
E96V Mutation in the Kdelr3 Gene Is Associated with Type 2 Diabetes Susceptibility in Obese NZO Mice

 , , , , , and
, , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
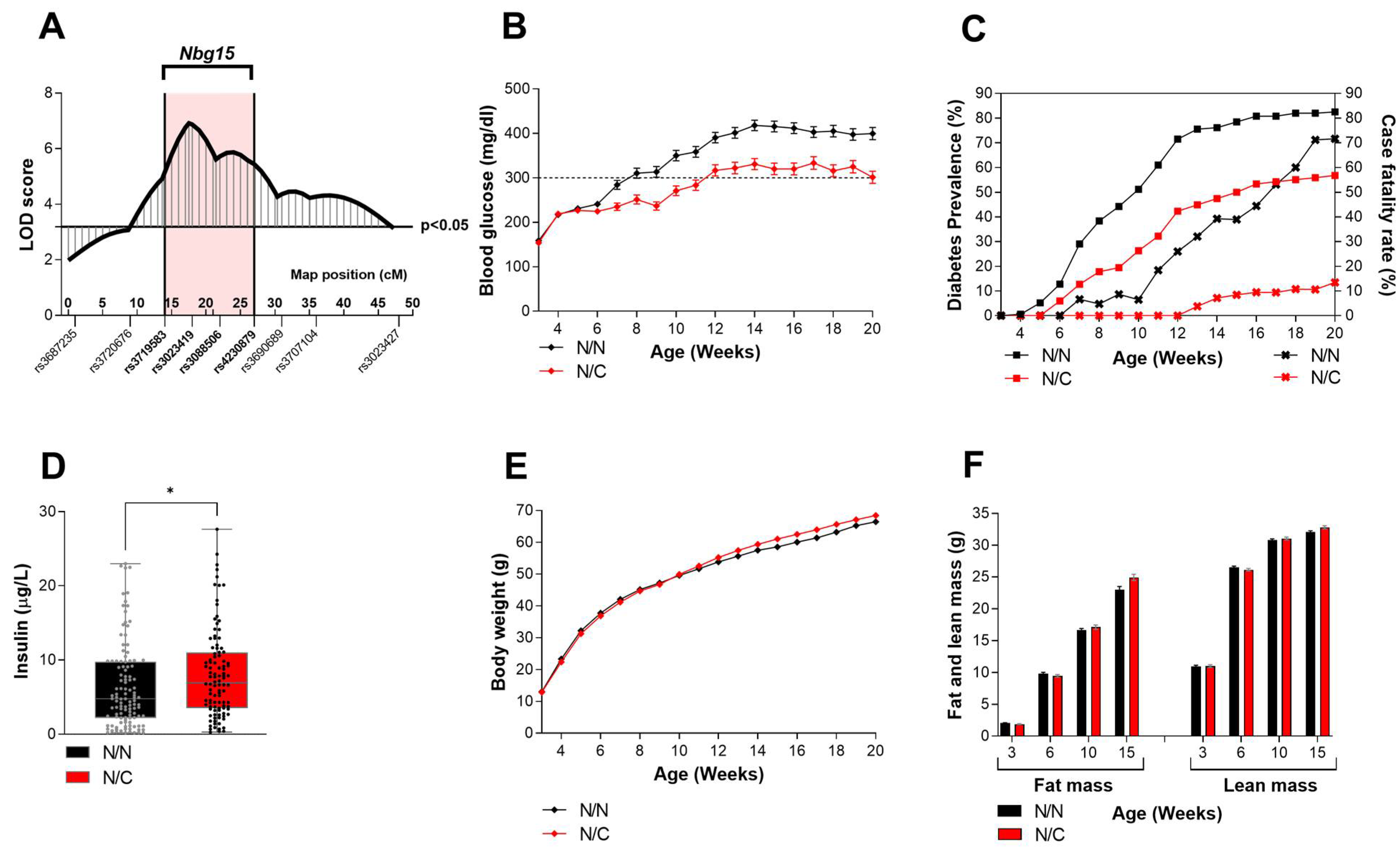
2.1. Heterozygous Carriers of the Nbg15-C3H Alleles Demonstrate Improved Blood Glucose Levels Independently of Their Body Weight
