
Amino Acid Nanofibers Improve Glycemia and Confer Cognitive Therapeutic Efficacy to Bound Insulin

 ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of AAC Compounds
2.3. Transmission Electron Microscopy (TEM)
2.4. Zeta Potential Measurement
2.5. Quartz Crystal Microbalance with Dissipation (QCM-D) Binding Assay
2.6. Spectroscopy
2.7. Confocal Fluorescence Microscopy
2.8. Glucose Uptake Assay
2.9. Transfected Cells
2.10. Animal Studies
2.10.1. Experimental Mouse Models
2.10.2. Body Composition and Activity Measurements
2.10.3. Glucose Tolerance Test (GTT) and Insulin Tolerance Tests (ITT)
2.10.4. Cognitive Test
2.11. Biochemical Analysis
2.12. mRNA Analysis
2.13. Affymetrix GeneChip
2.14. Statistical Analysis
3. Results and Discussion
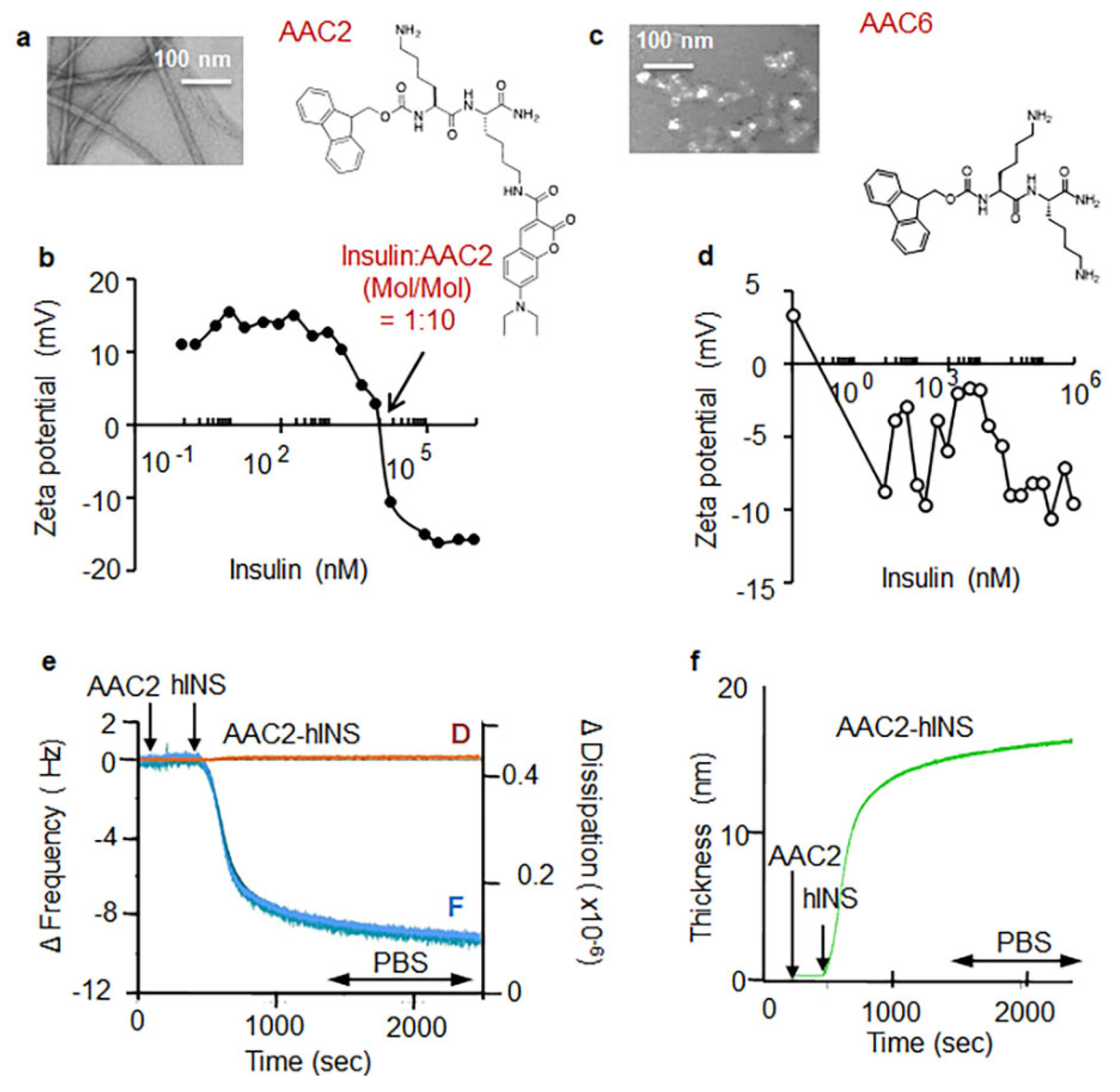
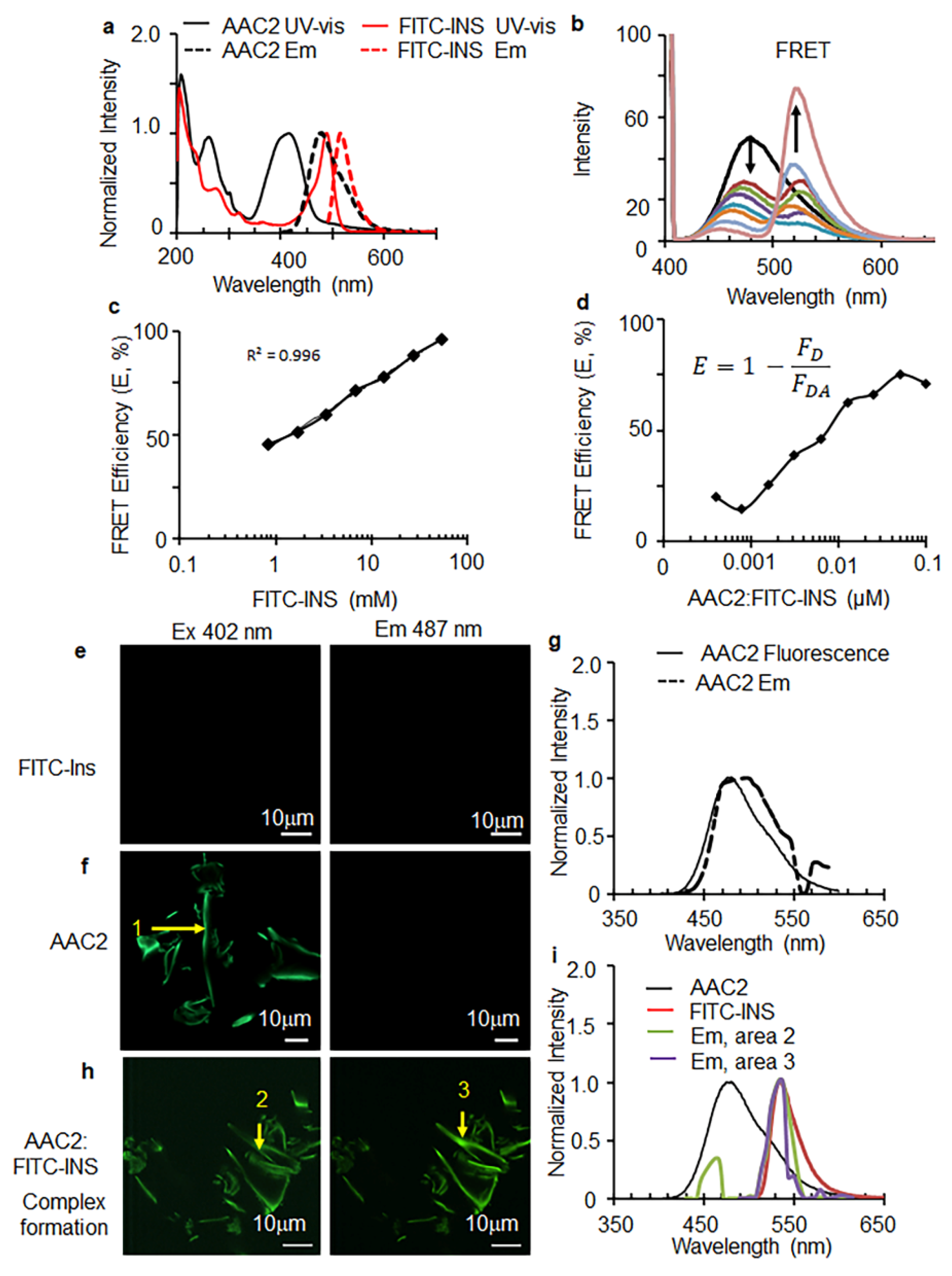
3.1. AAC2 and Insulin Form a Supramolecular Complex In Vitro
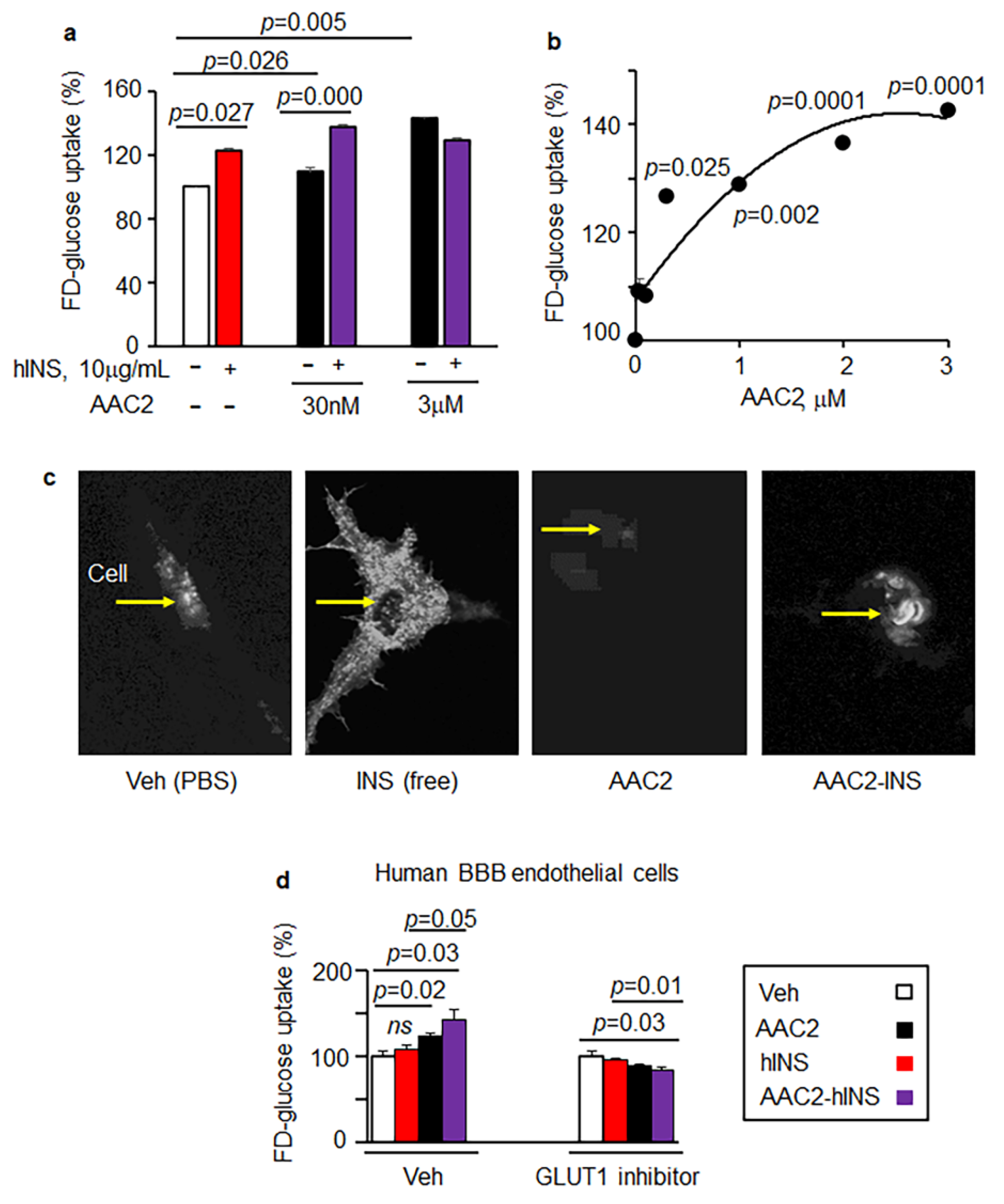
3.2. AAC2-hINS Complex Stimulated Glucose Uptake In Vitro in Peripheral and Nervous Tissues
3.3. Free AAC2, Free hINS, and AAC2-hINS Rescued STZ-Treated Mice from Death
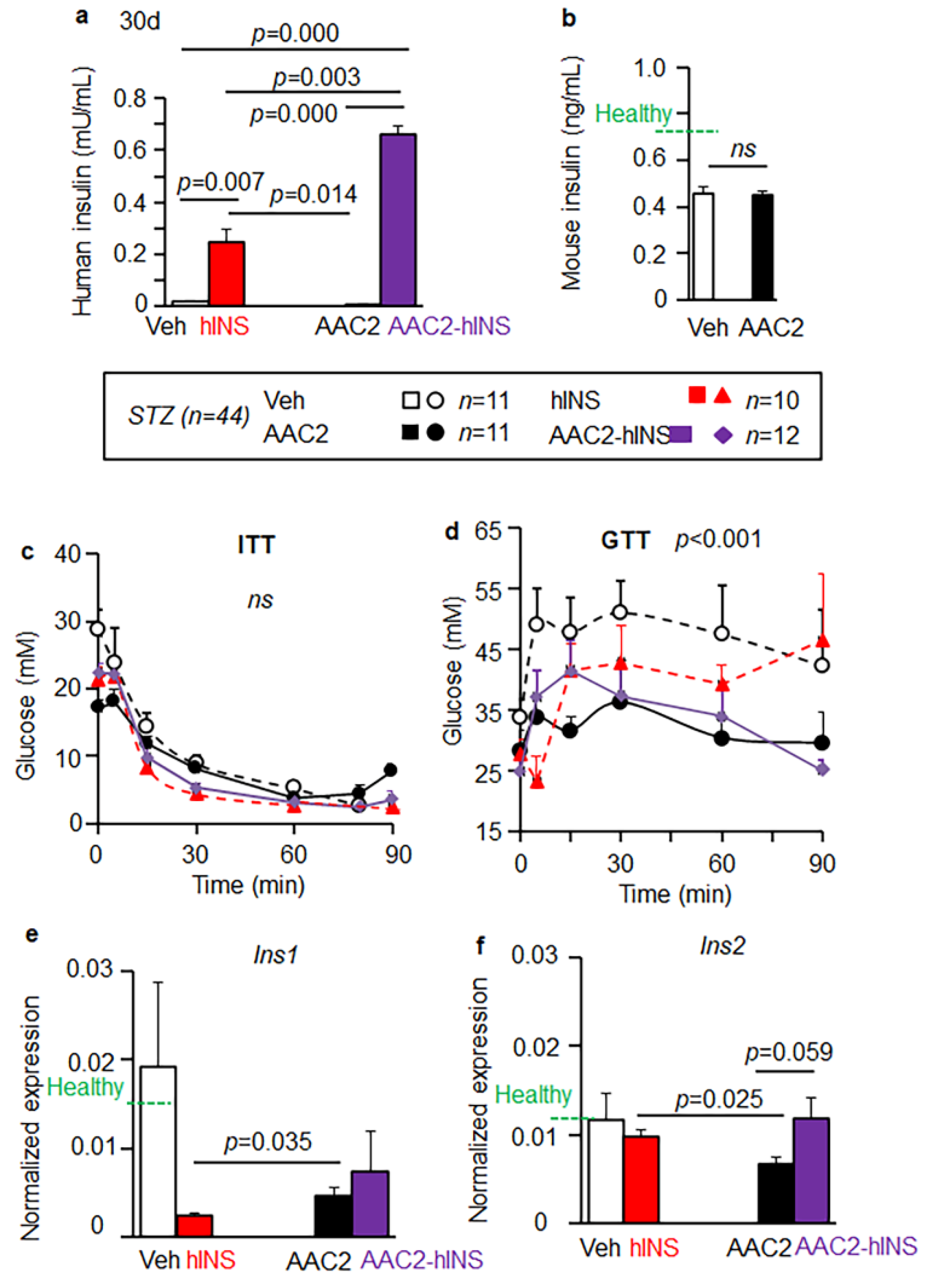
3.4. AAC2-hINS to Increases the Blood Levels of Insulin and Glucose Uptake in STZ Mice
3.5. Insulin Eexpression in the Brain
3.6. Distinct Regulatory Effects of AAC2-hINS Compared to Free AAC2 and hINS
3.7. AAC2-hINS Improved Cognitive Performance in T1D Mouse Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zaykov, A.N.; Mayer, J.P.; DiMarchi, R.D. Pursuit of a perfect insulin. Nat. Rev. Drug Discov. 2016, 15, 425–439. [Google Scholar] [CrossRef]
- Volpatti, L.R.; Matranga, M.A.; Cortinas, A.B.; Delcassian, D.; Daniel, K.B.; Langer, R.; Anderson, D.G. Glucose-Responsive Nanoparticles for Rapid and Extended Self-Regulated Insulin Delivery. ACS Nano 2020, 14, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Martens, T.; Beck, R.W.; Bailey, R.; Ruedy, K.J.; Calhoun, P.; Peters, A.L.; Pop-Busui, R.; Philis-Tsimikas, A.; Bao, S.; Umpierrez, G.; et al. Effect of Continuous Glucose Monitoring on Glycemic Control in Patients With Type 2 Diabetes Treated With Basal Insulin: A Randomized Clinical Trial. JAMA 2021, 325, 2262–2272. [Google Scholar] [CrossRef]
- Armstrong, D.G.; Swerdlow, M.A.; Armstrong, A.A.; Conte, M.S.; Padula, W.V.; Bus, S.A. Five year mortality and direct costs of care for people with diabetic foot complications are comparable to cancer. J. Foot Ankle Res. 2020, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Guarneri, A.M.; Hoffman, R.P. Nonglycemic Adverse Effects of Insulin. Curr. Diabetes Rev. 2021. E-pub Ahead of Print. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Lesort, M.; Johnson, G.V. Insulin-like growth factor-1 and insulin mediate transient site-selective increases in tau phosphorylation in primary cortical neurons. Neuroscience 2000, 99, 305–316. [Google Scholar] [CrossRef]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Asthana, S.; Cook, D.G.; Baker, L.D.; Cherrier, M.; Purganan, K.; Wait, C.; Petrova, A.; Latendresse, S.; Watson, G.S.; et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: Interactions with apolipoprotein E genotype. Psychoneuroendocrinology 2003, 28, 809–822. [Google Scholar] [CrossRef]
