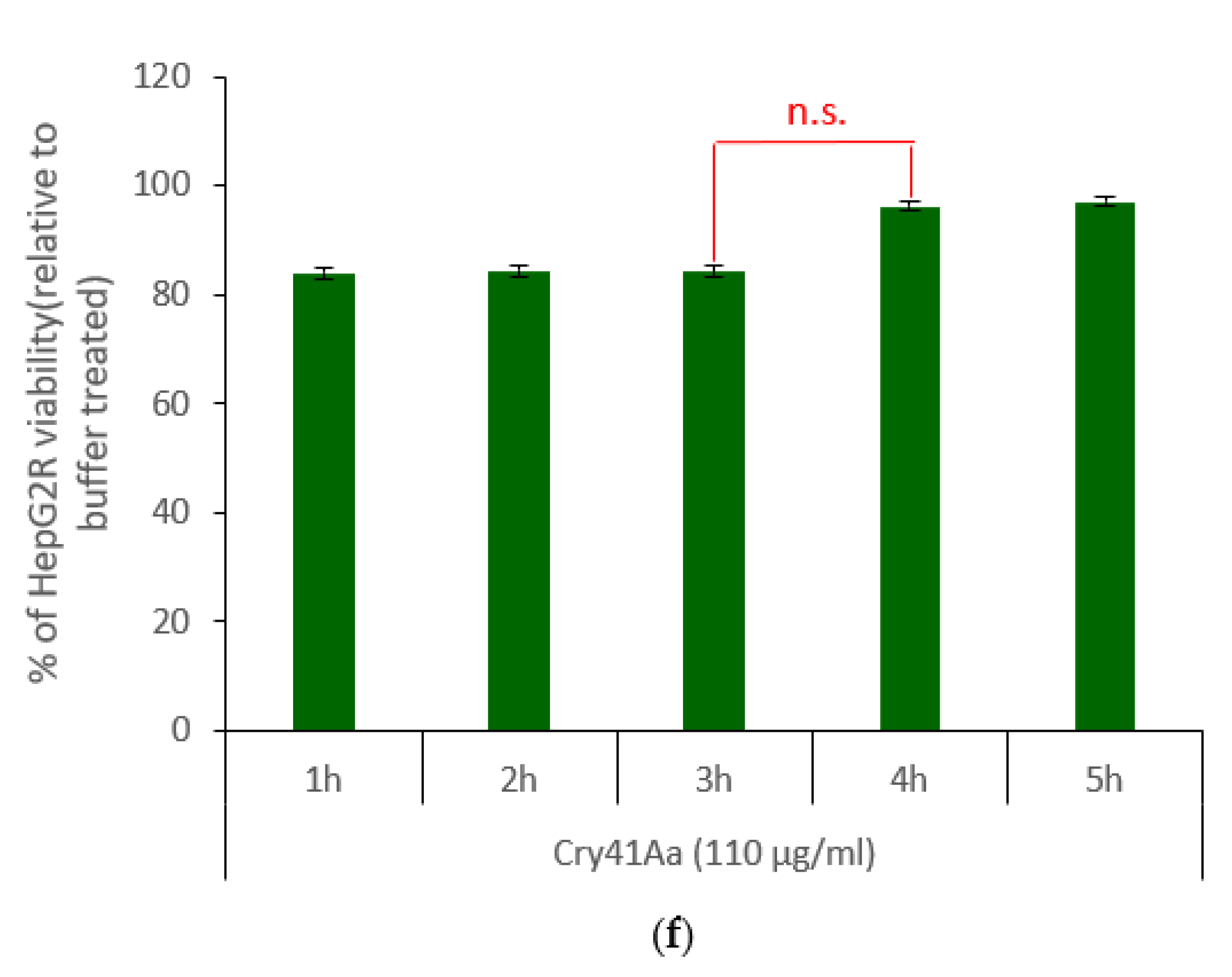
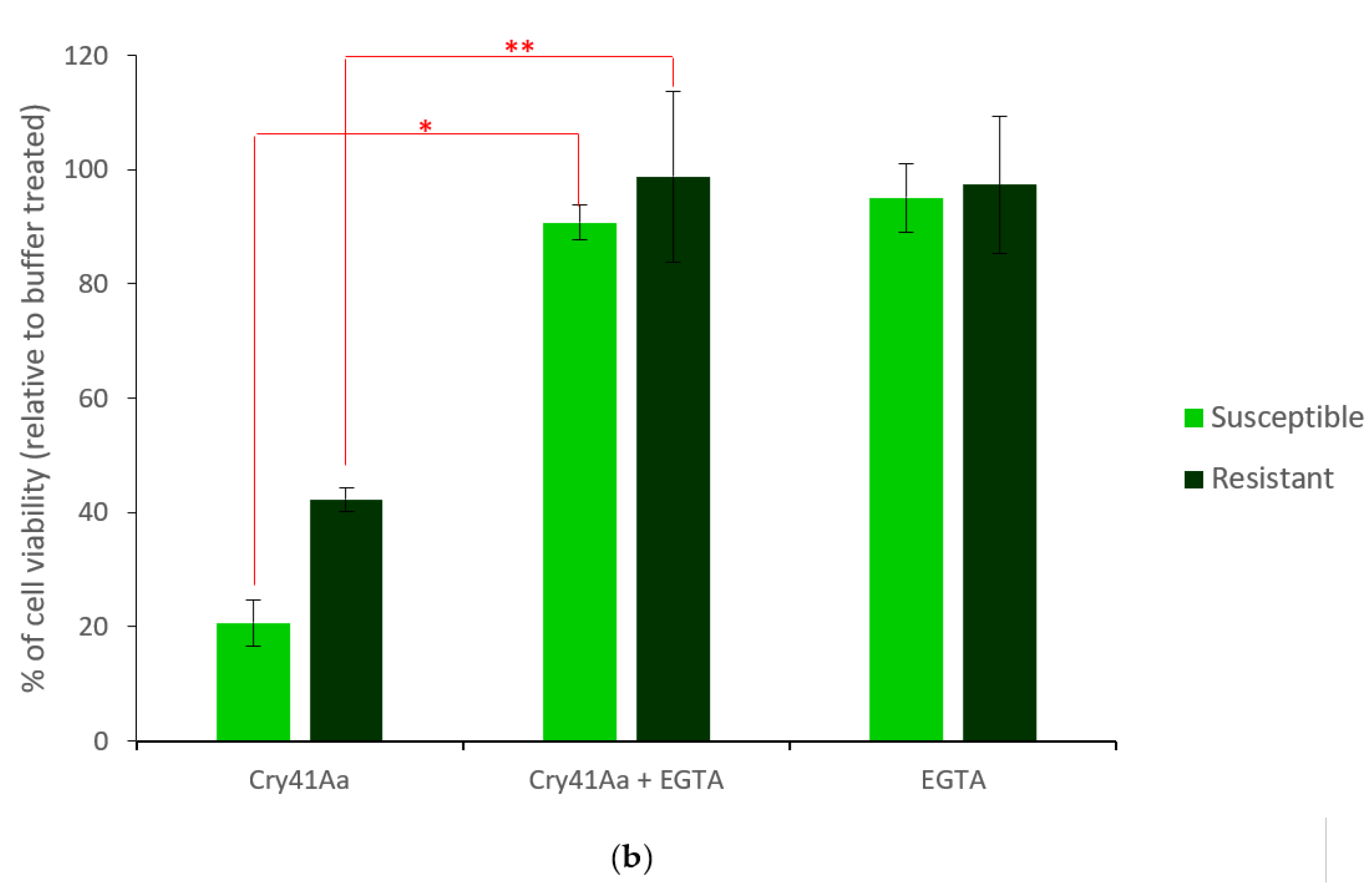
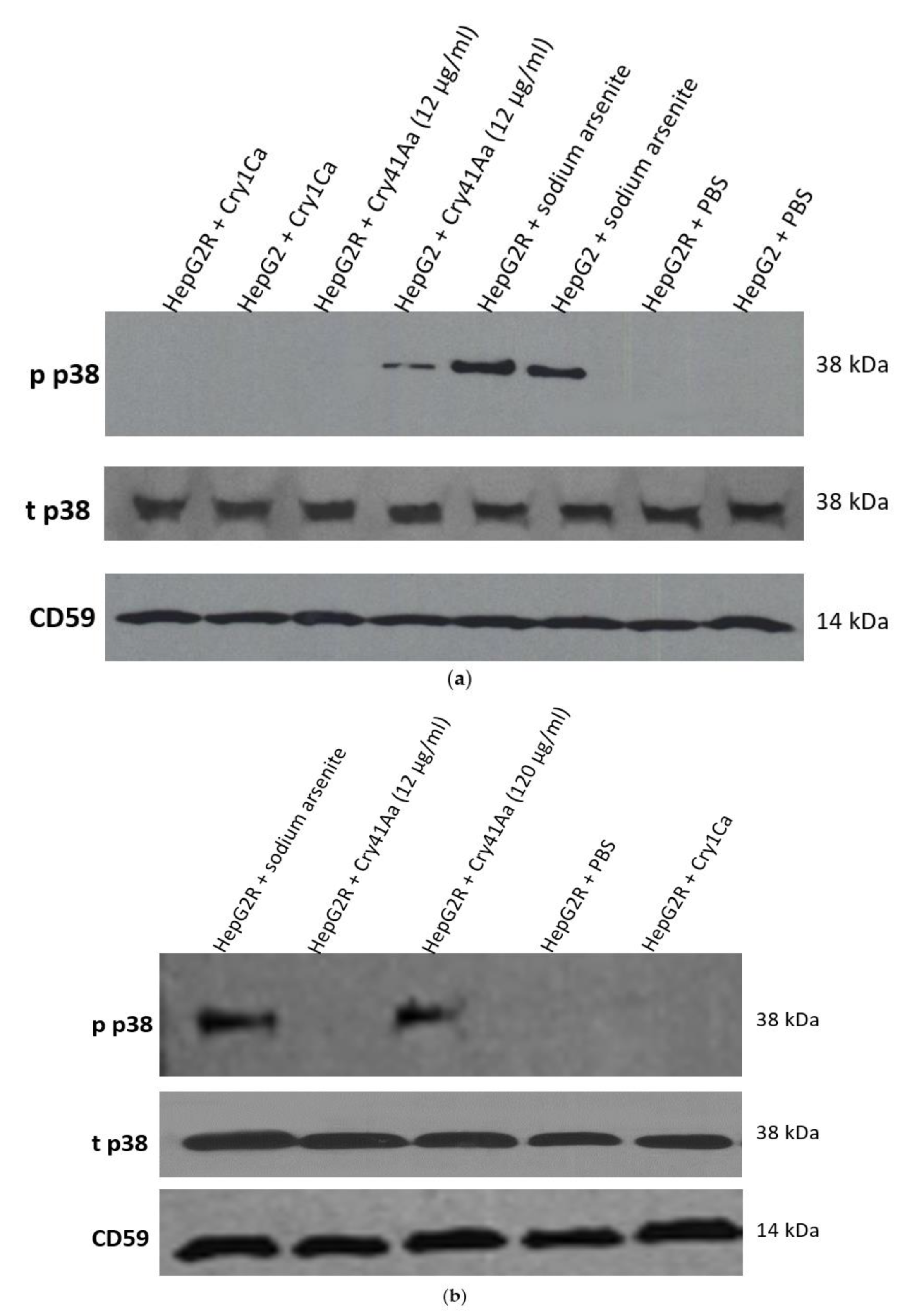
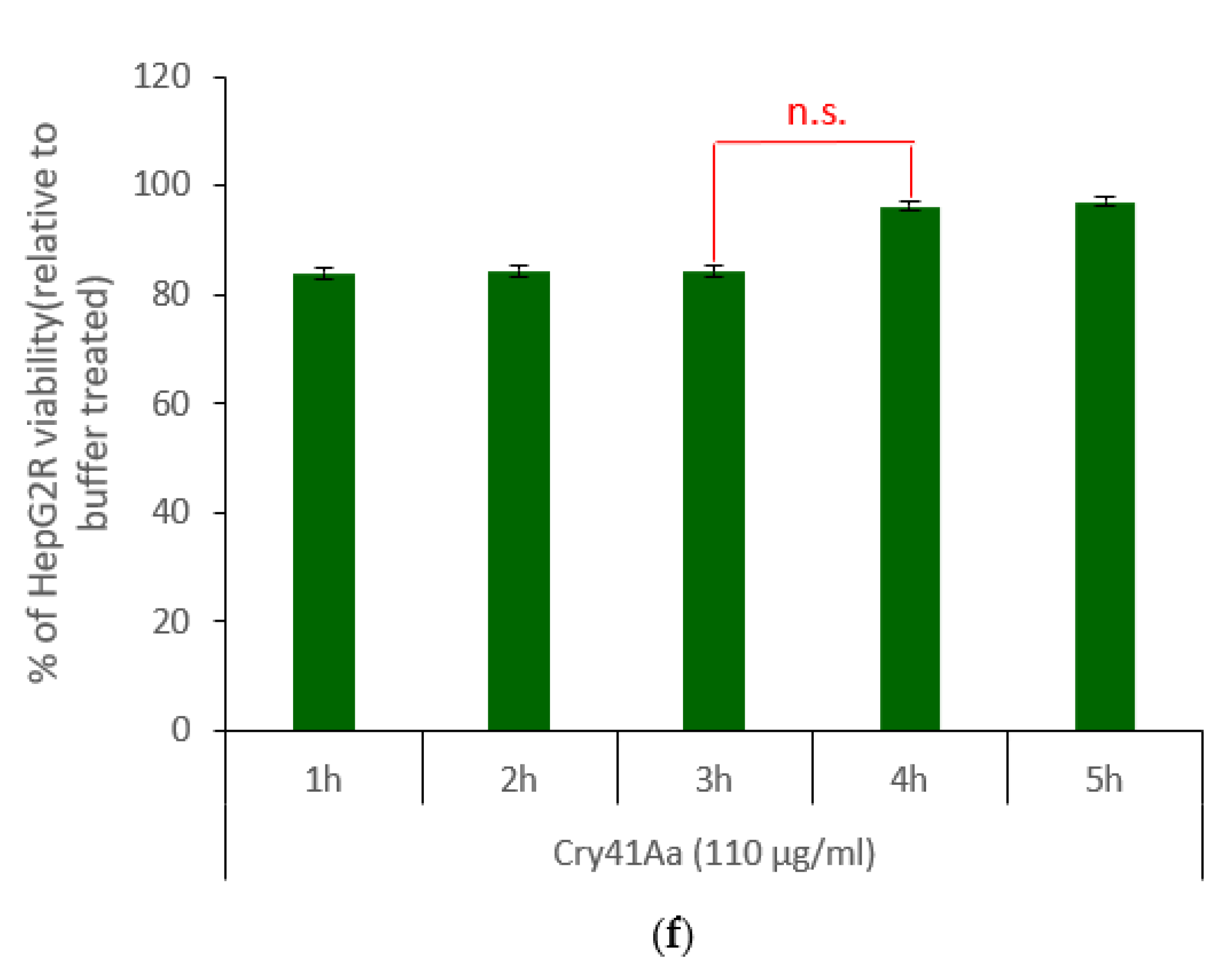
2.2. Characterisation of HepG2R Cell Response to Cry41Aa Compared with the Parental Cell Line
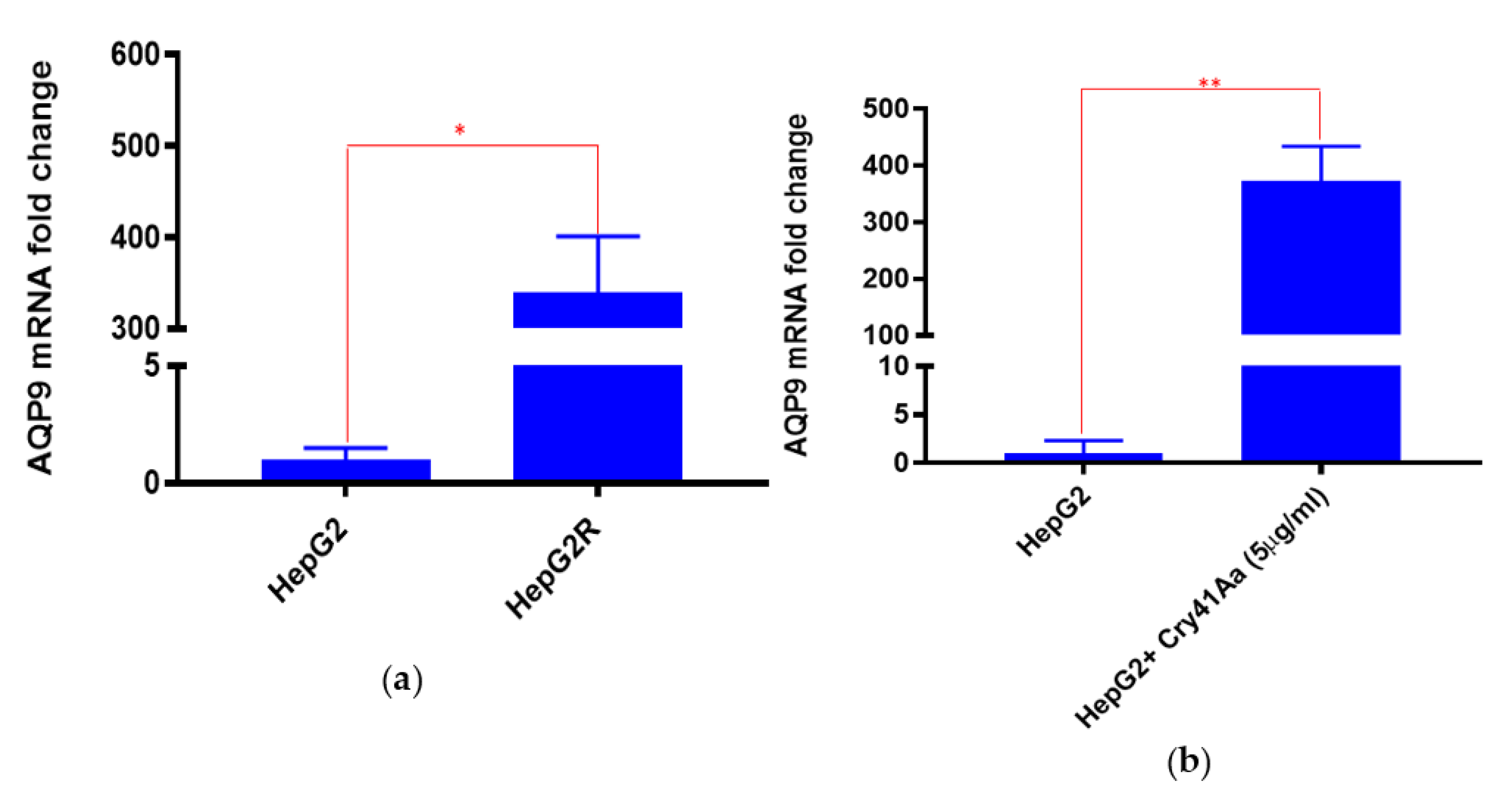
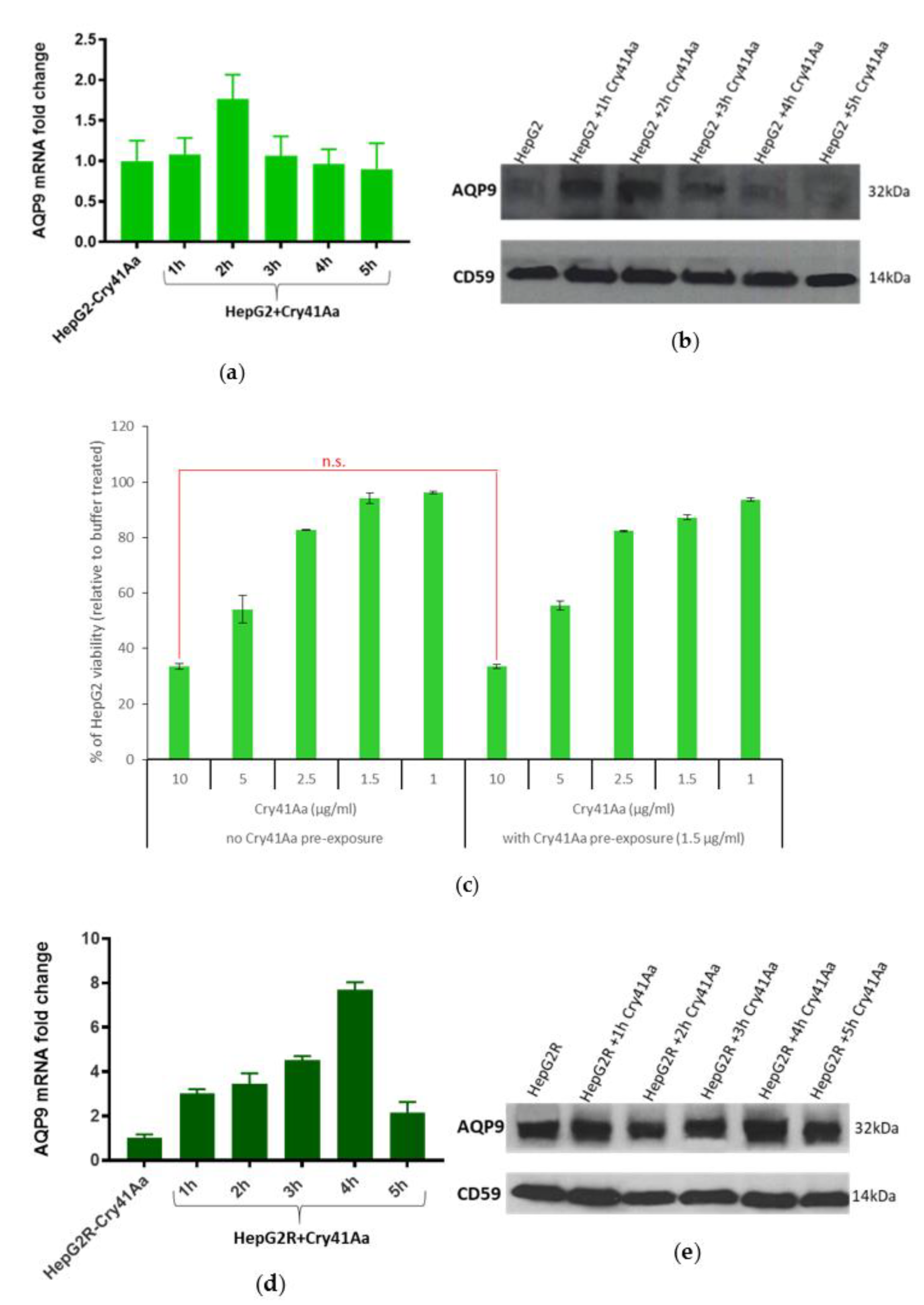
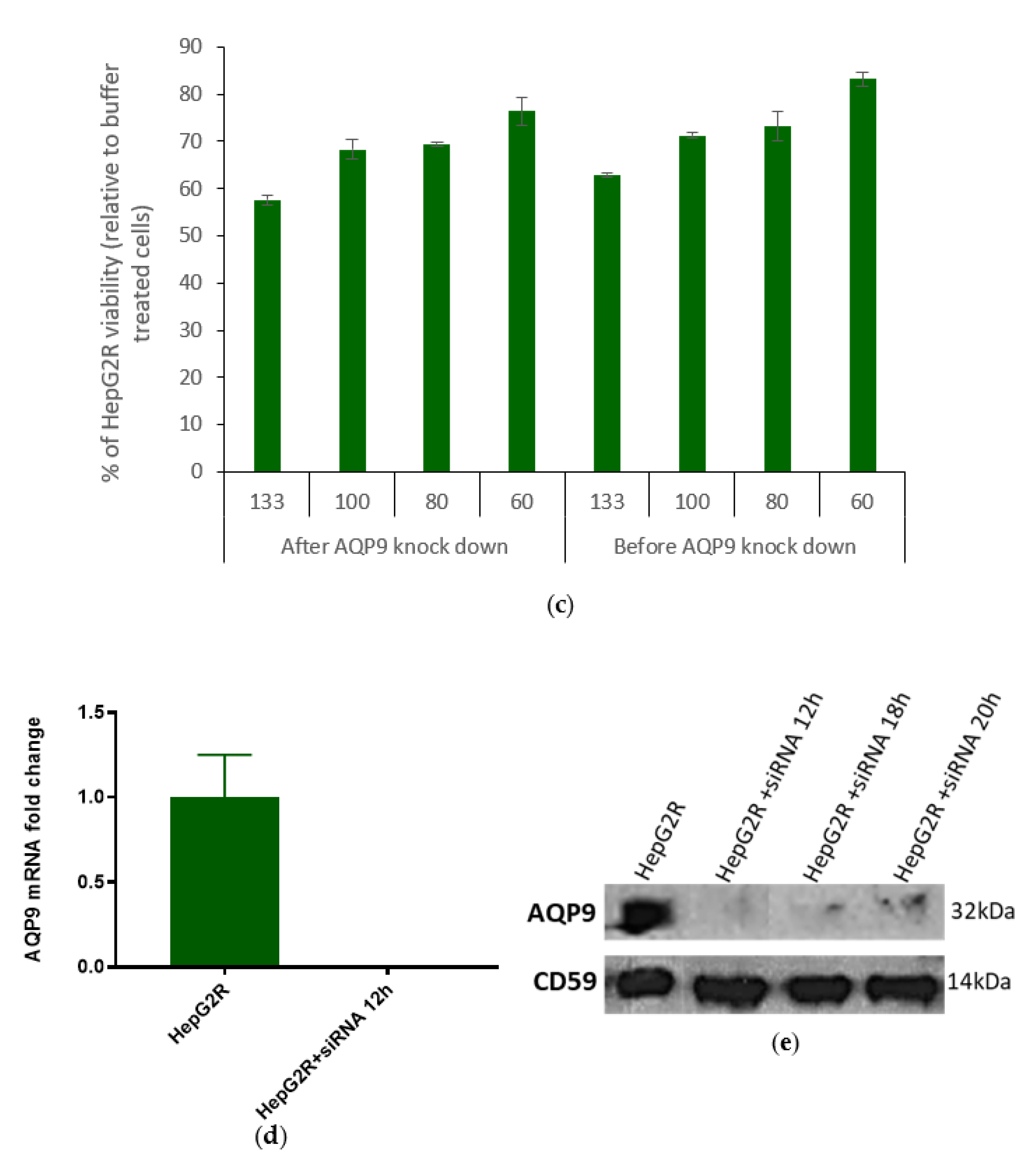
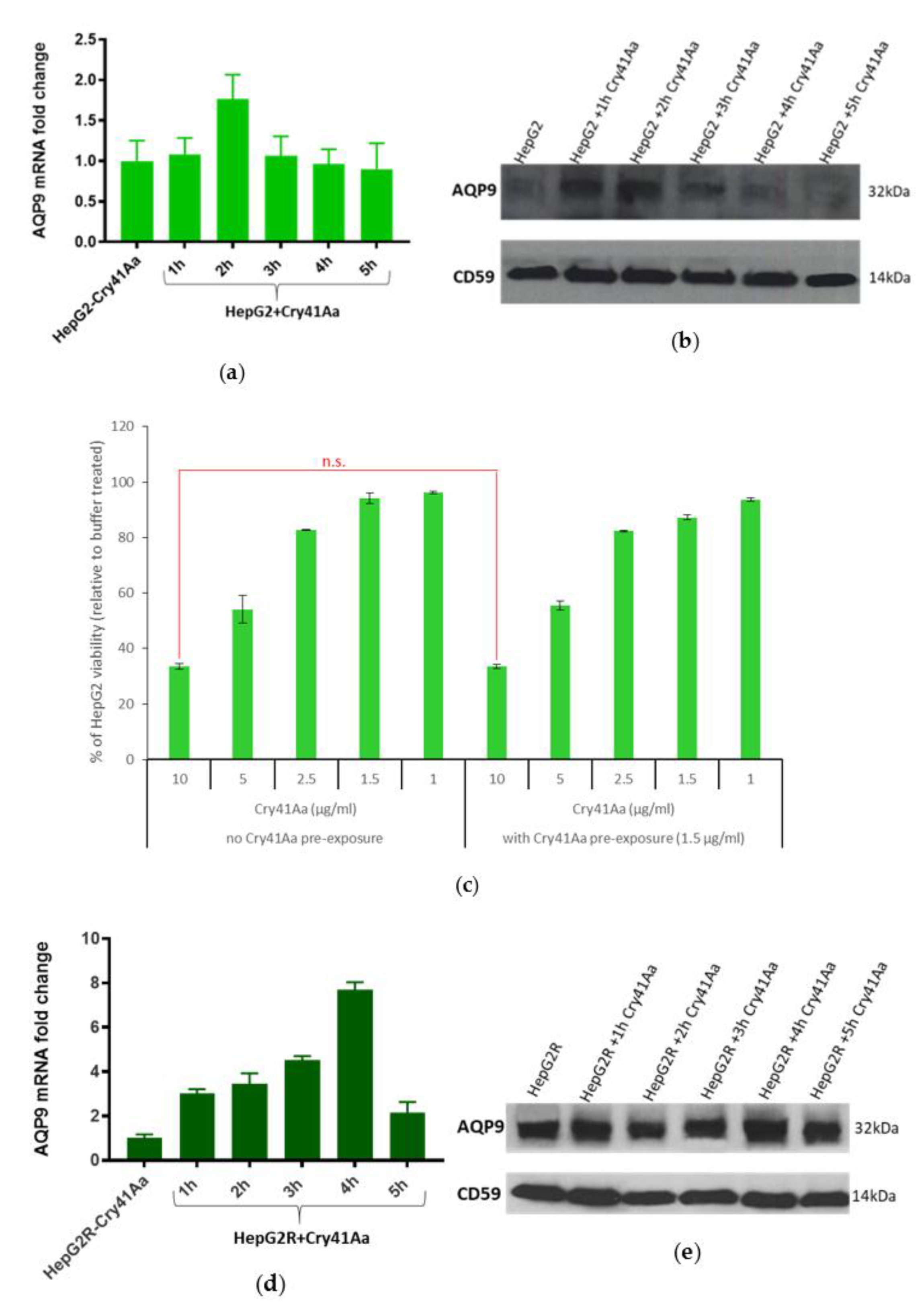
2.3. Differential Expression of AQP9 in the Resistant Cell Line
2.3.1. Validation of AQP9 Expression Levels
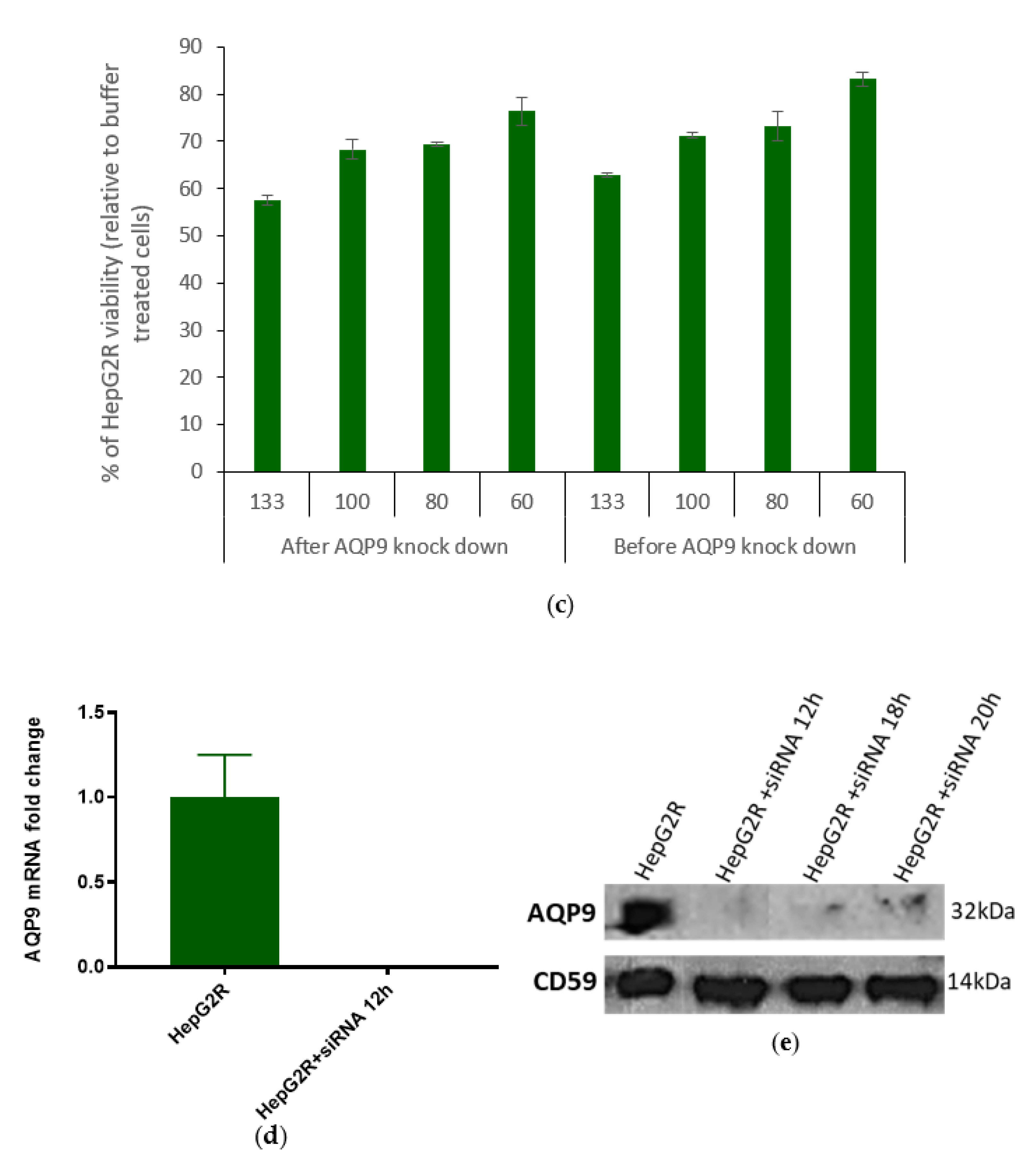
2.3.2. Correlation between AQP9 Expression and Cry41Aa Susceptibility
3. Discussion
4. Materials and Methods
4.1. Crystal Protein Harvesting and Activation
4.2. Protein Purification
4.3. Cell Cultures
4.4. Development of the Resistant HepG2R Cell Line
4.5. Cell Assays
4.6. Microscopy
4.7. Western Blots
4.8. Whole-Cell Patch-Clamp
4.9. RNA Extraction
4.10. Determination of RNA Concentration and RNA Integrity
