
The Potential Use of Isothermal Amplification Assays for In-Field Diagnostics of Plant Pathogens




Abstract
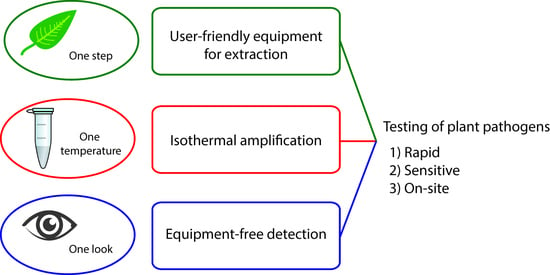
1. Introduction
2. Extraction of Pathogen Genome from Plants
2.1. Pre-Treatment of Plant Tissue for Nucleic Acid Extraction
2.2. Cell Disruption and Assessment of Inhibition Resistance for Isothermal Amplifications
2.3. Chemical Protocols for Homemade Extractions
2.4. Ready-to-Use Solutions (Commercial Kits) for Extraction
3. Isothermal Amplification
3.1. Recombinase Polymerase Amplification
3.2. Loop-Mediated Amplification
3.3. Rolling Circle Amplification
3.4. Nucleic Acid Sequence-Based Amplification
3.5. Helicase Dependent Amplification
4. Visualization of DNA Amplification Products
4.1. Coloration for Visual Detection and Fluorescence for UV Lamp Detection
4.1.1. Colorimetric Detection of RPA Amplicons
4.1.2. Colorimetric Detection of LAMP Amplicons
4.1.3. Colorimetric Detection of RCA Amplicon
4.2. Nanoparticle Aggregation
4.2.1. RPA Amplicon Detection with Nanoparticle Aggregation
4.2.2. RCA Amplicon Detection with Nanoparticle Aggregation
4.3. Lateral Flow Assays
4.3.1. RPA-Based Tests

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detected Target Specie, Gene | Host Organism | Detection Limit | Time of Detection, Min | Reference Method and Its Detection Limit | Method of RNA/DNA Extraction | Time of Extraction, Min | Ref |
|---|---|---|---|---|---|---|---|
| Potato virus X * (gp5 gene, 147 bp) | Potato leaves | 0.14 pg virus per gram of plant leaf (spiked samples) | 30 | RT–qPCR: 0.14 pg virus per gram of plant leaf | Syntol kit | 30 | [212] |
| Potato spindle tuber viroid * | Potato tuber | 106 copies of in vitro transcribed PSTV RNA, up to 107 dilution of infected plant | 30 | RT–qPCR: up to 107 dilution of infected plant | Syntol kit | 30 | [214] |
| Tomato spotted wilt virus ** (coat protein) | Pepper leaves | 10 fg/μL of transcribed TSWV RNA | 15 | RT–PCR: 10 fg/mcL of transcribed TSWV RNA | TRIzol (Thermo Fisher Scientific) extraction | 40 | [210] |
| Citrus tristeza virus * (coat protein) | Citrus aurantiifolia, C. sinensis, C. reticulata | For transcribed in vitro RNA: 141 fg (3.77 × 105 copies) For native RNA: 6.288 × 106 copies | 25 | RT–qPCR: for transcribed in vitro RNA: 141 fg (3.77 × 103 copies) | RNeasy Plant mini kit (Qiagen) | <20 | [211] |
| Milk vetch dwarf virus ** (coat protein) | Cowpea | Plasmid with cloned fragment of MDV spiked with crude extract: 10 copies/μL | 40 | RT–qPCR: plasmid with cloned fragment of MDV spiked with crude extract: 10 copies/mcL | E.Z.N.A.® Plant DNA Kit (Omega Bio-tek)/crude extraction | Approx. 40/<5 | [215] |
| Rice black-streaked dwarf virus * (P10 gene NC_003733.1, approx. 200 bp) | Rice leaves | 10-fold dilution of cDNA (Milenia test-strips) | 25 | RT–qPCR: 103 dilution of cDNA | RNAiso Plus Kit (TAKARA) | 60 | [227] |
| Hop stunt viroid 140 bp (101 labelled) | Leaves of hops, cucumbers, plums, grapes, and citrus | 2 × 109 copies transcript in crude extract (Agdia test-strips) | 40–50 | RT–PCR: 2 × 104 copies transcript in water | RNeasy Plant Mini Kit (QIAGEN), also crude homogenization | <20/<5 | [27] |
| Tomato chlorotic dwarf viroid * 228 bp (131 labelled) | Tomato seeds, leaves | 1 pg transcript, 1:25 dilution of leaf extract, 1:10 dilution of seed extract (Agdia test-strips) | 35 | RT–PCR: same | AmplifyRP® Acceler8™, crude extract | <5 | [216] |
| Plum pox virus * (coat protein, 147 bp) | Prunus leaves | 1 fg transcribed RNA, 1:10000 crude extract (Agdia test-strips, TNF probe) | 35 | Real-time RPA: 16 fg transcript. Real-time RT–PCR: 10 fg RNA | SurePrep™ Plant/FungiTotal RNA Purification Kit (Thermo Fisher Scientific)/crude plant extract | 30 | [218] |
| Little cherry virus 2 ** (coat protein, 134–295 bp) | Cherry budwood or leaf | Crude extract 1:100, 0.1 ng of pure total RNA (Agdia test-strips, nfo probe) | 25 | RT–PCR: crude extract 1:10K | RNeasy Plant Mini Kit/crude extract | <20/<5 | [92] |
| Dickeya solani * (SOL-C genomic region) | Potato tubers | 14000 CFU per gram of plant leaf (spiked samples) | 30 | qPCR | Syntol kit | 30 | [213] |
| Dickeya solani, D. chrysantemi, D. dianthicola, D. dadantii, D. paradisiaca, D. zeae–overall 34 strains (mglA/mglC genomic region) | Potato tubers, sweet potato tubers, taro corms | 1 CFU of D. dianthicola (purified bacteria or spiked samples) Real samples with other Dickeya species—pos/neg | 35 | PCR followed by sequencing (qualitative confirmation) | Wizard Genomic DNA Purification kit/crude extraction | 120 min | [51] |
| Genus Clavibacter and C. nebraskensis in particular * | Corn leaves | 3000 copies of genomic Clavibacter and 30 copies of genomic C. nebraskensis. 3000 copies of genomic Clavibacter and 300 copies of genomic C.nebraskensis in spiked samples | 35–40 | PCR for qualitative confirmation | Crude extract in TE buffer | 5–12 | [84] |
| Phytophthora hibernalis ** Ypt-1 gene, approx. 200 bp | Orange fruit crop | 0.2 ng (extracted from P. hibernalis), pos/neg for artificially inoculated plant (milenia test strips) | 25 | PCR: 2 ng extracted from P. hibernalis | DNAsecure Plant Kit (Tiangen Biotech) | 20 | [224] |
| Phytophthora sojae ** Ypt-1 gene, 217 bp | Soy seeds | 0.01 ng genomic DNA (milenia test strips) | 25 | LAMP: 0.1 ng genomic DNA [216] PCR: 1 ng genomic DNA [217] | DNAsecure Plant Kit (TIANGEN)/FastDNA SPIN Kit for Soil/NaOH lysis method [40] | 20/30/<10 | [225] |
| Phytophthora capsici Ypt-1 gene | Potato leaves | 10 pg genomic DNA, Pos/neg for infected plant (Milenia test strips) | 25/15 | LAMP: 100 pg genomic DNA, real-time qPCR: 100 fg | HP Fungal DNA Kit (Omega Bio-Tek)/Cellulose dipstick capture of DNA [49] | Approx. 25/<5 | [83] |
| Phytophthora infestans ** Ypt-1gene | Potato leaves | 500 fg of genomic DNA (approx. 2 genome copies) from the bacterial isolates Pos/neg for infected plants | 25–35 | Conventional PCR: 5 pg | PEG lysis | 5 | [208] |
| Phytophthora cactorum ** Ypt-1 gene | Strawberry leaves | 100 fg of genomic DNA | 35 | Conventional PCR: 1 pg | DNAsecure Plant Kit (Tiangen)/PEG lysis | 20 | [209] |
| Candidatus Liberibacter asiaticus ** 16S rRNA gene, 170 bp | Sweet orange fruit, acid lime leaves | <1 pg total DNA (with PCRD nucleic acid Detector and Agdia) | 30 | Real-time PCR: 10–100 fg of total DNA | DNeasy Plant mini kit/crude extraction | 20/<5 | [222] |
| Pectobacterium. Carotovorum * subsp. carotovorum; P. carotovorum subsp. odoriferum; P. carotovorum subsp. brasiliensis; P. atrosepticum; P. parmentieri | Tomato fruit, potato tuber | 10 fg DNA for either purified bacterial DNA or purified spiked samples (Milenia test-strips) | 35 | None | Wizard Genomic DNA Purification K/Plats with inoculated bacteria were homogenized in TE buffer | 120 | [228] |
| Gaeumannomyces avenae ** | Non-identified roots | 100 pg genomic DNA | 40 | LAMP: 1000 fg | Crude extract | <10 | [217] |
| Ophiosphaerella korrae ** | 100fg (Agdia test strips) | 1 fg | |||||
| Magnaporthiopsis poae ** | 1 fg | 100 fg | |||||
| Candidatus Phytoplasma oryzae ** imp gene KU820961 | Napier grass | 10–100 copies target DNA in water, pos/neg for plant extract (BioUSTAR test strips) | 25 | Real-time RPA: 1–10 copies of target DNA/PCR for plant extract | CTAB method/homemade homogenization method | Data not provided | [26] |
4.3.2. LAMP-Based Tests
| Detected Target | Host Organism | Detection Limit | Time of LAMP-LFA, Min | Reference Method | DNA Extraction | Time of Extraction | Ref |
|---|---|---|---|---|---|---|---|
| Cassava brown streak virus and Ugandan cassava brown streak virus ** (Coat protein) | Tobacco leaves | Pos/neg | 55 | Realtime PCR, LAMP, PCR | CTAB method | >8 h | [233] |
| Tobacco rattle virus and potato virus X ** | Potato | Positive/negative | 55 | RT-qLAMP–15 pg, RT-qPCR–15 pg, Pos/Neg tests: RT-PCR, RPA (TwistAmp Basic, AmplifyRP Acceler8 Discovery Kit), IsoAmp II Universal tHDA Kit (NEB), CRISDA | PureLink Plant RNA Reagent protocol/Modified PureLink Plant RNA Reagent protocol/Potato DNA/RNA rapid extraction set/InCus based on Monarch Total RNA Miniprep Kit (NEB)/crude extract | >60 min | [238] |
| Clavibacter michiganensis subsp. sepedonicus 16 S rDNA intergenic spacer region AF001266.1 | Potato tuber | Pos/neg test (validation of LAMP) | 70 | LAMP | SureFood PREP Advanced Kit (CONGEN) | 65 min | [235] |
| Leifsonia xyli subsp. Xyli ** ISLxx5 transposase gene NC_006087.1 | Sugarcane xylem sap and leaves | Pos/neg test, 1:5 diluted infected plant extract | 40 | LAMP | Homemade method | <20 min | [236] |
| Candidatus Liberibacter asiaticus ** | Sweet orange leaves, Diaphorina Citri fly | 10 pg purified DNA from infected plant | 45 | Real-time PCR: same | Wizard® Genomic DNA purification Kit (Promega) | Approx. 120 min | [197] |
| Xanthomonas citri ** Scheme of the complex looks like a padlock PthA4 gene XACb0065 | Lime leaves | 1 fg pure DNA, 5.2 CFU pure culture per reaction, 18.7 CFU from infected tissue per reaction | >30 | Conventional LAMP: same | Wizard® Genomic DNA purification Kit (Promega) | Approx. 120 min | [229] |