2.2. Introgression of the Nbg15/C3H Locus into the NZO Genome Protects Mice from Chronic Hyperglycemia
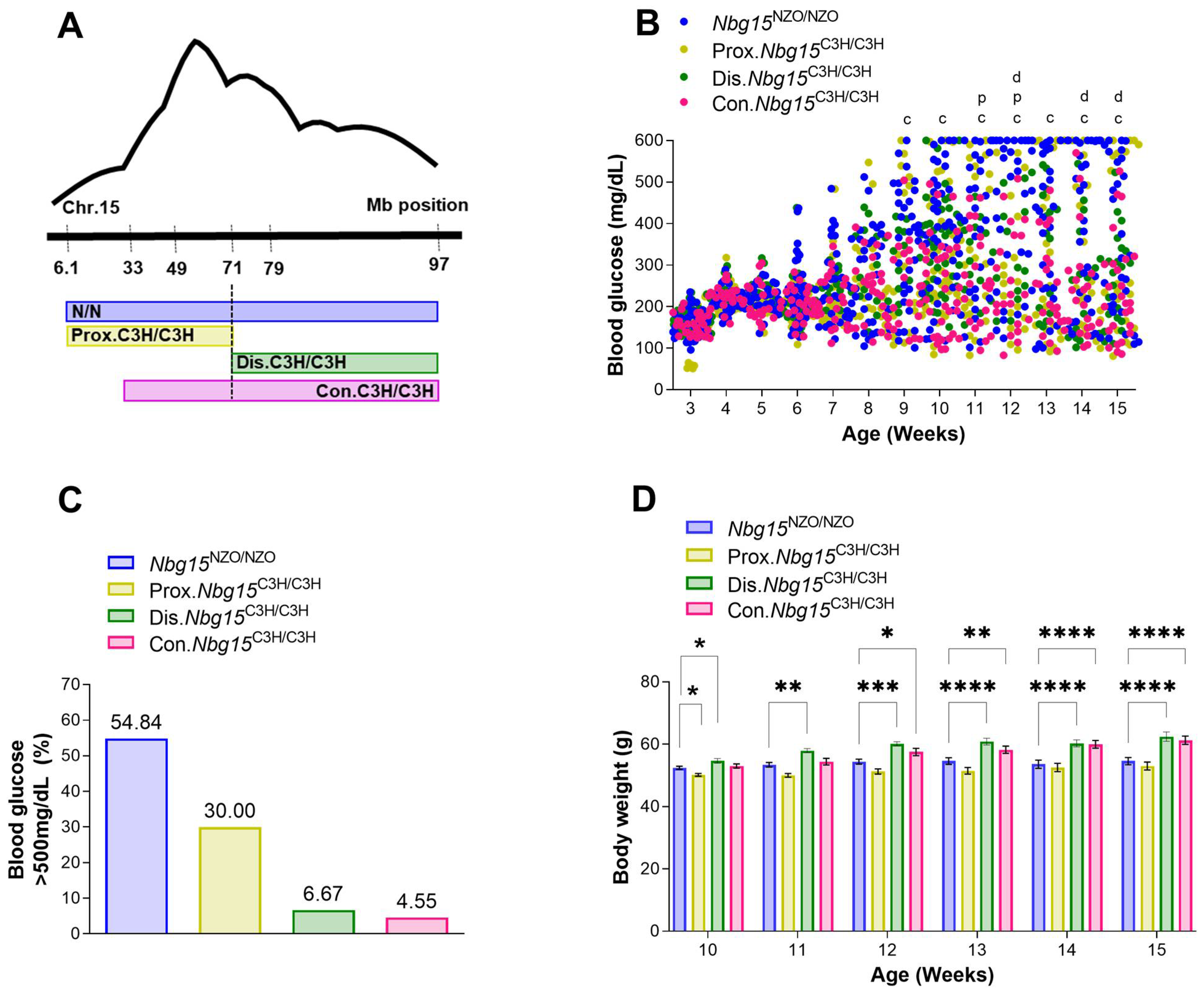
2.3. Distal but Not Proximal Nbg15 Sublocus Confers Protection from Hyperglycemia in NZO Congenic Mice
2.4. Distal Nbg15 Sublocus Protects NZO Congenic Mice from Pancreatic Islets Dysfunction
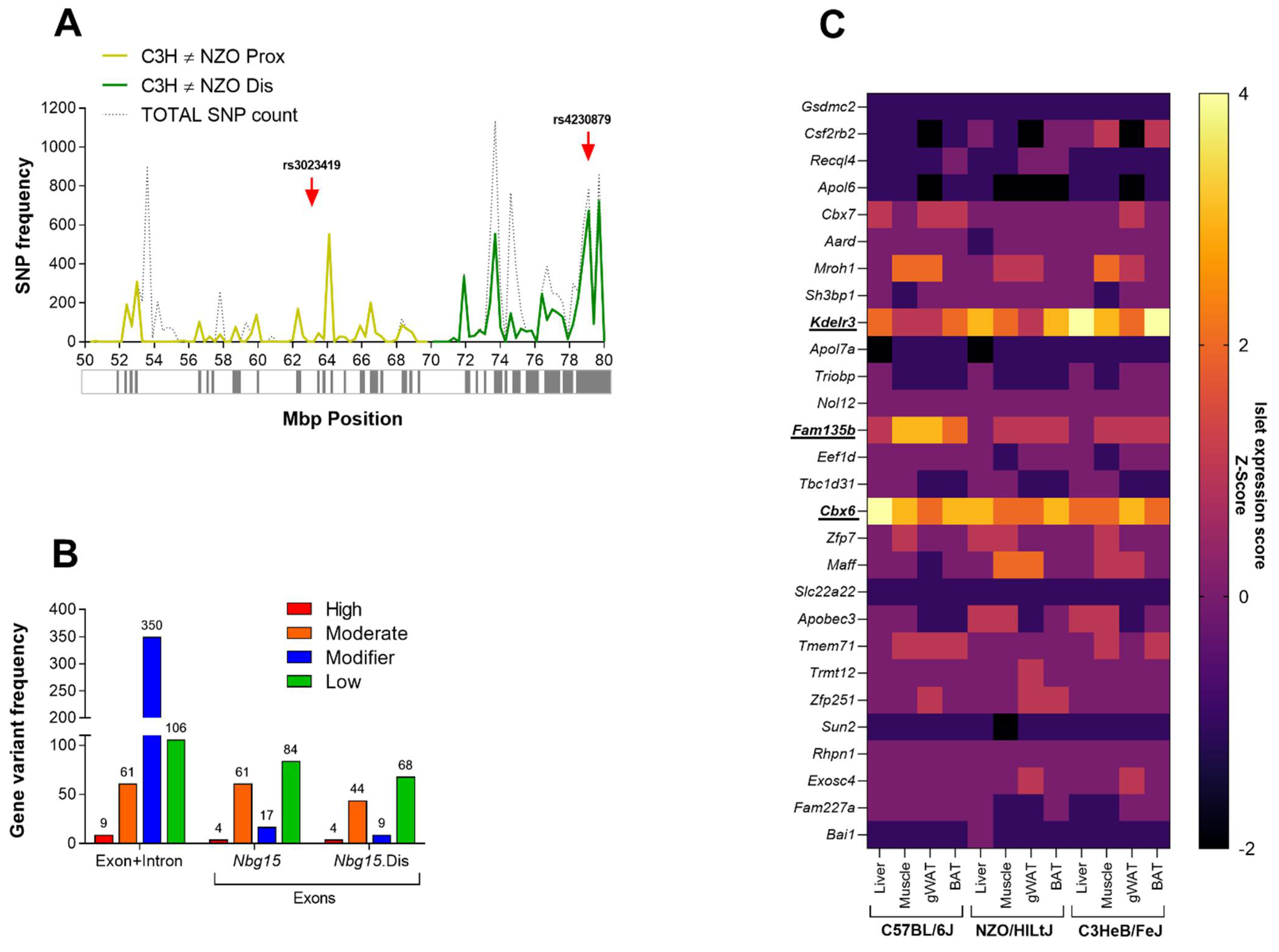