4.11. RNA-Seq and Analysis
4.12. cDNA Synthesis
4.13. qPCR
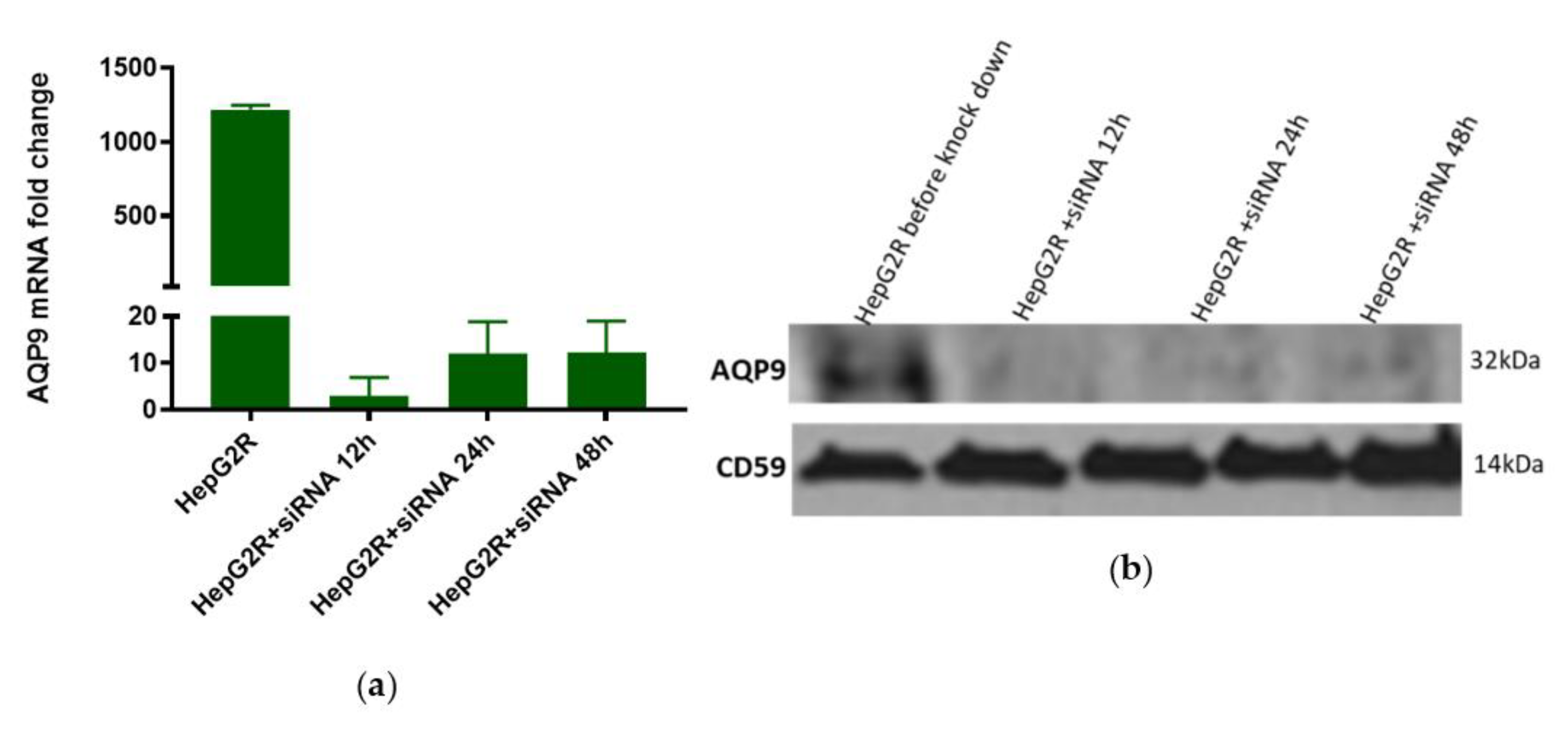
4.14. AQP9 Knockdown in HepG2R
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ferre, J.; van Rie, J. Biochemistry and genetics of insect resistance to Bacillus thuringiensis. Annu. Rev. Entomol. 2002, 47, 501–533. [Google Scholar] [CrossRef]
- Oppert, B.; Kramer, K.J.; Beeman, R.W.; Johnson, D.; McGaughey, W.H. Proteinase-mediated insect resistance to Bacillus thuringiensis toxins. J. Biol. Chem. 1997, 272, 23473–23476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.; Roberts, H.; Sarjan, M.; Featherstone, N.; Lahnstein, J.; Akhurst, R.; Schmidt, O. Is the mature endotoxin Cry1Ac from Bacillus thuringiensis inactivated by a coagulation reaction in the gut lumen of resistant Helicoverpa armigera larvae? Insect Biochem. Mol. Biol. 2005, 35, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Gunning, R.V.; Dang, H.T.; Kemp, F.C.; Nicholson, I.C.; Moores, G.D. New