| Aspergillus fumigatus ** anxC4 gene | No plant objects were tested | 100 fg of genomic DNA | 52 | Culture method and PCR: 100% correlation with the LAMP | QIAamp DNA Mini Kit | 20 | [237] |
| Phytophthora ramorum, P. kernoviae ** ITS 1 region of the nuclear ribosomal (nr)RNA gene | Rhododendron leaves | 17 pg purified genomic DNA (from fungi culture) | >60 | LAMP: 10 pg [220] | NucleoSpin Plant kit (Machery-Nagel)/homemade method based on lateral flow dipstick | 35 min/<10 min | [230] |
| Aspergillus sp. ** aflR gene | Different herbal samples | 10 copies of the gene in buffer | 30 min | LAMP: the same PCR: 100 copies of the gene | The Wizard® Magnetic DNA Purification System for Food | <50 min | [239] |
4.3.3. NASBA-Based Tests
4.4. Lab-on-a-Chip
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hulme, P.E. Unwelcome exchange: International trade as a direct and indirect driver of biological invasions worldwide. One Earth 2021, 4, 666–679. [Google Scholar] [CrossRef]
- Liebhold, A.M.; Brockerhoff, E.G.; Garrett, L.J.; Parke, J.L.; Britton, K.O. Live plant imports: The major pathway for forest insect and pathogen invasions of the US. Front. Ecol. Environ. 2012, 10, 135–143. [Google Scholar] [CrossRef]
- Zayan, S.A. Impact of climate change on plant diseases and IPM strategies. In Plant Diseases—Current Threats and Management Trends; Topolovec-Pintarić, S., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Vurro, M.; Bonciani, B.; Vannacci, G. Emerging infectious diseases of crop plants in developing countries: Impact on agriculture and socio-economic consequences. Food Secur. 2010, 2, 20. [Google Scholar] [CrossRef]
- Savary, S.; Ficke, A.; Aubertot, J.N.; Hollier, C. Crop losses due to diseases and their implications for global food production losses and food security. Food Secur. 2012, 4, 19. [Google Scholar] [CrossRef]
- Bebber, D.P.; Holmes, T.; Smith, D.; Gurr, S.J. Economic and physical determinants of the global distributions of crop pests and pathogens. New Phytol. 2014, 202, 901–910. [Google Scholar] [CrossRef]
- National Academy of Sciences. Plant-Disease Development and Control; National Academy of Sciences: Washington, DC, USA, 1968; p. 205. [Google Scholar]
- International Standards for Phytosanitary Measures (ISPMs). Materials of Commission on Phytosanitary Measures (CPM) of International Plant Protection Convention (IPPC) of Food and Agriculture Organization of the United Nations, ISSN 2521-7232. 2021. Available online: https://www.ippc.int/en/core-activities/standards-setting/ispms/ (accessed on 2 November 2021).
- Legislation: Phytosanitary Requirements/Restrictions/Prohibitions: EU Plant Health Import Requirements International Plant Protection Convention (IPPC) of Food and Agriculture Organization of the United Nations. 2013. Available online: https://www.ippc.int/en/countries/european-union/reportingobligation/2013/06/eu-plant-health-import-requirements/ (accessed on 2 November 2021).
- Maloy, O.C. Plant Disease Control: Principles and Practice; John and Wiley and Sons: Hoboken, NJ, USA, 1993; p. 364. [Google Scholar]
- Buja, I.; Sabella, E.; Monteduro, A.G.; Chiriacò, M.S.; De Bellis, L.; Luvisi, A.; Maruccio, G. Advances in plant disease detection and monitoring: From traditional assays to in-field diagnostics. Sensors 2021, 21, 2129. [Google Scholar] [CrossRef]
- Hirooka, H.I. Chemical control of plant diseases. J. Gen. Plant Pathol. 2013, 79, 390–401. [Google Scholar] [CrossRef]
- Mueller, D.S.; Wise, K.A.; Dufault, N.S.; Bradley, C.A. Fungicides for Field Crops; APS Publications: St. Paul, MN, USA, 2017. [Google Scholar]
- Rosa, S.; Pesaresi, P.; Mizzotti, C.; Bulone, V.; Mezzetti, B.; Baraldi, E.; Masiero, S. Game-changing alternatives to conventional fungicides: Small RNAs and short peptides. Trends Biotechnol. 2021. [Google Scholar] [CrossRef]
- Jones, J.B.; Jackson, L.E.; Balogh, B.; Obradovic, A.; Iriarte, F.B.; Momol, M.T. Bacteriophages for plant disease control. Annu. Rev. Phytopathol. 2007, 45, 245–262. [Google Scholar] [CrossRef]
- Pal, K.K.; Gardener, M. Biological control of plant pathogens. Plant Health Instr. 2006, 2, 1117–1142. [Google Scholar] [CrossRef]
- Wang, M.R.; Chen, L.; Zhang, Z.; Blystad, D.R.; Wang, Q.C. Cryotherapy: A novel method for virus eradication in economically important plant species. Methods Mol. Biol. 2018, 1815, 257–268. [Google Scholar] [CrossRef]
- Dong, O.X.; Ronald, P.C. Genetic engineering for disease resistance in plants: Recent progress and future perspectives. Plant Physiol. 2019, 180, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Aglave, B. Handbook of Plant Disease Identification and Management; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Agrios, G.N. Plant Pathology, 5th ed.; Elsevier Academic Press: Cambridge, MA, USA, 2005; p. 922. [Google Scholar]
- Fang, Y.; Ramasamy, R.P. Current and prospective methods for plant disease detection. Biosensors 2015, 5, 537–561. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.M.; Bertolini, E.; Olmos, A.; Caruso, P.; Gorris, M.T.; Llop, P.; Penyalver, R.; Cambra, M. Innovative tools for detection of plant pathogenic viruses and bacteria. Int. Microbiol. 2003, 6, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Broccanello, C.; Chiodi, C.; Funk, A.; McGrath, J.M.; Panella, L.; Stevanato, P. Comparison of three PCR-based assays for SNP genotyping in plants. Plant Methods 2018, 14, 28. [Google Scholar] [CrossRef]
- Gill, P.; Ghaemi, A. Nucleic acid isothermal amplification technologies: A review. Nucleosides Nucleotides Nucleic Acids 2008, 27, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Longchar, B.; Phukan, T.; Yadav, S.; Senthil-Kumar, M. An efficient low-cost xylem sap isolation method for bacterial wilt assays in tomato. Appl. Plant Sci. 2020, 8, e11335. [Google Scholar] [CrossRef] [PubMed]
- Wambua, L.; Schneider, B.; Okwaro, A.; Wanga, J.O.; Imali, O.; Wambua, P.N.; Agutu, L.; Olds, C.; Jones, C.S.; Masiga, D.; et al. Development of field-applicable tests for rapid and sensitive detection of Candidatus Phytoplasma oryzae. Mol. Cell. Probes 2017, 35, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Kappagantu, M.; Villamor, D.E.V.; Bullock, J.M.; Eastwell, K.C. A rapid isothermal assay for the detection of Hop stunt viroid in hop plants (Humulus lupulus), and its application in disease surveys. J. Virol. Methods 2017, 245, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Till, B.J.; Jankowicz-Cieslak, J.; Huynh, O.A.; Beshir, M.M.; Laport, R.G.; Hofinger, B.J. Low-cost DNA extraction. In Low-Cost Methods for Molecular Characterization of Mutant Plants: Tissue Desiccation, DNA Extraction and Mutation Discovery: Protocols; Springer International Publishing: Cham, Switzerland, 2015; pp. 13–17. [Google Scholar]
- Skubel, S.A.; Dushenkov, V.; Graf, B.L.; Niu, Q.; Poulev, A.; Kalariya, H.M.; Foxcroft, L.C.; Raskin, I. Rapid, field-deployable method for collecting and preserving plant metabolome for biochemical and functional characterization. PLoS ONE 2018, 13, e0203569. [Google Scholar] [CrossRef] [PubMed]
- Paranaiba, R.T.F.; Carvalho, C.B.V.; Paiva, R.S.; Trindade, B.R.; Barros, M.G.; Souza, E.P.; Gontijo, A.B.; Silveira, D. DNA from wood—A simple approach facing a challenging matrix—A preliminary study. Forensic Sci. Int. 2020, 314, 110371. [Google Scholar] [CrossRef] [PubMed]
- Rachmayanti, Y.; Leinemann, L.; Gailing, O.; Finkeldey, R. DNA from processed and unprocessed wood: Factors influencing the isolation success. Forensic Sci. Int. Genet. 2009, 3, 185–192. [Google Scholar] [CrossRef]
- Reynolds, M.M.; Williams, C.G. Extracting DNA from submerged pine wood. Genome 2004, 47, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Siregar, I.Z.; Ramdhani, M.J.; Karlinasari, L.; Adzkia, U.; Arifin, M.Z.; Dwiyanti, F.G. DNA isolation success rates from dried and fresh wood samples of selected 20 tropical wood tree species for possible consideration in forensic forestry. Sci. Justice 2021, 61, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.L.; Workman, J.N.; Nairn, C.J.; Fraedrich, S.W.; Villari, C. Rapid detection of raffaelea lauricola directly from host plant and beetle vector tissues using loop-mediated isothermal amplification. Plant Dis. 2020, 104, 3151–3158. [Google Scholar] [CrossRef] [PubMed]
- Rachmayanti, Y.; Leinemann, L.; Gailing, O.; Finkeldey, R. Extraction, amplification and characterization of wood DNA from dipterocarpaceae. Plant Mol. Biol. Report. 2006, 24, 45–55. [Google Scholar] [CrossRef]
- Green, M.J.; Thompson, D.A.; MacKenzie, D.J. Easy and efficient DNA extraction from woody plants for the detection of phytoplasmas by polymerase chain reaction. Plant Dis. 1999, 83, 482–485. [Google Scholar] [CrossRef]
- Yin, C.; Wang, Y.; Zhang, Y.; Wang, H.; Tao, R.; Li, Y.; Sung, C.K. A pine wood sample preparation method for high target and quality DNA extraction for detection of Esteya vermicola by PCR from living pine. J. Basic Microbiol. 2019, 59, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Fatima, T.; Srivastava, A.; Hanur, V.S.; Rao, A.M.S. An effective wood DNA extraction protocol for three economic important timber species of India. Am. J. Plant Sci. 2018, 9, 139–149. [Google Scholar] [CrossRef][Green Version]