- Kulstad, J.J.; Green, P.S.; Cook, D.G.; Watson, G.S.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Rhoads, K.; Mehta, P.D.; et al. Differential modulation of plasma beta-amyloid by insulin in patients with Alzheimer disease. Neurology 2006, 66, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Craft, S. Modulation of memory by insulin and glucose: Neuropsychological observations in Alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Gao, G.; Rueda, C.B.; Yu, H.; Thibodeaux, D.N.; Awano, T.; Engelstad, K.M.; Sanchez-Quintero, M.J.; Yang, H.; Li, F.; et al. Brain microvasculature defects and Glut1 deficiency syndrome averted by early repletion of the glucose transporter-1 protein. Nat. Commun. 2017, 8, 14152. [Google Scholar] [CrossRef]
- Reno, C.M.; Puente, E.C.; Sheng, Z.; Daphna-Iken, D.; Bree, A.J.; Routh, V.H.; Kahn, B.B.; Fisher, S.J. Brain GLUT4 Knockout Mice Have Impaired Glucose Tolerance, Decreased Insulin Sensitivity, and Impaired Hypoglycemic Counterregulation. Diabetes 2017, 66, 587–597. [Google Scholar] [CrossRef]
- Mazaika, P.K.; Weinzimer, S.A.; Mauras, N.; Buckingham, B.; White, N.H.; Tsalikian, E.; Hershey, T.; Cato, A.; Aye, T.; Fox, L.; et al. Variations in Brain Volume and Growth in Young Children With Type 1 Diabetes. Diabetes 2016, 65, 476–485. [Google Scholar] [CrossRef]
- Mauras, N.; Mazaika, P.; Buckingham, B.; Weinzimer, S.; White, N.H.; Tsalikian, E.; Hershey, T.; Cato, A.; Cheng, P.; Kollman, C.; et al. Longitudinal assessment of neuroanatomical and cognitive differences in young children with type 1 diabetes: Association with hyperglycemia. Diabetes 2015, 64, 1770–1779. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Gilsanz, P.; Quesenberry, C.P.; Karter, A.J.; Lacy, M.E. Association of Type 1 Diabetes and Hypoglycemic and Hyperglycemic Events and Risk of Dementia. Neurology 2021, 97, e275–e283. [Google Scholar] [CrossRef]
- Mittal, K.; Mani, R.J.; Katare, D.P. Type 3 Diabetes: Cross Talk between Differentially Regulated Proteins of Type 2 Diabetes Mellitus and Alzheimer’s Disease. Sci. Rep. 2016, 6, 25589. [Google Scholar] [CrossRef]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Williams, K.W.; Margatho, L.O.; Lee, C.E.; Choi, M.; Lee, S.; Scott, M.M.; Elias, C.F.; Elmquist, J.K. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 2472–2479. [Google Scholar] [CrossRef]