resistance mechanism in Helicoverpa armigera threatens transgenic crops expressing Bacillus thuringiensis Cry1Ac toxin. Appl. Environ. Microbiol. 2005, 71, 2558–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14 (Suppl. 1), 35–48. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ling, X.L.; Li, S.W.; Li, X.Q.; Yan, B. Establishment of a human hepatoma multidrug resistant cell line in vitro. World J. Gastroenterol. 2010, 16, 2291–2297. [Google Scholar] [CrossRef]
- Kuppen, P.J.K.; Schuitemaker, H.; van’t Veer, L.J.; de Bruijn, E.A.; van Oosterom, A.T.; Schrier, P.I. Cis-diamminedichloroplatinum (II)-resistant sublines derived from 2 human ovarian tumor-cell lines. Cancer Res. 1988, 10, 296. [Google Scholar]
- Okamura, M.; Shimada, J.; Sakagami, H. Comparative analysis of cell death induction by cisplatin and 5-FU in human oral squamous and hepatocellular carcinoma cell lines. Anticancer Res. 2008, 28, 253–259. [Google Scholar] [PubMed]
- Xie, B.S.; Zhao, H.C.; Yao, S.K.; Zhuo, D.X.; Jin, B.; Lv, D.C.; Wu, C.L.; Ma, D.L.; Gao, C.; Shu, X.M.; et al. Autophagy inhibition enhances etoposide-induced cell death in human hepatoma G2 cells. Int. J. Mol. Med. 2011, 27, 599–606. [Google Scholar] [PubMed] [Green Version]
- Domanska, B.; Fortea, E.; West, M.J.; Schwartz, J.L.; Crickmore, N. The role of membrane-bound metal ions in toxicity of a human cancer cell-active pore-forming toxin Cry41Aa from Bacillus thuringiensis. Toxicon 2019, 167, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ratner, A.J.; Hippe, K.R.; Aguilar, J.L.; Bender, M.H.; Nelson, A.L.; Weiser, J.N. Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J. Biol. Chem. 2006, 281, 12994–12998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffman, D.L.; Abrami, L.; Sasik, R.; Corbeil, J.; van der Goot, F.G.; Aroian, R.V. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 2004, 101, 10995–11000. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Zachary, J.F. Mechanisms and morphology of cellular injury, adaptation, and death. Pathol. Basis Vet. Dis. 2017, 2, 43.e19. [Google Scholar]