- Rizzo, D.; Da Lio, D.; Bartolini, L.; Salemi, C.; Del Nista, D.; Aronadio, A.; Pennacchio, F.; Binazzi, F.; Francardi, V.; Garonna, A.P.; et al. TaqMan probe assays on different biological samples for the identification of three ambrosia beetle species, Xylosandrus compactus (Eichoff), X. crassiusculus (Motschulsky) and X. germanus (Blandford) (Coleoptera Curculionidae Scolytinae). 3 Biotech 2021, 11, 259. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, D.; Luchi, N.; Da Lio, D.; Bartolini, L.; Nugnes, F.; Cappellini, G.; Bruscoli, T.; Salemi, C.; Griffo, R.V.; Garonna, A.P.; et al. Development of a loop-mediated isothermal amplification (LAMP) assay for the identification of the invasive wood borer Aromia bungii (Coleoptera: Cerambycidae) from frass. 3 Biotech 2021, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.O.; Bendich, A.J. Extraction of DNA from plant tissues. In Plant Molecular Biology Manual; Gelvin, S.B., Schilperoort, R.A., Verma, D.P.S., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1988; pp. 73–83. [Google Scholar]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors—Occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- John, M.E. An efficient method for isolation of RNA and DNA from plants containing polyphenolics. Nucleic Acids Res. 1992, 20, 2381. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Kawana, T.; Fukushima, E.; Suzutani, T. Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J. Biochem. Biophys. Methods 2007, 70, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Francois, P.; Tangomo, M.; Hibbs, J.; Bonetti, E.J.; Boehme, C.C.; Notomi, T.; Perkins, M.D.; Schrenzel, J. Robustness of a loop-mediated isothermal amplification reaction for diagnostic applications. FEMS Immunol. Med. Microbiol. 2011, 62, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kiddle, G.; Hardinge, P.; Buttigieg, N.; Gandelman, O.; Pereira, C.; McElgunn, C.J.; Rizzoli, M.; Jackson, R.; Appleton, N.; Moore, C.; et al. GMO detection using a bioluminescent real time reporter (BART) of loop mediated isothermal amplification (LAMP) suitable for field use. BMC Biotechnol. 2012, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Hadersdorfer, J.; Neumuller, M.; Treutter, D.; Fischer, T.C. Fast and reliable detection of Plum pox virus in woody host plants using the Blue LAMP protocol. Ann. Appl. Biol. 2011, 159, 456–466. [Google Scholar] [CrossRef]
- Elvira-Gonzalez, L.; Puchades, A.V.; Carpino, C.; Alfaro-Fernandez, A.; Font-San-Ambrosio, M.I.; Rubio, L.; Galipienso, L. Fast detection of Southern tomato virus by one-step transcription loop-mediated isothermal amplification (RT-LAMP). J. Virol. Methods 2017, 241, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Wilisiani, F.; Tomiyama, A.; Katoh, H.; Hartono, S.; Neriya, Y.; Nishigawa, H.; Natsuaki, T. Development of a LAMP assay with a portable device for real-time detection of begomoviruses under field conditions. J. Virol. Methods 2019, 265, 71–76. [Google Scholar] [CrossRef]
- Chandu, D.; Paul, S.; Parker, M.; Dudin, Y.; King-Sitzes, J.; Perez, T.; Mittanck, D.W.; Shah, M.; Glenn, K.C.; Piepenburg, O. Development of a rapid point-of-use DNA test for the screening of genuity(R) roundup ready 2 yield(R) soybean in seed samples. BioMed Res. Int. 2016, 2016, 3145921. [Google Scholar] [CrossRef] [PubMed]
- Boluk, G.; Dobhal, S.; Crockford, A.B.; Melzer, M.; Alvarez, A.M.; Arif, M. Genome-informed recombinase polymerase amplification assay coupled with a lateral flow device for in-field detection of dickeya species. Plant Dis. 2020, 104, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Srivastava, N.; Kumar, S.; Saritha, R.K.; Sharma, S.K.; Jain, R.K.; Baranwal, V.K. Development of a recombinase polymerase amplification assay for the diagnosis of banana bunchy top virus in different banana cultivars. Arch. Virol. 2017, 162, 2791–2796. [Google Scholar] [CrossRef] [PubMed]
- Brink, A.A.; Vervoort, M.B.; Middeldorp, J.M.; Meijer, C.J.; Van den Brule, A.J. Nucleic acid sequence-based amplification, a new method for analysis of spliced and unspliced Epstein-Barr virus latent transcripts, and its comparison with reverse transcriptase PCR. J. Clin. Microbiol. 1998, 36, 3164–3169. [Google Scholar] [CrossRef]
- Morre, S.A.; Sillekens, P.; Jacobs, M.V.; Van Aarle, P.; De Blok, S.; Van Gemen, B.; Walboomers, J.M.; Meijer, C.J.; Van den Brule, A.J. RNA amplification by nucleic acid sequence-based amplification with an internal standard enables reliable detection of Chlamydia trachomatis in cervical scrapings and urine samples. J. Clin. Microbiol. 1996, 34, 3108–3114. [Google Scholar] [CrossRef]
- Clancy, E.; Higgins, O.; Forrest, M.S.; Boo, T.W.; Cormican, M.; Barry, T.; Piepenburg, O.; Smith, T.J. Development of a rapid recombinase polymerase amplification assay for the detection of Streptococcus pneumoniae in whole blood. BMC Infect. Dis. 2015, 15, 481. [Google Scholar] [CrossRef]
- Rohrman, B.; Richards-Kortum, R. Inhibition of recombinase polymerase amplification by background DNA: A lateral flow-based method for enriching target DNA. Anal. Chem. 2015, 87, 1963–1967. [Google Scholar] [CrossRef] [PubMed]
- Birch, L.; Dawson, C.E.; Cornett, J.H.; Keer, J.T. A comparison of nucleic acid amplification techniques for the assessment of bacterial viability. Lett. Appl. Microbiol. 2001, 33, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Doseeva, V.; Forbes, T.; Wolff, J.; Khripin, Y.; O’Neil, D.; Rothmann, T.; Nazarenko, I. Multiplex isothermal helicase-dependent amplification assay for detection of Chlamydia trachomatis and Neisseria gonorrhoeae. Diagn. Microbiol. Infect. Dis. 2011, 71, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic. Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef]
- Allen, G.C.; Flores-Vergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Tussell, R.; Quijano-Ramayo, A.; Rojas-Herrera, R.; Larque-Saavedra, A.; Perez-Brito, D. A fast, simple, and reliable high-yielding method for DNA extraction from different plant species. Mol. Biotechnol. 2005, 31, 137–139. [Google Scholar] [CrossRef]
- White, E.J.; Venter, M.; Hiten, N.F.; Burger, J.T. Modified cetyltrimethylammonium bromide method improves robustness and versatility: The benchmark for plant RNA extraction. Biotechnol. J. 2008, 3, 1424–1428. [Google Scholar] [CrossRef]
- Gambino, G.; Perrone, I.; Gribaudo, I. A Rapid and effective method for RNA extraction from different tissues of grapevine and other woody plants. Phytochem. Anal. 2008, 19, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Mock, R.; Huang, Q.; Abad, J.; Hartung, J.; Kinard, G. A reliable and inexpensive method of nucleic acid extraction for the PCR-based detection of diverse plant pathogens. J. Virol. Methods 2008, 154, 48–55. [Google Scholar] [CrossRef]
- Logemann, J.; Schell, J.; Willmitzer, L. Improved method for the isolation of RNA from plant tissues. Anal. Biochem. 1987, 163, 16–20. [Google Scholar] [CrossRef]
- Rudbeck, L.; Dissing, J. Rapid, simple alkaline extraction of human genomic DNA from whole blood, buccal epithelial cells, semen and forensic stains for PCR. Biotechniques 1998, 25, 588–590. [Google Scholar] [CrossRef]
- Wang, H.; Qi, M.; Cutler, A.J. A simple method of preparing plant samples for PCR. Nucleic. Acids Res. 1993, 21, 4153–4154. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Bae, S.; Lee, S.; Lee, Y.; Chang, A. A rapid and simple genotyping method for various plants by direct-PCR. Plant Breed. Biotechnol. 2013, 1, 290–297. [Google Scholar] [CrossRef]
- MacKenzie, D.J.; McLean, M.A.; Mukerji, S.; Green, M. Improved RNA extraction from woody plants for the detection of viral pathogens by reverse transcription-polymerase chain reaction. Plant Dis. 1997, 81, 222–226. [Google Scholar] [CrossRef]
- Marengo, A.; Cagliero, C.; Sgorbini, B.; Anderson, J.L.; Emaus, M.N.; Bicchi, C.; Bertea, C.M.; Rubiolo, P. Development of an innovative and sustainable one-step method for rapid plant DNA isolation for targeted PCR using magnetic ionic liquids. Plant Methods 2019, 15, 23. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Uyemoto, J.K.; Kirkpatrick, B.C. A small-scale procedure for extracting nucleic acids from woody plants infected with various phytopathogens for PCR assay. J. Virol. Methods 1998, 71, 45–50. [Google Scholar] [CrossRef]
- Valasevich, N.; Schneider, B. Rapid detection of “Candidatus Phytoplasma mali” by recombinase polymerase amplification assays. J. Phytopathol. 2017, 165, 762–770. [Google Scholar] [CrossRef]
- Kersting, S.; Rausch, V.; Bier, F.F.; Von Nickisch-Rosenegk, M. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar. J. 2014, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Rabe, B.A.; Cepko, C. SARS-CoV-2 detection using isothermal amplification and a rapid, inexpensive protocol for sample inactivation and purification. Proc. Natl. Acad. Sci. USA 2020, 117, 24450–24458. [Google Scholar] [CrossRef] [PubMed]