- Barr, V.A.; Malide, D.; Zarnowski, M.J.; Taylor, S.I.; Cushman, S.W. Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology 1997, 138, 4463–4472. [Google Scholar] [CrossRef]
- Fuente-Martin, E.; Garcia-Caceres, C.; Granado, M.; de Ceballos, M.L.; Sanchez-Garrido, M.A.; Sarman, B.; Liu, Z.W.; Dietrich, M.O.; Tena-Sempere, M.; Argente-Arizon, P.; et al. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J. Clin. Investig. 2012, 122, 3900–3913. [Google Scholar] [CrossRef] [PubMed]
- Denisova, E.I.; Kozhevnikova, V.V.; Bazhan, N.M.; Makarova, E.N. Sex-specific effects of leptin administration to pregnant mice on the placentae and the metabolic phenotypes of offspring. FEBS Open Bio 2020, 10, 96–106. [Google Scholar] [CrossRef]
- Wauman, J.; Zabeau, L.; Tavernier, J. The Leptin Receptor Complex: Heavier Than Expected? Front. Endocrinol. 2017, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Thon, M.; Hosoi, T.; Ozawa, K. Insulin enhanced leptin-induced STAT3 signaling by inducing GRP78. Sci. Rep. 2016, 6, 34312. [Google Scholar] [CrossRef]
- Shaw, A.S.; Filbert, E.L. Scaffold proteins and immune-cell signalling. Nat. Rev. Immunol. 2009, 9, 47–56. [Google Scholar] [CrossRef]
- Good, M.C.; Zalatan, J.G.; Lim, W.A. Scaffold proteins: Hubs for controlling the flow of cellular information. Science 2011, 332, 680–686. [Google Scholar] [CrossRef]
- Lee, A.; Sun, Y.; Lin, T.; Song, N.J.; Mason, M.L.; Leung, J.H.; Kowdley, D.; Wall, J.; Brunetti, A.; Fitzgerald, J.; et al. Amino acid-based compound activates atypical PKC and leptin receptor pathways to improve glycemia and anxiety like behavior in diabetic mice. Biomaterials 2020, 239, 119839. [Google Scholar] [CrossRef]
- Voinova, M.V.; Rodahl, M.; Jonson, M.; Kasemo, B. Viscoelastic acoustic response of layered polymer films at fluid-solid interfaces: Continuum mechanics approach. Phys. Scr. 1999, 59, 391–396. [Google Scholar] [CrossRef]
- Reviakine, I.; Johannsmann, D.; Richter, R.P. Hearing what you cannot see and visualizing what you hear: Interpreting quartz crystal microbalance data from solvated interfaces. Anal. Chem. 2011, 83, 8838–8848. [Google Scholar] [CrossRef]
- Yasmeen, R.; Meyers, J.M.; Alvarez, C.E.; Thomas, J.L.; Bonnegarde-Bernard, A.; Alder, H.; Papenfuss, T.L.; Benson, D.M., Jr.; Boyaka, P.N.; Ziouzenkova, O. Aldehyde dehydrogenase-1a1 induces oncogene suppressor genes in B cell populations. Biochim. Biophys. Acta 2013, 1833, 3218–3227. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.A.; Lentz, S.I.; Roberts, J.L., Jr.; Feldman, E.L. Criteria for creating and assessing mouse models of diabetic neuropathy. Curr. Drug Targets 2008, 9, 3–13. [Google Scholar] [CrossRef]
- Ramos-Rodriguez, J.J.; Ortiz, O.; Jimenez-Palomares, M.; Kay, K.R.; Berrocoso, E.; Murillo-Carretero, M.I.; Perdomo, G.; Spires-Jones, T.; Cozar-Castellano, I.; Lechuga-Sancho, A.M.; et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 2013, 38, 2462–2475. [Google Scholar] [CrossRef] [PubMed]