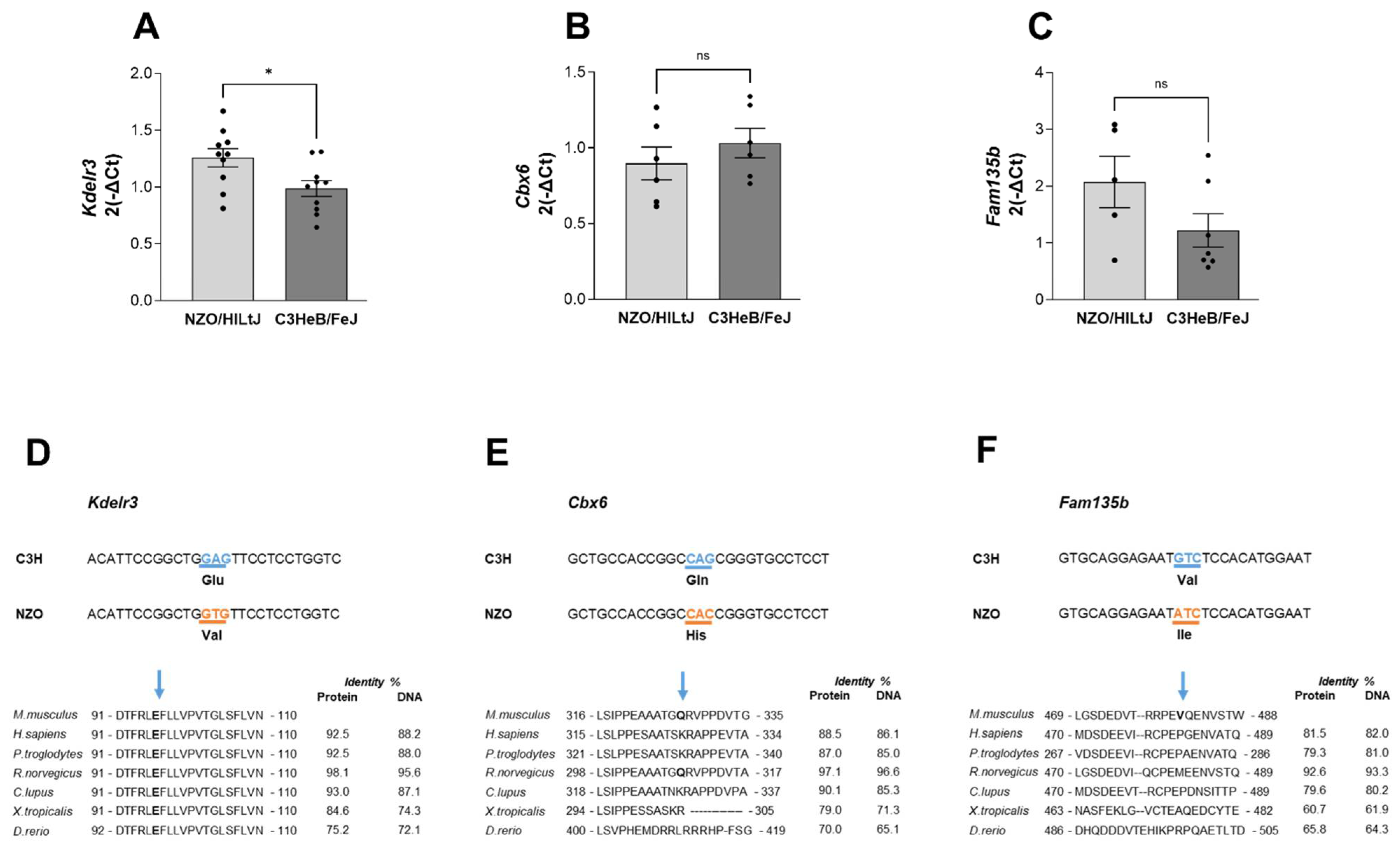
2.5. Kdelr3 Represents a Potentially Causal Gene for the Diabetes-Protective Distal Sublocus
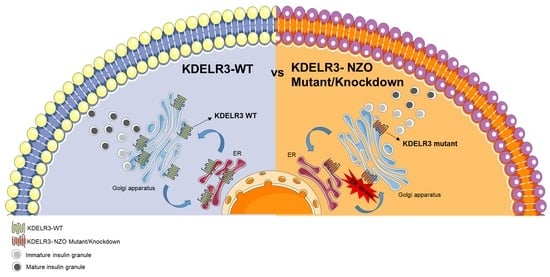
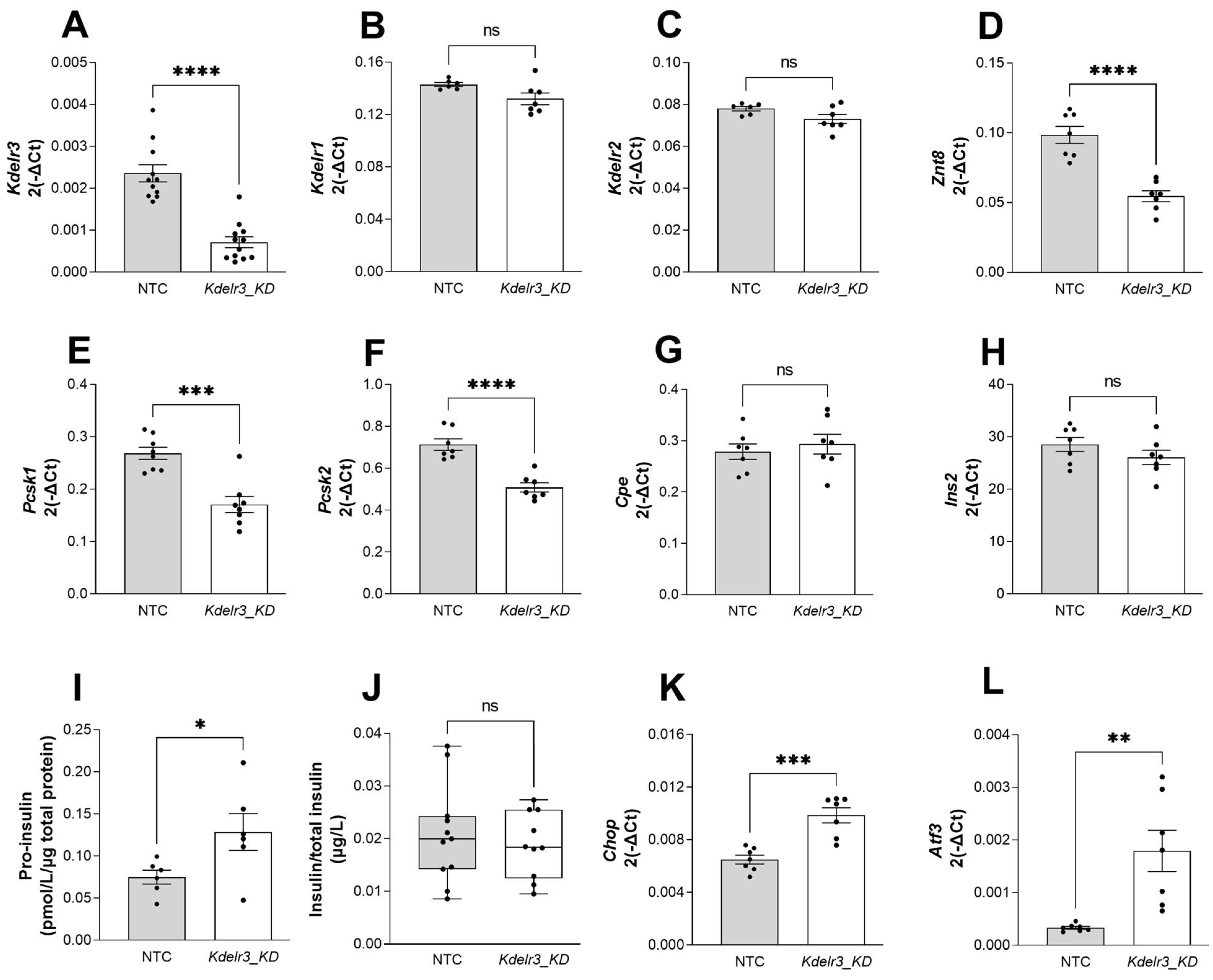
2.6. Changes in Kdelr3 Gene Expression Lead to Alterations in Insulin Granule Maturation and Upregulation of ER Stress Signals in Pancreatic Beta Cells
3. Discussion
Conclusion and Future Perspectives
4. Materials and Methods
4.1. Chemicals and Buffer
4.2. Experimental Animals
4.3. Blood Glucose, Body Weight and Body Composition
4.4. Fasting/Refeeding and Insulin Tolerance Test
4.5. Plasma Insulin
4.6. Pancreas Histology and Islets Isolation
4.7. Cell Culture and Knockdown Experiments
4.8. RNA Extraction, cDNA Synthesis and Quantitative Real-Time PCR
4.9. Sequence, Haplotype and Gene Variants Analysis
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- 2019, IDANe. IDF 2019 IDF DIABETES ATLAS Ninth Edition 2019. Available online: https://www.diabetesatlas.org/upload/resources/2019/IDF_Atlas_9th_Edition_2019.pdf (accessed on 10 December 2019).