- Drygin, Y.F.; Butenko, K.O.; Gasanova, T.V. Environmentally friendly method of RNA isolation. Anal. Biochem. 2021, 620, 114113. [Google Scholar] [CrossRef]
- Tomlinson, J.A.; Boonham, N.; Dickinson, M. Development and evaluation of a one-hour DNA extraction and loop-mediated isothermal amplification assay for rapid detection of phytoplasmas. Plant Pathol. 2010, 59, 465–471. [Google Scholar] [CrossRef]
- Zou, Y.; Mason, M.G.; Wang, Y.; Wee, E.; Turni, C.; Blackall, P.J.; Trau, M.; Botella, J.R. Nucleic acid purification from plants, animals and microbes in under 30 seconds. PLoS Biol. 2017, 15, e2003916. [Google Scholar] [CrossRef] [PubMed]
- Abu Almakarem, A.S.; Heilman, K.L.; Conger, H.L.; Shtarkman, Y.M.; Rogers, S.O. Extraction of DNA from plant and fungus tissues in situ. BMC Res. Notes 2012, 5, 266. [Google Scholar] [CrossRef] [PubMed]
- Baden, T.; Chagas, A.M.; Gage, G.J.; Marzullo, T.C.; Prieto-Godino, L.L.; Euler, T. Open Labware: 3-D printing your own lab equipment. PLoS Biol. 2015, 13, e1002086. [Google Scholar] [CrossRef] [PubMed]
- Byagathvalli, G.; Pomerantz, A.; Sinha, S.; Standeven, J.; Bhamla, M.S. A 3D-printed hand-powered centrifuge for molecular biology. PLoS Biol. 2019, 17, e3000251. [Google Scholar] [CrossRef] [PubMed]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ju, Y.; Wu, X.; Shen, P.; Cao, L.; Zhou, B.; Yan, X.; Pan, Y. Development of recombinase polymerase amplification combined with lateral flow detection assay for rapid and visual detection of Ralstonia solanacearum in tobacco. Plant Dis. 2021. [Google Scholar] [CrossRef]
- Yu, J.; Shen, D.; Dai, T.; Lu, X.; Xu, H.; Dou, D. Rapid and equipment-free detection of Phytophthora capsici using lateral flow strip-based recombinase polymerase amplification assay. Lett. Appl. Microbiol. 2019, 69, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Larrea-Sarmiento, A.; Stack, J.P.; Alvarez, A.M.; Arif, M. Multiplex recombinase polymerase amplification assay developed using unique genomic regions for rapid on-site detection of genus Clavibacter and C. nebraskensis. Sci. Rep. 2021, 11, 12017. [Google Scholar] [CrossRef] [PubMed]
- Crannell, Z.A.; Rohrman, B.; Richards-Kortum, R. Equipment-free incubation of recombinase polymerase amplification reactions using body heat. PLoS ONE 2014, 9, e112146. [Google Scholar] [CrossRef]
- Liu, L.; Wang, J.; Geng, Y.; Wang, J.; Li, R.; Shi, R.; Yuan, W. Equipment-free recombinase polymerase amplification assay using body heat for visual and rapid point-of-need detection of canine parvovirus 2. Mol. Cell. Probes 2018, 39, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Safenkova, I.V.; Ivanov, A.V.; Slutskaya, E.S.; Samokhvalov, A.V.; Zherdev, A.V.; Dzantiev, B.B. Key significance of DNA-target size in lateral flow assay coupled with recombinase polymerase amplification. Anal. Chim. Acta 2020, 1102, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Boyle, D.S.; Lehman, D.A.; Lillis, L.; Peterson, D.; Singhal, M.; Armes, N.; Parker, M.; Piepenburg, O.; Overbaugh, J. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. mBio 2013, 4, e00135-13. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Boudreau, D.K.; Bergeron, M.G. Influence of sequence mismatches on the specificity of recombinase polymerase amplification technology. Mol. Cell. Probes 2015, 29, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Lillis, L.; Siverson, J.; Lee, A.; Cantera, J.; Parker, M.; Piepenburg, O.; Lehman, D.A.; Boyle, D.S. Factors influencing Recombinase polymerase amplification (RPA) assay outcomes at point of care. Mol. Cell. Probes 2016, 30, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Euler, M.; Wang, Y.; Nentwich, O.; Piepenburg, O.; Hufert, F.T.; Weidmann, M. Recombinase polymerase amplification assay for rapid detection of Rift Valley fever virus. J. Clin. Virol. 2012, 54, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Mekuria, T.A.; Zhang, S.; Eastwell, K.C. Rapid and sensitive detection of Little cherry virus 2 using isothermal reverse transcription-recombinase polymerase amplification. J. Virol. Methods 2014, 205, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic. Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, K.; Hase, T.; Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probes 2002, 16, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Parida, M.; Posadas, G.; Inoue, S.; Hasebe, F.; Morita, K. Real-time reverse transcription loop-mediated isothermal amplification for rapid detection of West Nile virus. J. Clin. Microbiol. 2004, 42, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Rolando, J.C.; Jue, E.; Barlow, J.T.; Ismagilov, R.F. Real-time kinetics and high-resolution melt curves in single-molecule digital LAMP to differentiate and study specific and non-specific amplification. Nucleic. Acids Res. 2020, 48, e42. [Google Scholar] [CrossRef] [PubMed]
- Tanner, N.A.; Zhang, Y.; Evans, T.C., Jr. Simultaneous multiple target detection in real-time loop-mediated isothermal amplification. Biotechniques 2012, 53, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Becherer, L.; Borst, N.; Bakheit, M.; Frischmann, S.; Zengerle, R.; von Stetten, F. Loop-mediated isothermal amplification (LAMP)—Review and classification of methods for sequencespecific detection. Anal. Methods 2020, 12, 30. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.Q. Rolling replication of short DNA circles. Proc. Natl. Acad. Sci. USA 1995, 92, 4641–4645. [Google Scholar] [CrossRef] [PubMed]
- Dean, F.B.; Nelson, J.R.; Giesler, T.L.; Lasken, R.S. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001, 11, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Li, F.; Zhang, Z.; Zhang, K.; Kang, D.K.; Ankrum, J.A.; Le, X.C.; Zhao, W. Rolling circle amplification: A versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev. 2014, 43, 3324–3341. [Google Scholar] [CrossRef] [PubMed]
- Jeske, H. Barcoding of plant viruses with circular single-stranded DNA based on rolling circle amplification. Viruses 2018, 10, 469. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.; Habekuss, A.; Kazmaier, K.; Jeske, H. Surveying cereal-infecting geminiviruses in Germany—Diagnostics and direct sequencing using rolling circle amplification. Virus Res. 2007, 127, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Compton, J. Nucleic acid sequence-based amplification. Nature 1991, 350, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Deiman, B.; Van Aarle, P.; Sillekens, P. Characteristics and applications of nucleic acid sequence-based amplification (NASBA). Mol. Biotechnol. 2002, 20, 163–179. [Google Scholar] [CrossRef]
- Szemes, M.; Klerks, M.M.; van den Heuvel, J.F.; Schoen, C.D. Development of a multiplex AmpliDet RNA assay for simultaneous detection and typing of potato virus Y isolates. J. Virol. Methods 2002, 100, 83–96. [Google Scholar] [CrossRef]
- Scuderi, G.; Golmohammadi, M.; Cubero, J.; Lopez, M.M.; Cirvilleri, G.; Llop, P. Development of a simplified NASBA protocol for detecting viable cells of the citrus pathogen Xanthomonas citri subsp citri under different treatments. Plant Pathol. 2010, 59, 764–772. [Google Scholar] [CrossRef]
- Gonccalves, M.C.; Klerks, M.M.; Verbeek, M.; Vega, J.; Van den Heuvel, J.F.J.M. The use of molecular beacons combined with NASBA for the sensitive detection of Sugarcane yellow leaf virus. Eur. J. Plant. Pathol. 2002, 108, 401–407. [Google Scholar] [CrossRef]
- Heo, S.; Kim, H.R.; Lee, H.J. Development of a Quantitative Real-time Nucleic Acid Sequence based Amplification (NASBA) Assay for Early Detection of Apple scar skin viroid. Plant Pathol. J. 2019, 35, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, D.; Spak, J.; Klerks, M.M.; Schoen, C.D.; Thompson, J.R.; Jelkmann, W. Real-time NASBA for detection of Strawberry vein banding virus. Eur. J. Plant. Pathol. 2004, 110, 213–221. [Google Scholar] [CrossRef]
- Szemes, M.; Schoen, C.D. Design of molecular beacons for AmpliDet RNA assay—Characterization of binding stability and probe specificity. Anal. Biochem. 2003, 315, 189–201. [Google Scholar] [CrossRef]
- Klerks, M.M.; Leone, G.; Lindner, J.L.; Schoen, C.D.; Van den Heuvel, J.F. Rapid and Sensitive Detection of Apple stem pitting virus in Apple Trees Through RNA Amplification and Probing with Fluorescent Molecular Beacons. Phytopathology 2001, 91, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Xu, Y.; Kong, H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004, 5, 795–800. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Tang, W.; Ranalli, T.A.; Kim, H.J.; Wytiaz, J.; Kong, H. Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J. Biol. Chem. 2005, 280, 28952–28958. [Google Scholar] [CrossRef] [PubMed]