- Borniger, J.C.; Cisse, Y.M.; Cantemir-Stone, C.Z.; Bolon, B.; Nelson, R.J.; Marsh, C.B. Behavioral abnormalities in mice lacking mesenchyme-specific Pten. Behav. Brain Res. 2016, 304, 80–85. [Google Scholar] [CrossRef]
- Walton, J.C.; Chen, Z.; Weil, Z.M.; Pyter, L.M.; Travers, J.B.; Nelson, R.J. Photoperiod-mediated impairment of long-term potention and learning and memory in male white-footed mice. Neuroscience 2011, 175, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Jeworrek, C.; Hollmann, O.; Steitz, R.; Winter, R.; Czeslik, C. Interaction of IAPP and insulin with model interfaces studied using neutron reflectometry. Biophys. J. 2009, 96, 1115–1123. [Google Scholar] [CrossRef]
- Rodenhausen, K.B.; Kasputis, T.; Pannier, A.K.; Gerasimov, J.Y.; Lai, R.Y.; Solinsky, M.; Tiwald, T.E.; Wang, H.; Sarkar, A.; Hofmann, T.; et al. Combined optical and acoustical method for determination of thickness and porosity of transparent organic layers below the ultra-thin film limit. Rev. Sci. Instrum. 2011, 82, 103111. [Google Scholar] [CrossRef]
- Takakusa, H.; Kikuchi, K.; Urano, Y.; Kojima, H.; Nagano, T. A novel design method of ratiometric fluorescent probes based on fluorescence resonance energy transfer switching by spectral overlap integral. Chemistry 2003, 9, 1479–1485. [Google Scholar] [CrossRef]
- Ohta, Y.; Wakita, H.; Kawaguchi, M.; Ieda, N.; Osada, S.; Nakagawa, H. Ratiometric assay of CARM1 activity using a FRET-based fluorescent probe. Bioorg. Med. Chem. Lett. 2019, 29, 126728. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Shah, B.P.; Garfunkel, E.; Lee, K.B. Versatile fluorescence resonance energy transfer-based mesoporous silica nanoparticles for real-time monitoring of drug release. ACS Nano 2013, 7, 2741–2750. [Google Scholar] [CrossRef]
- Virgintino, D.; Robertson, D.; Monaghan, P.; Errede, M.; Bertossi, M.; Ambrosi, G.; Roncali, L. Glucose transporter GLUT1 in human brain microvessels revealed by ultrastructural immunocytochemistry. J. Submicrosc. Cytol. Pathol. 1997, 29, 365–370. [Google Scholar] [PubMed]
- Van Belle, T.L.; Taylor, P.; von Herrath, M.G. Mouse Models for Type 1 Diabetes. Drug Discov. Today Dis. Models 2009, 6, 41–45. [Google Scholar] [CrossRef][Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.W.; Docherty, K. Comparative analysis of insulin gene promoters: Implications for diabetes research. Diabetes 2006, 55, 3201–3213. [Google Scholar] [CrossRef]
- Bialoszewska, A.; Malejczyk, J. Biological and Clinical Significance of Human NKRP1A/LLT1 Receptor/Ligand Interactions. Crit. Rev. Immunol. 2018, 38, 479–489. [Google Scholar] [CrossRef]