- Riddle, M.C.; Buse, J.B.; Franks, P.W.; Knowler, W.C.; Ratner, R.E.; Selvin, E.; Wexler, D.J.; Kahn, S.E. COVID-19 in People With Diabetes: Urgently Needed Lessons From Early Reports. Diabetes Care 2020, 43, 1378–1381. [Google Scholar] [CrossRef]
- Prasad, R.B.; Groop, L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes 2015, 6, 87–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; Wu, Y.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, M.; Maeda, S. Genetics of type 2 diabetes: The GWAS era and future perspectives [Review]. Endocr. J. 2011, 58, 723–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manchia, M.; Cullis, J.; Turecki, G.; Rouleau, G.A.; Uher, R.; Alda, M. The impact of phenotypic and genetic heterogeneity on results of genome wide association studies of complex diseases. PLoS ONE 2013, 8, e76295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liley, J.; Todd, J.A.; Wallace, C. A method for identifying genetic heterogeneity within phenotypically defined disease subgroups. Nat. Genet. 2017, 49, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Broman, K.W. Review of statistical methods for QTL mapping in experimental crosses. Lab. Anim. 2001, 30, 44–52. [Google Scholar]
- Joost, H.G.; Schurmann, A. The genetic basis of obesity-associated type 2 diabetes (diabesity) in polygenic mouse models. Mamm. Genome 2014, 25, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Jurgens, H.S.; Schurmann, A.; Kluge, R.; Ortmann, S.; Klaus, S.; Joost, H.G.; Tschop, M.H. Hyperphagia, lower body temperature, and reduced running wheel activity precede development of morbid obesity in New Zealand obese mice. Physiol. Genom. 2006, 25, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Chadt, A.; Leicht, K.; Deshmukh, A.; Jiang, L.Q.; Scherneck, S.; Bernhardt, U.; Dreja, T.; Vogel, H.; Schmolz, K.; Kluge, R.; et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat. Genet. 2008, 40, 1354–1359. [Google Scholar] [CrossRef]
- Park, S.Y.; Jin, W.; Woo, J.R.; Shoelson, S.E. Crystal structures of human TBC1D1 and TBC1D4 (AS160) RabGTPase-activating protein (RabGAP) domains reveal critical elements for GLUT4 translocation. J. Biol. Chem. 2011, 286, 18130–18138. [Google Scholar] [CrossRef] [PubMed]
- Middelbeek, R.J.; Chambers, M.A.; Tantiwong, P.; Treebak, J.T.; An, D.; Hirshman, M.F.; Musi, N.; Goodyear, L.J. Insulin stimulation regulates AS160 and TBC1D1 phosphorylation sites in human skeletal muscle. Nutr. Diabetes 2013, 3, e74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, H.; Kamitz, A.; Hallahan, N.; Lebek, S.; Schallschmidt, T.; Jonas, W.; Jahnert, M.; Gottmann, P.; Zellner, L.; Kanzleiter, T.; et al. A collective diabetes cross in combination with a computational framework to dissect the genetics of human obesity and Type 2 diabetes. Hum. Mol. Genet. 2018, 27, 3099–3112. [Google Scholar] [CrossRef] [PubMed]
- Schallschmidt, T.; Lebek, S.; Altenhofen, D.; Damen, M.; Schulte, Y.; Knebel, B.; Herwig, R.; Rasche, A.; Stermann, T.; Kamitz, A.; et al. Two Novel Candidate Genes for Insulin Secretion Identified by Comparative Genomics of Multiple Backcross Mouse Populations. Genetics 2018, 210, 1527–1542. [Google Scholar] [CrossRef] [Green Version]
- Jonas, W.; Kluth, O.; Helms, A.; Voss, S.; Jahnert, M.; Gottmann, P.; Speckmann, T.; Knebel, B.; Chadt, A.; Al-Hasani, H.; et al. Identification of Novel Genes Involved in Hyperglycemia in Mice. Int. J. Mol. Sci. 2022, 23, 3205. [Google Scholar] [CrossRef]
- Chung, B.; Stadion, M.; Schulz, N.; Jain, D.; Scherneck, S.; Joost, H.G.; Schurmann, A. The diabetes gene Zfp69 modulates hepatic insulin sensitivity in mice. Diabetologia 2015, 58, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Scherneck, S.; Nestler, M.; Vogel, H.; Bluher, M.; Block, M.D.; Berriel Diaz, M.; Herzig, S.; Schulz, N.; Teichert, M.; Tischer, S.; et al. Positional cloning of zinc finger domain transcription factor Zfp69, a candidate gene for obesity-associated diabetes contributed by mouse locus Nidd/SJL. PLoS Genet. 2009, 5, e1000541. [Google Scholar] [CrossRef] [Green Version]