- Artiushin, S.; Tong, Y.; Timoney, J.; Lemieux, B.; Schlegel, A.; Kong, H. Thermophilic helicase-dependent DNA amplification using the IsoAmp SE experimental kit for rapid detection of Streptococcus equi subspecies equi in clinical samples. J. Vet. Diagn. Invest. 2011, 23, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Du, X.J.; Zhou, T.J.; Li, P.; Wang, S. A rapid Salmonella detection method involving thermophilic helicase-dependent amplification and a lateral flow assay. Mol. Cell. Probes 2017, 34, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Horst, A.L.; Rosenbohm, J.M.; Kolluri, N.; Hardick, J.; Gaydos, C.A.; Cabodi, M.; Klapperich, C.M.; Linnes, J.C. A paperfluidic platform to detect Neisseria gonorrhoeae in clinical samples. Biomed. Microdevices 2018, 20, 35. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Yang, H.; Gong, Y.; You, M.; Liu, Z.; Choi, J.R.; Wen, T.; Qu, Z.; Mei, Q.; Xu, F. A fully disposable and integrated paper-based device for nucleic acid extraction, amplification and detection. Lab Chip 2017, 17, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Alvandi, A.H.; Abdul-Tehrani, H.; Sadeghizadeh, M. Colorimetric detection of Helicobacter pylori DNA using isothermal helicase-dependent amplification and gold nanoparticle probes. Diagn. Microbiol. Infect. Dis. 2008, 62, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Schwenkbier, L.; Pollok, S.; Rudloff, A.; Sailer, S.; Cialla-May, D.; Weber, K.; Popp, J. Non-instrumented DNA isolation, amplification and microarray-based hybridization for a rapid on-site detection of devastating Phytophthora kernoviae. Analyst 2015, 140, 6610–6618. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Aguilar-Moreno, G.S.; Wayadande, A.; Fletcher, J.; Ochoa-Corona, F.M. Primer modification improves rapid and sensitive in vitro and field-deployable assays for detection of high plains virus variants. Appl. Environ. Microbiol. 2014, 80, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, L.; Hong, S.; Sun, Q.; Yao, W.; Pei, R. Label-free DNA-based biosensors using structure-selective light-up dyes. Analyst 2016, 141, 6481–6489. [Google Scholar] [CrossRef]
- Dragan, A.I.; Pavlovic, R.; McGivney, J.B.; Casas-Finet, J.R.; Bishop, E.S.; Strouse, R.J.; Schenerman, M.A.; Geddes, C.D. SYBR Green I: Fluorescence properties and interaction with DNA. J. Fluoresc. 2012, 22, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Karsai, A.; Muller, S.; Platz, S.; Hauser, M.T. Evaluation of a homemade SYBR green I reaction mixture for real-time PCR quantification of gene expression. Biotechniques 2002, 32, 790–796. [Google Scholar] [CrossRef]
- Abbasi, I.; Kirstein, O.D.; Hailu, A.; Warburg, A. Optimization of loop-mediated isothermal amplification (LAMP) assays for the detection of Leishmania DNA in human blood samples. Acta Trop. 2016, 162, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Monis, P.T.; Giglio, S.; Saint, C.P. Comparison of SYTO9 and SYBR Green I for real-time polymerase chain reaction and investigation of the effect of dye concentration on amplification and DNA melting curve analysis. Anal. Biochem. 2005, 340, 24–34. [Google Scholar] [CrossRef]
- Jiang, H.X.; Zhao, M.Y.; Niu, C.D.; Kong, D.M. Real-time monitoring of rolling circle amplification using aggregation-induced emission: Applications in biological detection. Chem. Commun. 2015, 51, 16518–16521. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.H.; Zhong, H.J.; Lu, L.; Chan, D.S.; Ma, D.L. Luminescent and colorimetric strategies for the label-free DNA-based detection of enzyme activity. Brief. Funct. Genom. 2013, 12, 525–535. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, J.; Li, K.; Tian, H.; Xu, W. Label-free visual biosensor based on cascade amplification for the detection of Salmonella. Anal. Chim. Acta 2019, 1075, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Moriam, S.; Umer, M.; Phan, H.P.; Salomon, C.; Kline, R.; Nguyen, N.T.; Shiddiky, M.J.A. Naked-eye and electrochemical detection of isothermally amplified HOTAIR long non-coding RNA. Analyst 2018, 143, 3021–3028. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.; Miller, N.S.; Smolina, I. Visual detection of bacterial pathogens via PNA-based padlock probe assembly and isothermal amplification of DNAzymes. Anal. Chem. 2014, 86, 11992–11998. [Google Scholar] [CrossRef]
- Lu, X.; Shi, X.; Wu, G.; Wu, T.; Qin, R.; Wang, Y. Visual detection and differentiation of Classic Swine Fever Virus strains using nucleic acid sequence-based amplification (NASBA) and G-quadruplex DNAzyme assay. Sci. Rep. 2017, 7, 44211. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Honda, E.; Ogura, A.; Nomoto, A.; Hanaki, K. Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. Biotechniques 2009, 46, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Nagamine, K.; Tomita, N.; Notomi, T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 2001, 289, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Tanner, N.A.; Zhang, Y.; Evans, T.C., Jr. Visual detection of isothermal nucleic acid amplification using pH-sensitive dyes. Biotechniques 2015, 58, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Almassian, D.R.; Cockrell, L.M.; Nelson, W.M. Portable nucleic acid thermocyclers. Chem. Soc. Rev. 2013, 42, 8769–8798. [Google Scholar] [CrossRef] [PubMed]
- Cha, D.; Kim, D.; Choi, W.; Park, S.; Han, H. Point-of-care diagnostic (POCD) method for detecting Bursaphelenchus xylophilus in pinewood using recombinase polymerase amplification (RPA) with the portable optical isothermal device (POID). PLoS ONE 2020, 15, e0227476. [Google Scholar] [CrossRef] [PubMed]
- Chiriaco, M.S.; Luvisi, A.; Primiceri, E.; Sabella, E.; De Bellis, L.; Maruccio, G. Development of a lab-on-a-chip method for rapid assay of Xylella fastidiosa subsp. pauca strain CoDiRO. Sci. Rep. 2018, 8, 7376. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, T.; Drago, S.; Valentini, F.; Elbeaino, T.; Stampone, G.; Digiaro, M.; D’onghia, A.M.A. On-site detection of Xylella fastidiosa in host plants and in ‘spy insects’ using the real-time loop-mediated isothermal amplification method. Phytopathol. Mediterr. 2015, 54, 488–496. [Google Scholar] [CrossRef]
- Ravindran, A.; Levy, J.; Pierson, E.; Gross, D.C. Loop-mediated isothermal amplification procedure (LAMP) for detection of the potato zebra chip pathogen “Candidatus Liberibacter solanacearum”. Methods Mol. Biol. 2015, 1302, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, S.Y.; Back, C.G.; Ten, L.N.; Jung, H.Y. Loop-Mediated Isothermal Amplification for the Detection of Xanthomonas arboricola pv. pruni in Peaches. Plant Pathol. J. 2019, 35, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Ocenar, J.; Arizala, D.; Boluk, G.; Dhakal, U.; Gunarathne, S.; Paudel, S.; Dobhal, S.; Arif, M. Development of a robust, field-deployable loop-mediated isothermal amplification (LAMP) assay for specific detection of potato pathogen Dickeya dianthicola targeting a unique genomic region. PLoS ONE 2019, 14, e0218868. [Google Scholar] [CrossRef] [PubMed]
- Dobhal, S.; Larrea-Sarmiento, A.; Alvarez, A.M.; Arif, M. Development of a loop-mediated isothermal amplification assay for specific detection of all known subspecies of Clavibacter michiganensis. J. Appl. Microbiol. 2019, 126, 388–401. [Google Scholar] [CrossRef]
- Verma, G.; Sharma, S.; Raigond, B.; Pathania, S.; Naga, K.; Chakrabarti, S.K. Development and application of fluorescent loop mediated isothermal amplification technique to detect Phytophthora infestans from potato tubers targeting ITS-1 region. 3 Biotech 2019, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, L.; Li, H.; Gao, Y.; Mu, W.; Liu, F. Development of a LAMP method for detecting the N75S mutant in SDHI-resistant Corynespora cassiicola. Anal. Biochem. 2020, 597, 113687. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, H.; Moradi, A.; Hamedi, J.; Basiri, M. Development of a loop-mediated isothermal amplification assay for rapid and specific identification of ACT producing alternaria alternata, the agent of brown spot disease in tangerine. Appl. Biochem. Biotechnol. 2016, 178, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Al-Sheikh, H.M. LAMP-PCR detection of ochratoxigenic Aspergillus species collected from peanut kernel. Genet. Mol. Res. 2015, 14, 634–644. [Google Scholar] [CrossRef]
- Chandra, A.; Keizerweerd, A.T.; Que, Y.; Grisham, M.P. Loop-mediated isothermal amplification (LAMP) based detection of Colletotrichum falcatum causing red rot in sugarcane. Mol. Biol. Rep. 2015, 42, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Niessen, L.; Vogel, R.F. Detection of Fusarium graminearum DNA using a loop-mediated isothermal amplification (LAMP) assay. Int. J. Food Microbiol. 2010, 140, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jung, Y.; Kil, E.J.; Kim, J.; Thi Tran, D.; Choi, S.K.; Yoon, J.Y.; Cho, W.K.; Lee, S. Loop-mediated isothermal amplification for the rapid detection of Chrysanthemum chlorotic mottle viroid (CChMVd). J. Virol. Methods 2013, 193, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Thanarajoo, S.S.; Kong, L.L.; Kadir, J.; Lau, W.H.; Vadamalai, G. Detection of Coconut cadang-cadang viroid (CCCVd) in oil palm by reverse transcription loop-mediated isothermal amplification (RT-LAMP). J. Virol. Methods 2014, 202, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Panno, S.; Matic, S.; Tiberini, A.; Caruso, A.G.; Bella, P.; Torta, L.; Stassi, R.; Davino, A.S. Loop mediated isothermal amplification: Principles and applications in plant virology. Plants 2020, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, Z.; Fletcher, J.D.; Wang, Y.; Wang, R.; Guo, Z.; He, Y. Rapid and sensitive detection of lettuce necrotic yellows virus and cucumber mosaic virus infecting lettuce (lactuca sativa L.) by reverse transcription loop-mediated isothermal amplification. Plant Pathol. J. 2020, 36, 76–86. [Google Scholar] [CrossRef]