- Cox, A.R.; Lam, C.J.; Bonnyman, C.W.; Chavez, J.; Rios, J.S.; Kushner, J.A. Angiopoietin-like protein 8 (ANGPTL8)/betatrophin overexpression does not increase beta cell proliferation in mice. Diabetologia 2015, 58, 1523–1531. [Google Scholar] [CrossRef]
- Nidhina Haridas, P.A.; Soronen, J.; Sadevirta, S.; Mysore, R.; Quagliarini, F.; Pasternack, A.; Metso, J.; Perttila, J.; Leivonen, M.; Smas, C.M.; et al. Regulation of Angiopoietin-Like Proteins (ANGPTLs) 3 and 8 by Insulin. J. Clin. Endocrinol. Metab. 2015, 100, E1299–E1307. [Google Scholar] [CrossRef] [PubMed]
- Schwebach, C.L.; Agrawal, R.; Lindert, S.; Kudryashova, E.; Kudryashov, D.S. The Roles of Actin-Binding Domains 1 and 2 in the Calcium-Dependent Regulation of Actin Filament Bundling by Human Plastins. J. Mol. Biol. 2017, 429, 2490–2508. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.H.; Tu, H.C.; Wilkens, S.; Loh, S.N. Loss of bound zinc facilitates amyloid fibril formation of leukocyte-cell-derived chemotaxin 2 (LECT2). J. Biol. Chem. 2021, 296, 100446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kang, H.; Xiao, J.; Shi, B.; Li, X.; Chen, G. LHPP Inhibits the Proliferation and Metastasis of Renal Cell Carcinoma. BioMed Res. Int. 2020, 2020, 7020924. [Google Scholar] [CrossRef] [PubMed]
- Pleasure, D.; Guo, F.; Chechneva, O.; Bannerman, P.; McDonough, J.; Burns, T.; Wang, Y.; Hull, V. Pathophysiology and Treatment of Canavan Disease. Neurochem. Res. 2020, 45, 561–565. [Google Scholar] [CrossRef]
- Laksitorini, M.D.; Yathindranath, V.; Xiong, W.; Parkinson, F.E.; Thliveris, J.A.; Miller, D.W. Impact of Wnt/beta-catenin signaling on ethanol-induced changes in brain endothelial cell permeability. J. Neurochem. 2021, 157, 1118–1137. [Google Scholar] [CrossRef]
- Heller-Stilb, B.; van Roeyen, C.; Rascher, K.; Hartwig, H.G.; Huth, A.; Seeliger, M.W.; Warskulat, U.; Haussinger, D. Disruption of the taurine transporter gene (taut) leads to retinal degeneration in mice. FASEB J. 2002, 16, 231–233. [Google Scholar] [CrossRef]
- Baliou, S.; Kyriakopoulos, A.M.; Goulielmaki, M.; Panayiotidis, M.I.; Spandidos, D.A.; Zoumpourlis, V. Significance of taurine transporter (TauT) in homeostasis and its layers of regulation (Review). Mol. Med. Rep. 2020, 22, 2163–2173. [Google Scholar] [CrossRef]
- Warskulat, U.; Borsch, E.; Reinehr, R.; Heller-Stilb, B.; Monnighoff, I.; Buchczyk, D.; Donner, M.; Flogel, U.; Kappert, G.; Soboll, S.; et al. Chronic liver disease is triggered by taurine transporter knockout in the mouse. FASEB J. 2006, 20, 574–576. [Google Scholar] [CrossRef]
- Ito, T.; Yoshikawa, N.; Inui, T.; Miyazaki, N.; Schaffer, S.W.; Azuma, J. Tissue depletion of taurine accelerates skeletal muscle senescence and leads to early death in mice. PLoS ONE 2014, 9, e107409. [Google Scholar] [CrossRef]