- Knowles, B.H.; Ellar, D. Colloid-osmotic lysis is a general feature of the mechanism of action of Bacillus thuringiensis-endotoxins with different insect specificity. Biochim. Biophys. Acta 1987, 924, 509–518. [Google Scholar] [CrossRef]
- Endo, H.; Azuma, M.; Adegawa, S.; Kikuta, S.; Sato, R. Water influx via aquaporin directly determines necrotic cell death induced by the Bacillus thuringiensis cry toxin. FEBS Lett. 2017, 591, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souissi, W. Cytocidal Activity of Cry41Aa, an Anticancer Toxin from Bacillus thuringiensis. Ph.D. Thesis, University of Sussex, Falmer, UK, 2018. [Google Scholar]
- Odii, B.; Coussons, P. Pharmacological isolation of experimental models of drug-resistant hepatocellular carcinoma cell line. J. Cancer Ther. 2012, 3, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.; Chu, A.C.; Fung, K.P. Inhibition of P-glycoprotein expression and reversal of drug resistance of human hepatoma HepG2 cells by multidrug resistance gene (mdr1) antisense RNA. Life Sci. 2000, 67, 2117–2124. [Google Scholar] [CrossRef]
- Zhai, B.J.; Shao, Z.Y.; Zhao, C.L.; Hu, K.; Wu, F. Development and characterization of multidrug resistant human hepatocarcinoma cell line in nude mice. World J. Gastroenterol. 2006, 12, 6614–6619. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, K.; Zhang, D.; Gong, L.; He, F.; Soberón, M.; Bravo, A.; Tabashnik, B.E.; Wu, K. Resistance to Bacillus thuringiensis mediated by an ABC transporter mutation increases susceptibility to toxins from other bacteria in an invasive insect. PLoS Pathog. 2016, 12, e1005450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Ellar, D.J. Analysis of the large aqueous pores produced by a Bacillus thuringiensis protein insecticide in Manduca sexta midgut-brush-border-membrane vesicles. Eur. J. Biochem. 1997, 245, 797–804. [Google Scholar] [CrossRef]