- Choi, C.W.; Hyun, J.W.; Hwang, R.Y.; Powell, C.A. Loop-mediated isothermal amplification assay for detection of candidatus liberibacter asiaticus, a causal agent of citrus huanglongbing. Plant Pathol. J. 2018, 34, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Xie, Z.; Wang, R.; Guo, Z.; He, Y. Rapid Detection of Lily mottle virus and Arabis mosaic virus infecting Lily (Lilium spp.) using reverse transcription loop-mediated isothermal amplification. Plant Pathol. J. 2020, 36, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, S.; Wu, Z.; Ling, K.S. Reverse transcription loop-mediated isothermal amplification for species-specific detection of tomato chlorotic spot orthotospovirus. J. Virol. Methods 2018, 253, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Neubauer, J.; Spanner, R.; Natwick, M.; Rios, J.; Metz, N.; Secor, G.A.; Bolton, M.D. Rapid detection of cercospora beticola in sugar beet and mutations associated with fungicide resistance using LAMP or probe-based qPCR. Plant Dis. 2020, 104, 1654–1661. [Google Scholar] [CrossRef]
- Romero, J.L.; Carver, G.D.; Arce Johnson, P.; Perry, K.L.; Thompson, J.R. A rapid, sensitive and inexpensive method for detection of grapevine red blotch virus without tissue extraction using loop-mediated isothermal amplification. Arch. Virol. 2019, 164, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Sarkes, A.; Fu, H.; Feindel, D.; Harding, M.; Feng, J. Development and evaluation of a loop-mediated isothermal amplification (LAMP) assay for the detection of Tomato brown rugose fruit virus (ToBRFV). PLoS ONE 2020, 15, e0230403. [Google Scholar] [CrossRef] [PubMed]
- Tegli, S.; Biancalani, C.; Ignatov, A.N.; Osdaghi, E. A powerful LAMP weapon against the threat of the quarantine plant pathogen curtobacterium flaccumfaciens pv. flaccumfaciens. Microorganisms 2020, 8, 1705. [Google Scholar] [CrossRef]
- De Paiva, B.A.R.; Wendland, A.; Teixeira, N.C.; Ferreira, M. Rapid detection of xanthomonas citri pv. fuscans and xanthomonas phaseoli pv. phaseoli in common bean by loop-mediated isothermal amplification. Plant Dis. 2020, 104, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Frisch, L.M.; Mann, M.A.; Marek, D.N.; Niessen, L. Development and optimization of a loop-mediated isothermal amplification (LAMP) assay for the species-specific detection of Penicillium expansum. Food Microbiol. 2021, 95, 103681. [Google Scholar] [CrossRef]
- Wigmann, E.F.; Meyer, K.; Cendoya, E.; Maul, R.; Vogel, R.F.; Niessen, L. A loop-mediated isothermal amplification (LAMP) based assay for the rapid and sensitive group-specific detection of fumonisin producing Fusarium spp. Int. J. Food Microbiol. 2020, 325, 108627. [Google Scholar] [CrossRef] [PubMed]
- Almasi, M.A. Development of a colorimetric reverse transcription loop-mediated isothermal amplification assay for the detection of Mirafiori lettuce big-vein virus. Arch. Virol. 2017, 162, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- Almasi, M.A.; Almasi, G. Development and evaluation of a reverse transcription loop-mediated isothermal amplification assay for detection of beet necrotic yellow vein virus. Arch. Virol. 2017, 162, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, Z.; Chen, X.; Peng, C.; Xu, X.; Wei, W.; Li, F.; Xu, J. Use of a novel metal indicator to judge loop-mediated isothermal amplification for detecting the 35S promoter. Anal. Bioanal. Chem. 2017, 409, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, D.; Da Lio, D.; Panattoni, A.; Salemi, C.; Cappellini, G.; Bartolini, L.; Parrella, G. Rapid and sensitive detection of tomato brown rugose fruit virus in tomato and pepper seeds by reverse transcription loop-mediated isothermal amplification assays (real time and visual) and comparison with RT-PCR end-point and RT-qPCR methods. Front. Microbiol. 2021, 12, 640932. [Google Scholar] [CrossRef]
- Venkataravanappa, V.; Ashwathappa, K.V.; Reddy, C.N.L.; Shankarappa, K.S.; Reddy, M.K. Characterization of Tomato leaf curl New Delhi virus associated with leaf curl and yellowing disease of Watermelon and development of LAMP assay for its detection. 3 Biotech 2020, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Siemonsmeier, A.; Hadersdorfer, J.; Neumuller, M.; Schwab, W.; Treutter, D. A LAMP protocol for the detection of ‘Candidatus Phytoplasma pyri’, the causal agent of pear decline. Plant Dis. 2019, 103, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Ward, L.I.; Clover, G.R. Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 2010, 100, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Vielba-Fernandez, A.; de Vicente, A.; Perez-Garcia, A.; Fernandez-Ortuno, D. Monitoring methyl benzimidazole carbamate-resistant isolates of the cucurbit powdery mildew pathogen, podosphaera xanthii, using loop-mediated isothermal amplification. Plant Dis. 2019, 103, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Hu, X.R.; Zhang, C.Q. Molecular detection of QoI resistance in colletotrichum gloeosporioides causing strawberry anthracnose based on loop-mediated isothermal amplification assay. Plant Dis. 2019, 103, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Zhang, L.; Zheng, X.; Qian, Y.; Zhang, Y.; Zhao, L.; Cheng, Q. Rapid and specific detection of the poplar black spot disease caused by marssonina brunnea using loop-mediated isothermal amplification assay. Plants 2021, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Vettraino, A.M.; Luchi, N.; Rizzo, D.; Pepori, A.L.; Pecori, F.; Santini, A. Rapid diagnostics for Gnomoniopsis smithogilvyi (syn. Gnomoniopsis castaneae) in chestnut nuts: New challenges by using LAMP and real-time PCR methods. AMB Express 2021, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Kubota, R.; Vine, B.G.; Alvarez, A.M.; Jenkins, D.M. Detection of Ralstonia solanacearum by loop-mediated isothermal amplification. Phytopathology 2008, 98, 1045–1051. [Google Scholar] [CrossRef]
- Lin, H.; Jiang, X.; Yi, J.; Wang, X.; Zuo, R.; Jiang, Z.; Wang, W.; Zhou, E. Molecular identification of Neofabraea species associated with bull’s-eye rot on apple using rolling-circle amplification of partial EF-1alpha sequence. Can. J. Microbiol. 2018, 64, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Davari, M.; Van Diepeningen, A.D.; Babai-Ahari, A.; Arzanlou, M.; Najafzadeh, M.J.; Van der Lee, T.A.J.; De Hoog, G.S. Rapid identification of Fusarium graminearum species complex using rolling circle amplification (RCA). J. Microbiol. Meth. 2012, 89, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Liu, W.; Liu, Y.; Zhan, F.; Chen, H.; Lei, H.; Liu, Y. Colorimetric detection of nucleic acid sequences in plant pathogens based on CRISPR/Cas9 triggered signal amplification. Mikrochim. Acta 2019, 186, 243. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wu, Q.; Feng, X.; Liao, Z.; Peng, W.; Liu, Y.; Peng, D.; Liu, Z.; Mo, M. Interfacing DNA with nanoparticles: Surface science and its applications in biosensing. Int. J. Biol. Macromol. 2020, 151, 757–780. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Jin, B. Prospects of nanoparticle-DNA binding and its implications in medical biotechnology. Biotechnol. Adv. 2012, 30, 1721–1732. [Google Scholar] [CrossRef]
- Capek, I. Dispersions based on noble metal nanoparticles-DNA conjugates. Adv. Colloid Interface Sci. 2011, 163, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.S.; Rogowski, J.L.; Jones, L.; Gu, F.X. Colorimetric biosensing of pathogens using gold nanoparticles. Biotechnol. Adv. 2015, 33, 666–680. [Google Scholar] [CrossRef] [PubMed]
- Khater, M.; De la Escosura-Muniz, A.; Merkoci, A. Biosensors for plant pathogen detection. Biosens. Bioelectron. 2017, 93, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.M.; Yang, J.T. Visual DNA diagnosis of tomato yellow leaf curl virus with integrated recombinase polymerase amplification and a gold-nanoparticle probe. Sci. Rep. 2019, 9, 15146. [Google Scholar] [CrossRef]
- Dharanivasan, G.; Jesse, D.M.I.; Rajamuthuramalingam, T.; Rajendran, G.; Shanthi, S.; Kathiravan, K. Scanometric detection of tomato leaf curl new delhi viral DNA using mono- and bifunctional AuNP-conjugated oligonucleotide probes. ACS Omega 2019, 4, 10094–10107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Ahmed, R.; Damayantharan, M.; Unal, B.; Butt, H.; Yetisen, A.K. Lateral and vertical flow assays for point-of-care diagnostics. Adv. Healthc. Mater. 2019, 8, e1900244. [Google Scholar] [CrossRef] [PubMed]