- Fujikawa-Adachi, K.; Nishimori, I.; Taguchi, T.; Onishi, S. Human carbonic anhydrase XIV (CA14): cDNA cloning, mRNA expression, and mapping to chromosome 1. Genomics 1999, 61, 74–81. [Google Scholar] [CrossRef]
- Svichar, N.; Waheed, A.; Sly, W.S.; Hennings, J.C.; Hubner, C.A.; Chesler, M. Carbonic anhydrases CA4 and CA14 both enhance AE3-mediated Cl−-HCO3− exchange in hippocampal neurons. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 3252–3258. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem. 2006, 387, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Wi, S.; Lee, J.W.; Kim, M.; Park, C.H.; Cho, S.R. An Enriched Environment Ameliorates Oxidative Stress and Olfactory Dysfunction in Parkinson’s Disease with alpha-Synucleinopathy. Cell Transpl. 2018, 27, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Borcari, N.R.; Dos Santos, J.F.; Reigado, G.R.; Freitas, B.L.; Araujo, M.D.S.; Nunes, V.A. Vitamins Modulate the Expression of Antioxidant Genes in Progesterone-Treated Pancreatic beta Cells: Perspectives for Gestational Diabetes Management. Int. J. Endocrinol. 2020, 2020, 8745120. [Google Scholar] [CrossRef]
- Willer, C.J.; Sanna, S.; Jackson, A.U.; Scuteri, A.; Bonnycastle, L.L.; Clarke, R.; Heath, S.C.; Timpson, N.J.; Najjar, S.S.; Stringham, H.M.; et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat. Genet. 2008, 40, 161–169. [Google Scholar] [CrossRef]
- Ozyilmaz, B.; Kirbiyik, O.; Koc, A.; Ozdemir, T.R.; Kaya Ozer, O.; Kutbay, Y.B.; Erdogan, K.M.; Saka Guvenc, M.; Ozturk, C. Molecular genetic evaluation of NLRP3, MVK and TNFRSF1A associated periodic fever syndromes. Int. J. Immunogenet. 2019, 46, 232–240. [Google Scholar] [CrossRef]
- Lim, S.; Jung, G.A.; Glover, D.J.; Clark, D.S. Enhanced Enzyme Activity through Scaffolding on Customizable Self-Assembling Protein Filaments. Small 2019, 15, e1805558. [Google Scholar] [CrossRef]
- Cusick, M.E.; Klitgord, N.; Vidal, M.; Hill, D.E. Interactome: Gateway into systems biology. Hum. Mol. Genet. 2005, 14, R171–R181. [Google Scholar] [CrossRef]
- Han, X.; Patters, A.B.; Ito, T.; Azuma, J.; Schaffer, S.W.; Chesney, R.W. Knockout of the TauT gene predisposes C57BL/6 mice to streptozotocin-induced diabetic nephropathy. PLoS ONE 2015, 10, e0117718. [Google Scholar] [CrossRef]
- Warskulat, U.; Heller-Stilb, B.; Oermann, E.; Zilles, K.; Haas, H.; Lang, F.; Haussinger, D. Phenotype of the taurine transporter knockout mouse. Methods Enzym. 2007, 428, 439–458. [Google Scholar] [CrossRef]
- Hong, B.N.; Kang, T.H. Auditory neuropathy in streptozotocin-induced diabetic mouse. Neurosci. Lett. 2008, 431, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Gupta, R.C.; Lee, H.W. Taurine-diabetes interaction: From involvement to protection. Curr. Diabetes Rev. 2007, 3, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zheng, Y.; Zhao, L.; Chen, M.; Bai, G.; Hu, Y.; Hu, W.; Yan, Z.; Gao, H. Cognitive decline in type 2 diabetic db/db mice may be associated with brain region-specific metabolic disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Yokogoshi, H.; Mochizuki, H.; Nanami, K.; Hida, Y.; Miyachi, F.; Oda, H. Dietary taurine enhances cholesterol degradation and reduces serum and liver cholesterol concentrations in rats fed a high-cholesterol diet. J. Nutr. 1999, 129, 1705–1712. [Google Scholar] [CrossRef]