- Aga, H.; Hallahan, N.; Gottmann, P.; Jaehnert, M.; Osburg, S.; Schulze, G.; Kamitz, A.; Arends, D.; Brockmann, G.; Schallschmidt, T.; et al. Identification of Novel Potential Type 2 Diabetes Genes Mediating beta-Cell Loss and Hyperglycemia Using Positional Cloning. Front. Genet. 2020, 11, 567191. [Google Scholar] [CrossRef]
- Takeshita, S.; Moritani, M.; Kunika, K.; Inoue, H.; Itakura, M. Diabetic modifier QTLs identified in F2 intercrosses between Akita and A/J mice. Mamm. Genome 2006, 17, 927–940. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, S.; Kitayama, S.; Suzuki, T.; Moritani, M.; Inoue, H.; Itakura, M. Diabetic modifier QTL, Dbm4, affecting elevated fasting blood glucose concentrations in congenic mice. Genes Genet. Syst. 2012, 87, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Hillebrandt, S.; Goos, C.; Matern, S.; Lammert, F. Genome-wide analysis of hepatic fibrosis in inbred mice identifies the susceptibility locus Hfib1 on chromosome 15. Gastroenterology 2002, 123, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Newell, B.L.; Kechris, K.; McQueen, M.B.; Johnson, T.E. Genetic analysis of a murine QTL for diet restriction on chromosome 15. Age 2015, 37, 9740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togawa, K.; Moritani, M.; Yaguchi, H.; Itakura, M. Multidimensional genome scans identify the combinations of genetic loci linked to diabetes-related phenotypes in mice. Hum. Mol. Genet. 2006, 15, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoag, K.A.; Clise-Dwyer, K.; Lim, Y.H.; Nashold, F.E.; Gestwicki, J.; Cancro, M.P.; Hayes, C.E. A quantitative-trait locus controlling peripheral B-cell deficiency maps to mouse Chromosome 15. Immunogenetics 2000, 51, 924–929. [Google Scholar] [CrossRef]
- Reifsnyder, P.C.; Churchill, G.; Leiter, E.H. Maternal environment and genotype interact to establish diabesity in mice. Genome Res. 2000, 10, 1568–1578. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Korstanje, R.; Higgins, D.; Paigen, B. Haplotype analysis in multiple crosses to identify a QTL gene. Genome Res. 2004, 14, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, A. A Strategy for Identifying Quantitative Trait Genes Using Gene Expression Analysis and Causal Analysis. Genes 2017, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Brauer, P.; Parker, J.L.; Gerondopoulos, A.; Zimmermann, I.; Seeger, M.A.; Barr, F.A.; Newstead, S. Structural basis for pH-dependent retrieval of ER proteins from the Golgi by the KDEL receptor. Science 2019, 363, 1103–1107. [Google Scholar] [CrossRef]
- Tao, Y.; Cheung, L.S.; Li, S.; Eom, J.S.; Chen, L.Q.; Xu, Y.; Perry, K.; Frommer, W.B.; Feng, L. Structure of a eukaryotic SWEET transporter in a homotrimeric complex. Nature 2015, 527, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Raykhel, I.; Alanen, H.; Salo, K.; Jurvansuu, J.; Nguyen, V.D.; Latva-Ranta, M.; Ruddock, L. A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 2007, 179, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Capitani, M.; Sallese, M. The KDEL receptor: New functions for an old protein. FEBS Lett. 2009, 583, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Trychta, K.A.; Back, S.; Henderson, M.J.; Harvey, B.K. KDEL Receptors Are Differentially Regulated to Maintain the ER Proteome under Calcium Deficiency. Cell Rep. 2018, 25, 1829–1840.e6. [Google Scholar] [CrossRef] [PubMed]
- Saudek, V. Cystinosin, MPDU1, SWEETs and KDELR belong to a well-defined protein family with putative function of cargo receptors involved in vesicle trafficking. PLoS ONE 2012, 7, e30876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.K.; Kim, H.S.; Lee, I.K.; Park, K.G. Endoplasmic reticulum stress and insulin biosynthesis: A review. Exp. Diabetes Res. 2012, 2012, 509437. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Calvo, T.; Zapardiel-Gonzalo, J.; Amirian, N.; Castillo, E.; Lajevardi, Y.; Krogvold, L.; Dahl-Jorgensen, K.; von Herrath, M.G. Increase in Pancreatic Proinsulin and Preservation of beta-Cell Mass in Autoantibody-Positive Donors Prior to Type 1 Diabetes Onset. Diabetes 2017, 66, 1334–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Calvo, T.; Chen, Y.C.; Verchere, C.B.; Haataja, L.; Arvan, P.; Leete, P.; Richardson, S.J.; Morgan, N.G.; Qian, W.J.; Pugliese, A.; et al. Altered beta-Cell Prohormone Processing and Secretion in Type 1 Diabetes. Diabetes 2021, 70, 1038–1050. [Google Scholar] [CrossRef]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [Green Version]