- Ngom, B.; Guo, Y.; Wang, X.; Bi, D. Development and application of lateral flow test strip technology for detection of infectious agents and chemical contaminants: A review. Anal. Bioanal. Chem. 2010, 397, 1113–1135. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, B. Lateral Flow Technology for Field-Based Applications-Basics and Advanced Developments. Top. Companion Anim. Med. 2015, 30, 139–147. [Google Scholar] [CrossRef]
- Koczula, K.M.; Gallotta, A. Lateral flow assays. Essays Biochem. 2016, 60, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Parolo, C.; Sena-Torralba, A.; Bergua, J.F.; Calucho, E.; Fuentes-Chust, C.; Hu, L.; Rivas, L.; Alvarez-Diduk, R.; Nguyen, E.P.; Cinti, S.; et al. Tutorial: Design and fabrication of nanoparticle-based lateral-flow immunoassays. Nat. Protoc. 2020, 15, 3788–3816. [Google Scholar] [CrossRef]
- Huang, X.; Aguilar, Z.P.; Xu, H.; Lai, W.; Xiong, Y. Membrane-based lateral flow immunochromatographic strip with nanoparticles as reporters for detection: A review. Biosens. Bioelectron. 2016, 75, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Bahadır, E.B.; Sezgintürk, M.K. Lateral flow assays: Principles, designs and labels. TrAC Trends Anal. Chem. 2016, 82, 20. [Google Scholar] [CrossRef]
- Mao, X.; Ma, Y.; Zhang, A.; Zhang, L.; Zeng, L.; Liu, G. Disposable nucleic acid biosensors based on gold nanoparticle probes and lateral flow strip. Anal. Chem. 2009, 81, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Xu, H.; Takalkar, S.; Gurung, A.S.; Liu, B.; Zheng, Y.; Guo, Z.; Baloda, M.; Baryeh, K.; Liu, G. Carbon nanotube-based lateral flow biosensor for sensitive and rapid detection of DNA sequence. Biosens. Bioelectron. 2015, 64, 367–372. [Google Scholar] [CrossRef]
- Jauset-Rubio, M.; Svobodova, M.; Mairal, T.; McNeil, C.; Keegan, N.; Saeed, A.; Abbas, M.N.; El-Shahawi, M.S.; Bashammakh, A.S.; Alyoubi, A.O.; et al. Ultrasensitive, rapid and inexpensive detection of DNA using paper based lateral flow assay. Sci. Rep. 2016, 6, 37732. [Google Scholar] [CrossRef]
- Jahanpeyma, F.; Forouzandeh, M.; Rasaee, M.J.; Shoaie, N. An enzymatic paper-based biosensor for ultrasensitive detection of DNA. Front. Biosci. 2019, 11, 122–135. [Google Scholar]
- Rigano, L.A.; Malamud, F.; Orce, I.G.; Filippone, M.P.; Marano, M.R.; Do Amaral, A.M.; Castagnaro, A.P.; Vojnov, A.A. Rapid and sensitive detection of Candidatus Liberibacter asiaticus by loop mediated isothermal amplification combined with a lateral flow dipstick. BMC Microbiol. 2014, 14, 86. [Google Scholar] [CrossRef]
- Kiatpathomchai, W.; Jaroenram, W.; Arunrut, N.; Jitrapakdee, S.; Flegel, T.W. Shrimp Taura syndrome virus detection by reverse transcription loop-mediated isothermal amplification combined with a lateral flow dipstick. J. Virol. Methods 2008, 153, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.V.; Dantzler, J.L.; Weigl, B.H. Analytical tools to improve optimization procedures for lateral flow assays. Diagnostics 2017, 7, 29. [Google Scholar] [CrossRef]
- Urusov, A.E.; Zherdev, A.V.; Dzantiev, B.B. Towards lateral flow quantitative assays: Detection approaches. Biosensors 2019, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shikha, S.; Mei, Q.; Liu, J.; Zhang, Y. Fluorescent microbeads for point-of-care testing: A review. Mikrochim. Acta 2019, 186, 361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, G.; Wang, Y.; Zhou, J.; Li, C. Establishment and application of hyperbranched rolling circle amplification coupled with lateral flow dipstick for the sensitive detection of Karenia mikimotoi. Harmful Algae. 2019, 84, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.T.; Haegeman, A.; Danchin, E.G.; Gaur, H.S.; Helder, J.; Jones, M.G.; Kikuchi, T.; Manzanilla-Lopez, R.; Palomares-Rius, J.E.; Wesemael, W.M.; et al. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant Pathol. 2013, 14, 946–961. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, S.; Furzer, O.; Jones, J.D.; Judelson, H.S.; Ali, G.S.; Dalio, R.J.; Roy, S.G.; Schena, L.; Zambounis, A.; Panabieres, F.; et al. The Top 10 oomycete pathogens in molecular plant pathology. Mol. Plant Pathol. 2015, 16, 413–434. [Google Scholar] [CrossRef]
- Mansfield, J.; Genin, S.; Magori, S.; Citovsky, V.; Sriariyanum, M.; Ronald, P.; Dow, M.; Verdier, V.; Beer, S.V.; Machado, M.A.; et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Scholthof, K.B.; Adkins, S.; Czosnek, H.; Palukaitis, P.; Jacquot, E.; Hohn, T.; Hohn, B.; Saunders, K.; Candresse, T.; Ahlquist, P.; et al. Top 10 plant viruses in molecular plant pathology. Mol. Plant Pathol. 2011, 12, 938–954. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zheng, Y.; Zhang, F.; Yu, J.; Dai, T.; Wang, R.; Tian, Y.; Xu, H.; Shen, D.; Dou, D. A rapid, equipment-free method for detecting phytophthora infestans in the field using a lateral flow strip-based recombinase polymerase amplification assay. Plant Dis. 2020, 104, 2774–2778. [Google Scholar] [CrossRef]
- Lu, X.; Xu, H.; Song, W.; Yang, Z.; Yu, J.; Tian, Y.; Jiang, M.; Shen, D.; Dou, D. Rapid and simple detection of Phytophthora cactorum in strawberry using a coupled recombinase polymerase amplification-lateral flow strip assay. Phytopathol. Res. 2021, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cho, I.S.; Ju, H.J.; Jeong, R.D. Rapid and visual detection of tomato spotted wilt virus using recombinase polymerase amplification combined with lateral flow strips. Mol. Cell. Probes 2021, 57, 101727. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.K.; Kokane, S.B.; Gowda, S. Development of a reverse transcription recombinase polymerase based isothermal amplification coupled with lateral flow immunochromatographic assay (CTV-RT-RPA-LFICA) for rapid detection of Citrus tristeza virus. Sci. Rep. 2020, 10, 20593. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Safenkova, I.V.; Zherdev, A.V.; Dzantiev, B.B. Nucleic acid lateral flow assay with recombinase polymerase amplification: Solutions for highly sensitive detection of RNA virus. Talanta 2020, 210, 120616. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Safenkova, I.V.; Drenova, N.V.; Zherdev, A.V.; Dzantiev, B.B. Development of lateral flow assay combined with recombinase polymerase amplification for highly sensitive detection of Dickeya solani. Mol. Cell. Probes 2020, 53, 101622. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Shmyglya, I.V.; Zherdev, A.V.; Dzantiev, B.B.; Safenkova, I.V. The challenge for rapid detection of high-structured circular RNA: Assay of potato spindle tuber viroid based on recombinase polymerase amplification and lateral flow tests. Plants 2020, 9, 1369. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yan, D.; Wu, X.; Chen, Z.; Lai, Y.; Lv, L.; Yan, F.; Chen, J.; Zheng, H.; Song, X. Rapid and visual detection of milk vetch dwarf virus using recombinase polymerase amplification combined with lateral flow strips. Virol. J. 2020, 17, 102. [Google Scholar] [CrossRef]
- Hammond, R.W.; Zhang, S. Development of a rapid diagnostic assay for the detection of tomato chlorotic dwarf viroid based on isothermal reverse-transcription-recombinase polymerase amplification. J. Virol. Methods 2016, 236, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Karakkat, B.B.; Hockemeyer, K.; Franchett, M.; Olson, M.; Mullenberg, C.; Koch, P.L. Detection of root-infecting fungi on cool-season turfgrasses using loop-mediated isothermal amplification and recombinase polymerase amplification. J. Microbiol. Methods 2018, 151, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ravelonandro, M.; Russell, P.; McOwen, N.; Briard, P.; Bohannon, S.; Vrient, A. Rapid diagnostic detection of plum pox virus in Prunus plants by isothermal AmplifyRP (R) using reverse transcription-recombinase polymerase amplification. J. Virol. Methods 2014, 207, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Abrescia, N.G.; Bamford, D.H.; Grimes, J.M.; Stuart, D.I. Structure unifies the viral universe. Annu. Rev. Biochem. 2012, 81, 795–822. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Caetano-Anolles, G. Identification of capsid/coat related protein folds and their utility for virus classification. Front. Microbiol. 2017, 8, 380. [Google Scholar] [CrossRef] [PubMed]
- Hull, R. Plant Virology, 5th ed.; Elsevier Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2014; p. 1104. [Google Scholar]
- Ghosh, D.K.; Kokane, S.B.; Kokane, A.D.; Warghane, A.J.; Motghare, M.R.; Bhose, S.; Sharma, A.K.; Reddy, M.K. Development of a recombinase polymerase based isothermal amplification combined with lateral flow assay (HLB-RPA-LFA) for rapid detection of “Candidatus Liberibacter asiaticus”. PLoS ONE 2018, 13, e0208530. [Google Scholar] [CrossRef] [PubMed]