- Borck, P.C.; Vettorazzi, J.F.; Branco, R.C.S.; Batista, T.M.; Santos-Silva, J.C.; Nakanishi, V.Y.; Boschero, A.C.; Ribeiro, R.A.; Carneiro, E.M. Taurine supplementation induces long-term beneficial effects on glucose homeostasis in ob/ob mice. Amino Acids 2018, 50, 765–774. [Google Scholar] [CrossRef]
- Nakatsuru, Y.; Murase-Mishiba, Y.; Bessho-Tachibana, M.; Terasaki, J.; Hanafusa, T.; Imagawa, A. Taurine improves glucose tolerance in STZ-induced insulin-deficient diabetic mice. Diabetol. Int. 2018, 9, 234–242. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, X.; Luo, M.; Lan, Q.; Ullah, H.; Zhang, C.; Li, S.; Chen, X.; Wang, Y.; Piao, F. Taurine ameliorates axonal damage in sciatic nerve of diabetic rats and high glucose exposed DRG neuron by PI3K/Akt/mTOR-dependent pathway. Amino Acids 2021, 53, 395–406. [Google Scholar] [CrossRef]
- Yun, K.U.; Ryu, C.S.; Lee, J.Y.; Noh, J.R.; Lee, C.H.; Lee, H.S.; Kang, J.S.; Park, S.K.; Kim, B.H.; Kim, S.K. Hepatic metabolism of sulfur amino acids in db/db mice. Food Chem. Toxicol. 2013, 53, 180–186. [Google Scholar] [CrossRef]
- Santos-Silva, J.C.; Ribeiro, R.A.; Vettorazzi, J.F.; Irles, E.; Rickli, S.; Borck, P.C.; Porciuncula, P.M.; Quesada, I.; Nadal, A.; Boschero, A.C.; et al. Taurine supplementation ameliorates glucose homeostasis, prevents insulin and glucagon hypersecretion, and controls beta, alpha, and delta-cell masses in genetic obese mice. Amino Acids 2015, 47, 1533–1548. [Google Scholar] [CrossRef]
- Hsuchou, H.; Kastin, A.J.; Tu, H.; Joan Abbott, N.; Couraud, P.O.; Pan, W. Role of astrocytic leptin receptor subtypes on leptin permeation across hCMEC/D3 human brain endothelial cells. J. Neurochem. 2010, 115, 1288–1298. [Google Scholar] [CrossRef]
- Castelblanco, E.; Hernandez, M.; Castelblanco, A.; Gratacos, M.; Esquerda, A.; Mollo, A.; Ramirez-Morros, A.; Real, J.; Franch-Nadal, J.; Fernandez-Real, J.M.; et al. Low-grade Inflammatory Marker Profile May Help to Differentiate Patients With LADA, Classic Adult-Onset Type 1 and Type 2 Diabetes. Diabetes Care 2018, 41, 862–868. [Google Scholar] [CrossRef]