- Asplund, O.; Storm, P.; Chandra, V.; Hatem, G.; Ottosson-Laakso, E.; Mansour-Aly, D.; Krus, U.; Ibrahim, H.; Ahlqvist, E.; Tuomi, T.; et al. Islet Gene View-a tool to facilitate islet research. Life Sci. Alliance 2022, 5, e202201376. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, F.; Wei, K.; Mao, E.; Yin, G.; Wu, C. Identification of Novel Gene Signature Associated with Cell Glycolysis to Predict Survival in Hepatocellular Carcinoma Patients. J. Oncol. 2021, 2021, 5564525. [Google Scholar] [CrossRef]
- Arunagiri, A.; Haataja, L.; Pottekat, A.; Pamenan, F.; Kim, S.; Zeltser, L.M.; Paton, A.W.; Paton, J.C.; Tsai, B.; Itkin-Ansari, P.; et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife 2019, 8, e44532. [Google Scholar] [CrossRef] [PubMed]
- Reza, M.I.; Syed, A.A.; Kumariya, S.; Singh, P.; Husain, A.; Gayen, J.R. Pancreastatin induces islet amyloid peptide aggregation in the pancreas, liver, and skeletal muscle: An implication for type 2 diabetes. Int. J. Biol. Macromol. 2021, 182, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; De Franco, E.; Arvan, P.; Cnop, M. Pathological beta-Cell Endoplasmic Reticulum Stress in Type 2 Diabetes: Current Evidence. Front. Endocrinol. 2021, 12, 650158. [Google Scholar] [CrossRef] [PubMed]
- Stermann, T.; Menzel, F.; Weidlich, C.; Jeruschke, K.; Weiss, J.; Altenhofen, D.; Benninghoff, T.; Pujol, A.; Bosch, F.; Rustenbeck, I.; et al. Deletion of the RabGAP TBC1D1 Leads to Enhanced Insulin Secretion and Fatty Acid Oxidation in Islets From Male Mice. Endocrinology 2018, 159, 1748–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Goodstadt, L.; Danecek, P.; White, M.A.; Wong, K.; Yalcin, B.; Heger, A.; Agam, A.; Slater, G.; Goodson, M.; et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011, 477, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Doran, A.G.; Wong, K.; Flint, J.; Adams, D.J.; Hunter, K.W.; Keane, T.M. Deep genome sequencing and variation analysis of 13 inbred mouse strains defines candidate phenotypic alleles, private variation and homozygous truncating mutations. Genome Biol. 2016, 17, 167. [Google Scholar] [CrossRef] [Green Version]
- Lilue, J.; Doran, A.G.; Fiddes, I.T.; Abrudan, M.; Armstrong, J.; Bennett, R.; Chow, W.; Collins, J.; Collins, S.; Czechanski, A.; et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat. Genet. 2018, 50, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Gonzaludo, N.P.; Strunk, S.; Dahm, S.; Schuchhardt, J.; Kleinjung, F.; Wuschke, S.; Joost, H.G.; Al-Hasani, H. A meta-analysis of QTL for diabetes-related traits in rodents. Physiol. Genom. 2008, 34, 42–53. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altenhofen, D.; Khuong, J.M.-A.; Kuhn, T.; Lebek, S.; Görigk, S.; Kaiser, K.; Binsch, C.; Griess, K.; Knebel, B.; Belgardt, B.-F.; et al. E96V Mutation in the Kdelr3 Gene Is Associated with Type 2 Diabetes Susceptibility in Obese NZO Mice. Int. J. Mol. Sci. 2023, 24, 845. https://doi.org/10.3390/ijms24010845
Altenhofen D, Khuong JM-A, Kuhn T, Lebek S, Görigk S, Kaiser K, Binsch C, Griess K, Knebel B, Belgardt B-F, et al. E96V Mutation in the Kdelr3 Gene Is Associated with Type 2 Diabetes Susceptibility in Obese NZO Mice. International Journal of Molecular Sciences. 2023; 24(1):845. https://doi.org/10.3390/ijms24010845
Chicago/Turabian StyleAltenhofen, Delsi, Jenny Minh-An Khuong, Tanja Kuhn, Sandra Lebek, Sarah Görigk, Katharina Kaiser, Christian Binsch, Kerstin Griess, Birgit Knebel, Bengt-Frederik Belgardt, and et al. 2023. "E96V Mutation in the Kdelr3 Gene Is Associated with Type 2 Diabetes Susceptibility in Obese NZO Mice" International Journal of Molecular Sciences 24, no. 1: 845. https://doi.org/10.3390/ijms24010845