- El-Tholoth, M.; Branavan, M.; Naveenathayalan, A.; Balachandran, W. Recombinase polymerase amplification-nucleic acid lateral flow immunoassays for Newcastle disease virus and infectious bronchitis virus detection. Mol. Biol. Rep. 2019, 46, 6391–6397. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Hu, T.; Yang, X.; Shen, D.; Jiao, B.; Tian, W.; Xu, Y. A recombinase polymerase amplification-lateral flow dipstick assay for rapid detection of the quarantine citrus pathogen in China, Phytophthora hibernalis. PeerJ 2019, 7, e8083. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Yang, X.; Hu, T.; Jiao, B.; Xu, Y.; Zheng, X.; Shen, D. Comparative evaluation of a novel recombinase polymerase amplification-lateral flow dipstick (RPA-LFD) assay, LAMP, conventional PCR, and leaf-disc baiting methods for detection of phytophthora sojae. Front. Microbiol. 2019, 10, 1884. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2018, 144, 31–67. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Sun, F.; Li, X.; Lan, Y.; Du, L.; Zhou, T.; Zhou, Y. Reverse transcription-recombinase polymerase amplification combined with lateral flow strip for detection of rice black-streaked dwarf virus in plants. J. Virol. Methods 2019, 263, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.A.; Larrea-Sarmiento, A.; Alvarez, A.M.; Arif, M. Genome-informed diagnostics for specific and rapid detection of Pectobacterium species using recombinase polymerase amplification coupled with a lateral flow device. Sci. Rep. 2018, 8, 15972. [Google Scholar] [CrossRef] [PubMed]
- Rigano, L.A.; Marano, M.R.; Castagnaro, A.P.; Do Amaral, A.M.; Vojnov, A.A. Rapid and sensitive detection of Citrus Bacterial Canker by loop-mediated isothermal amplification combined with simple visual evaluation methods. BMC Microbiol. 2010, 10, 176. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.A.; Dickinson, M.J.; Boonham, N. Rapid detection of Phytophthora ramorum and P. kernoviae by two-minute DNA extraction followed by isothermal amplification and amplicon detection by generic lateral flow device. Phytopathology 2010, 100, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Wang, Y.; Zhang, L.; Xu, J.; Ye, C. Loop-mediated isothermal amplification label-based gold nanoparticles lateral flow biosensor for detection of enterococcus faecalis and staphylococcus aureus. Front. Microbiol. 2017, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Li, D.; Xu, J.; Ye, C. Detection of nucleic acids and elimination of carryover contamination by using loop-mediated isothermal amplification and antarctic thermal sensitive uracil-DNA-glycosylase in a lateral flow biosensor: Application to the detection of Streptococcus pneumoniae. Mikrochim. Acta 2018, 185, 212. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.A.; Ostoja-Starzewska, S.; Adams, I.P.; Miano, D.W.; Abidrabo, P.; Kinyua, Z.; Alicai, T.; Dickinson, M.J.; Peters, D.; Boonham, N.; et al. Loop-mediated isothermal amplification for rapid detection of the causal agents of cassava brown streak disease. J. Virol. Methods 2013, 191, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Long, H.; Huang, W.; Liu, J.; Cui, J.; Kong, L.; Hu, X.; Gu, J.; Peng, D. Rapid, simple and direct detection of Meloidogyne hapla from infected root galls using loop-mediated isothermal amplification combined with FTA technology. Sci. Rep. 2017, 7, 44853. [Google Scholar] [CrossRef]
- Sagcan, H.; Turgut Kara, N. Detection of Potato ring rot Pathogen Clavibacter michiganensis subsp. sepedonicus by Loop-mediated isothermal amplification (LAMP) assay. Sci. Rep. 2019, 9, 20393. [Google Scholar] [CrossRef]
- Naidoo, N.; Ghai, M.; Moodley, K.; Mkize, L.; Martin, L.; McFarlane, S.; Rutherford, S. Modified RS-LAMP assay and use of lateral flow devices for rapid detection of Leifsonia xyli subsp. xyli. Lett. Appl. Microbiol. 2017, 65, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Gu, R.; Li, X.; Mu, D. Simple and rapid detection Aspergillus fumigatus by loop-mediated isothermal amplification coupled with lateral flow biosensor assay. J. Appl. Microbiol. 2021, 131, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Edgu, G.; Freund, L.J.; Hartje, S.; Tacke, E.; Hofferbert, H.R.; Twyman, R.M.; Noll, G.A.; Muth, J.; Prufer, D. Fast, Precise, and Reliable Multiplex Detection of Potato Viruses by Loop-Mediated Isothermal Amplification. Int. J. Mol. Sci. 2020, 21, 8741. [Google Scholar] [CrossRef] [PubMed]
- Chaumpluk, P.; Plubcharoensook, P.; Prasongsuk, S. Rapid detection of aflatoxigenic Aspergillus sp. in herbal specimens by a simple, bendable, paper-based lab-on-a-chip. Biotechnol. J. 2016, 11, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Van Beckhoven, J.R.; Stead, D.E.; Van der Wolf, J.M. Detection of Clavibacter michiganensis subsp. sepedonicus by AmpliDet RNA, a new technology based on real time monitoring of NASBA amplicons with a molecular beacon. J. Appl. Microbiol. 2002, 93, 840–849. [Google Scholar] [CrossRef]
- Bentsink, L.; Leone, G.O.; Van Beckhoven, J.R.; Van Schijndel, H.B.; Van Gemen, B.; Van der Wolf, J.M. Amplification of RNA by NASBA allows direct detection of viable cells of Ralstonia solanacearum in potato. J. Appl. Microbiol. 2002, 93, 647–655. [Google Scholar] [CrossRef]
- Leone, G.; Van Schijndel, H.; Van Gemen, B.; Kramer, F.R.; Schoen, C.D. Molecular beacon probes combined with amplification by NASBA enable homogeneous, real-time detection of RNA. Nucleic. Acids Res. 1998, 26, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Olmos, A.; Bertolini, E.; Cambra, M. Isothermal amplification coupled with rapid flow-through hybridisation for sensitive diagnosis of Plum pox virus. J. Virol. Methods 2007, 139, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Yrad, F.M.; Castanares, J.M.; Alocilja, E.C. Visual detection of dengue-1 RNA using gold nanoparticle-based lateral flow biosensor. Diagnostics 2019, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Rohrman, B.A.; Leautaud, V.; Molyneux, E.; Richards-Kortum, R.R. A lateral flow assay for quantitative detection of amplified HIV-1 RNA. PLoS ONE 2012, 7, e45611. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, J.; Xu, J.J.; Zhang, S.; Chen, H.Y. Advances in DNA/RNA detection using nanotechnology. Adv. Clin. Chem. 2019, 91, 31–98. [Google Scholar] [CrossRef]
- Dutse, S.W.; Yusof, N.A. Microfluidics-based lab-on-chip systems in DNA-based biosensing: An overview. Sensors 2011, 11, 5754–5768. [Google Scholar] [CrossRef]
- Ansari, M.I.H.; Hassan, S.; Qurashi, A.; Khanday, F.A. Microfluidic-integrated DNA nanobiosensors. Biosens. Bioelectron. 2016, 85, 247–260. [Google Scholar] [CrossRef]
- Peter, H.; Wienke, J.; Bier, F.F. Lab-on-a-Chip Multiplex Assays. Methods Mol. Biol. 2017, 1546, 283–294. [Google Scholar] [CrossRef]
- Mao, K.; Min, X.; Zhang, H.; Zhang, K.; Cao, H.; Guo, Y.; Yang, Z. Paper-based microfluidics for rapid diagnostics and drug delivery. J. Control. Release 2020, 322, 187–199. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, Y.; Fohlerova, Z.; Chang, H.; Iliescu, C.; Neuzil, P. LAMP-on-a-chip: Revising microfluidic platforms for loop-mediated DNA amplification. Trends Anal. Chem. 2019, 113, 44–53. [Google Scholar] [CrossRef]
- Li, R.; Chen, J.; Zhang, X.; Cui, J.; Tao, S.; Yang, L. Mini-disk capillary array coupling with LAMP for visual detection of multiple nucleic acids using genetically modified organism analysis as an example. J. Agric. Food Chem. 2020, 68, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Yang, S.Y.; Lin, C.L.; Wang, C.H.; Li, P.C.; Chen, T.Y.; Jan, F.J.; Lee, G.B. Detection of viruses directly from the fresh leaves of a Phalaenopsis orchid using a microfluidic system. Nanomedicine 2013, 9, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Chidambara, V.A.; Andreasen, S.Z.; Golabi, M.; Huynh, V.N.; Linh, Q.T.; Bang, D.D.; Wolff, A. Point-of-care devices for pathogen detections: The three most important factors to realise towards commercialization. TrAC Trends Anal. Chem. 2020, 131, 116004. [Google Scholar] [CrossRef]





| Detection | Rapid | Minimal Equipment | High Specificity | High Sensitivity | Pronounced and Constant Signal | Easy to Perform in Field |
|---|---|---|---|---|---|---|
| SYBR Green | + | +/− | +/− | +/− | − | + |
| Coloration | + | + | − | − | − | + |
| GNP | +/− | + | − | − | +/− | + |
| LFA | + | + | +/− | + | + | + |
| Lab-on-chip | + | +/− | + | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanov, A.V.; Safenkova, I.V.; Zherdev, A.V.; Dzantiev, B.B. The Potential Use of Isothermal Amplification Assays for In-Field Diagnostics of Plant Pathogens. Plants 2021, 10, 2424. https://doi.org/10.3390/plants10112424
Ivanov AV, Safenkova IV, Zherdev AV, Dzantiev BB. The Potential Use of Isothermal Amplification Assays for In-Field Diagnostics of Plant Pathogens. Plants. 2021; 10(11):2424. https://doi.org/10.3390/plants10112424
Chicago/Turabian StyleIvanov, Aleksandr V., Irina V. Safenkova, Anatoly V. Zherdev, and Boris B. Dzantiev. 2021. "The Potential Use of Isothermal Amplification Assays for In-Field Diagnostics of Plant Pathogens" Plants 10, no. 11: 2424. https://doi.org/10.3390/plants10112424
APA StyleIvanov, A. V., Safenkova, I. V., Zherdev, A. V., & Dzantiev, B. B. (2021). The Potential Use of Isothermal Amplification Assays for In-Field Diagnostics of Plant Pathogens. Plants, 10(11), 2424. https://doi.org/10.3390/plants10112424