- Peyronnet, O.; Nieman, B.; Généreux, F.; Vachon, V.; Laprade, R.; Schwartz, J.L. Estimation of the radius of the pores formed by the Bacillus thuringiensis Cry1C delta-endotoxin in planar lipid bilayers. Biochim. Biophys. Acta 2002, 1567, 113–122. [Google Scholar] [CrossRef]
- Cooper, S.T.; McNeil, P.L. Membrane repair: Mechanisms and pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los, F.C.O.; Kao, C.Y.; Smitham, J.; McDonald, K.L.; Ha, C.; Peixoto, C.A.; Aroian, R.V. RAB-5- and RAB-11-dependent vesicle-trafficking pathways are required for plasma membrane repair after attack by bacterial pore-forming toxin. Cell Host Microbe 2011, 9, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Chang, S.H.; Becker, E.B.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souissi, W.; Alistair, T.; Domanska, B.; Fortea, E.; West, M.J.; Schwartz, J.-L.; Crickmore, N. Probing the Mechanism of Action of Cry41Aa on HepG2 through the Establishment of a Resistant Subline. Toxins 2022, 14, 319. https://doi.org/10.3390/toxins14050319
Souissi W, Alistair T, Domanska B, Fortea E, West MJ, Schwartz J-L, Crickmore N. Probing the Mechanism of Action of Cry41Aa on HepG2 through the Establishment of a Resistant Subline. Toxins. 2022; 14(5):319. https://doi.org/10.3390/toxins14050319
Chicago/Turabian StyleSouissi, Wided, Tweedie Alistair, Barbara Domanska, Eva Fortea, Michelle J. West, Jean-Louis Schwartz, and Neil Crickmore. 2022. "Probing the Mechanism of Action of Cry41Aa on HepG2 through the Establishment of a Resistant Subline" Toxins 14, no. 5: 319. https://doi.org/10.3390/toxins14050319
APA StyleSouissi, W., Alistair, T., Domanska, B., Fortea, E., West, M. J., Schwartz, J.-L., & Crickmore, N. (2022). Probing the Mechanism of Action of Cry41Aa on HepG2 through the Establishment of a Resistant Subline. Toxins, 14(5), 319. https://doi.org/10.3390/toxins14050319