- Smolina, K.; Wotton, C.J.; Goldacre, M.J. Risk of dementia in patients hospitalised with type 1 and type 2 diabetes in England, 1998–2011: A retrospective national record linkage cohort study. Diabetologia 2015, 58, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, A.L.; Nieuwenhoff, M.D.; Klaassen, E.S.; Verma, A.; Kramer, M.H.; Strijers, R.; Vrancken, A.F.; Eringa, E.; Groeneveld, G.J.; Serne, E.H. Relationships Between Type 2 Diabetes, Neuropathy, and Microvascular Dysfunction: Evidence From Patients With Cryptogenic Axonal Polyneuropathy. Diabetes Care 2017, 40, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Rodill, L.G.; Exalto, L.G.; Gilsanz, P.; Biessels, G.J.; Quesenberry, C.P., Jr.; Whitmer, R.A. Diabetic Retinopathy and Dementia in Type 1 Diabetes. Alzheimer Dis. Assoc. Disord. 2017, 32, 125. [Google Scholar] [CrossRef]
- Gottesman, R.F.; Albert, M.S.; Alonso, A.; Coker, L.H.; Coresh, J.; Davis, S.M.; Deal, J.A.; McKhann, G.M.; Mosley, T.H.; Sharrett, A.R.; et al. Associations Between Midlife Vascular Risk Factors and 25-Year Incident Dementia in the Atherosclerosis Risk in Communities (ARIC) Cohort. JAMA Neurol. 2017, 74, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Bedinger, D.H.; Adams, S.H. Metabolic, anabolic, and mitogenic insulin responses: A tissue-specific perspective for insulin receptor activators. Mol. Cell. Endocrinol. 2015, 415, 143–156. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Picconi, F.; Parravano, M.; Ylli, D.; Pasqualetti, P.; Coluzzi, S.; Giordani, I.; Malandrucco, I.; Lauro, D.; Scarinci, F.; Giorno, P.; et al. Retinal neurodegeneration in patients with type 1 diabetes mellitus: The role of glycemic variability. Acta Diabetol. 2017, 54, 489–497. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, A.; Mason, M.L.; Lin, T.; Kumar, S.B.; Kowdley, D.; Leung, J.H.; Muhanna, D.; Sun, Y.; Ortega-Anaya, J.; Yu, L.; et al. Amino Acid Nanofibers Improve Glycemia and Confer Cognitive Therapeutic Efficacy to Bound Insulin. Pharmaceutics 2022, 14, 81. https://doi.org/10.3390/pharmaceutics14010081
Lee A, Mason ML, Lin T, Kumar SB, Kowdley D, Leung JH, Muhanna D, Sun Y, Ortega-Anaya J, Yu L, et al. Amino Acid Nanofibers Improve Glycemia and Confer Cognitive Therapeutic Efficacy to Bound Insulin. Pharmaceutics. 2022; 14(1):81. https://doi.org/10.3390/pharmaceutics14010081
Chicago/Turabian StyleLee, Aejin, McKensie L. Mason, Tao Lin, Shashi Bhushan Kumar, Devan Kowdley, Jacob H. Leung, Danah Muhanna, Yuan Sun, Joana Ortega-Anaya, Lianbo Yu, and et al. 2022. "Amino Acid Nanofibers Improve Glycemia and Confer Cognitive Therapeutic Efficacy to Bound Insulin" Pharmaceutics 14, no. 1: 81. https://doi.org/10.3390/pharmaceutics14010081
APA StyleLee, A., Mason, M. L., Lin, T., Kumar, S. B., Kowdley, D., Leung, J. H., Muhanna, D., Sun, Y., Ortega-Anaya, J., Yu, L., Fitzgerald, J., DeVries, A. C., Nelson, R. J., Weil, Z. M., Jiménez-Flores, R., Parquette, J. R., & Ziouzenkova, O. (2022). Amino Acid Nanofibers Improve Glycemia and Confer Cognitive Therapeutic Efficacy to Bound Insulin. Pharmaceutics, 14(1), 81. https://doi.org/10.3390/pharmaceutics14010081