Abstract
The NO-cGMP signal transduction pathway plays a crucial role in tone regulation in hepatic sinusoids and peripheral blood vessels. In a cirrhotic liver, the key enzymes endothelial NO synthase (eNOS), soluble guanylate cyclase (sGC), and phosphodiesterase-5 (PDE-5) are overexpressed, leading to decreased cyclic guanosine-monophosphate (cGMP). This results in constriction of hepatic sinusoids, contributing about 30% of portal pressure. In contrast, in peripheral arteries, dilation prevails with excess cGMP due to low PDE-5. Both effects eventually lead to circulatory dysfunction in progressed liver cirrhosis. The conventional view of portal hypertension (PH) pathophysiology has been described using the “NO-paradox”, referring to reduced NO availability inside the liver and elevated NO production in the peripheral systemic circulation. However, recent data suggest that an altered availability of cGMP could better elucidate the contrasting findings of intrahepatic vasoconstriction and peripheral systemic vasodilation than mere focus on NO availability. Preclinical and clinical data have demonstrated that targeting the NO-cGMP pathway in liver cirrhosis using PDE-5 inhibitors or sGC stimulators/activators decreases intrahepatic resistance through dilation of sinusoids, lowering portal pressure, and increasing portal venous blood flow. These results suggest further clinical applications in liver cirrhosis. Targeting the NO-cGMP system plays a role in possible reversal of liver fibrosis or cirrhosis. PDE-5 inhibitors may have therapeutic potential for hepatic encephalopathy. Serum/plasma levels of cGMP can be used as a non-invasive marker of clinically significant portal hypertension. This manuscript reviews new data about the role of the NO-cGMP signal transduction system in pathophysiology of cirrhotic portal hypertension and provides perspective for further studies.
1. Introduction
Portal hypertension (PH) is defined as portal venous pressure exceeding 5 mm Hg. Significant sequelae of PH occur when pressure exceeds 10–12 mmHg [1,2,3,4,5,6]. It develops gradually from asymptomatic abnormalities to life-threatening complications. The impairment of portal blood flow may occur in different anatomical locations. PH is classified as prehepatic (e.g., portal venous thrombosis), intrahepatic, and posthepatic (e.g., hepatic vein thrombosis, chronic right-sided heart failure) [2,7,8,9]. Intrahepatic PH is further differentiated into presinusoidal, sinusoidal, and postsinusoidal. Sinusoidal PH occurs most frequently as a direct consequence of liver cirrhosis and is the focus in this review. Successful therapy of PH aims to decrease the hepatic-venous pressure gradient (HVPG) by ≥20% of baseline or ≤12 mmHg [1,2,10,11]. A short-term response of HVPG to acute drug application (i.e., propranolol) by ≥10% of baseline or a reduction of ≤12 mmHg predicts a beneficial effect for prophylaxis of variceal bleeding [11,12,13,14].
An increase in hepatic vascular resistance to portal blood flow is the main factor of intrahepatic PH. The structural component contributing about 70% to the intrahepatic vascular resistance is caused by the disturbed liver architecture, fibrotic tissue, regenerative nodules, angiogenesis, narrowing of intrahepatic blood vessels by extracellular matrix proteins, and vascular occlusion. A dynamic component accounts for about 30%. It is caused by an impaired interplay of the sinusoidal cells, i.e., hepatocytes, liver sinusoidal endothelial cells (LSECs), and hepatic stellate cells (HSCs), characterized by transformation of contractile HSCs to myofibroblasts, synthesis of extracellular matrix proteins, and capillarization of LSECs. Eventually, this leads to an increased sinusoidal tone caused by an imbalance of dilating and constricting factors [7,15,16,17,18,19,20,21]. Moreover, elevated splanchnic blood flow due to dilation of splanchnic blood vessels contributes to the development and/or worsening of PH [21,22,23].
The main regulator of vascular and sinusoidal tone is the NO-cGMP pathway [19,20,24]. Endothelial NO-synthase (eNOS) generates NO from L-arginine, which diffuses to smooth muscle cells in peripheral blood vessels and HSCs in liver sinusoids. NO activates soluble guanylate cyclase (sGC), catalyzing the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). cGMP, in turn, activates protein kinases (e.g., protein kinase G, (PKG)), PDE-5, and cGMP-gated ion channels. cGMP induces a decrease of intracellular calcium ions and vascular dilation. cGMP is then converted to functionally inactive 5′-GMP by PDE-5 [25,26,27,28]. Among other regulatory pathways, the NO-cGMP signal transduction system is characteristically altered in liver cirrhosis. Hence, structural alterations and functional dysregulations play a crucial role in the pathogenesis of PH. For years, research in PH has been focused on NO and led to the theory of the “NO-paradox”—intrahepatic circulation is marked by an NO deficiency, in contrast to excess NO in the peripheral systemic circulation [29,30,31,32]. It states that vasoconstriction prevails in intrahepatic vasculature, whereas extrahepatic vasculature is characterized by dilation, leading to a hyperdynamic circulatory state.
This manuscript reviews recent studies on localization and expression of the enzymes eNOS, sGC, and PDE-5, as well as the molecule cGMP in a healthy and cirrhotic liver. It proposes shifting the focus to cGMP as the key component responsible for sinusoidal constriction and peripheral vasodilation in liver cirrhosis. We discuss study recommendations on the effects of sGC modulators and PDE-5-inhibitors in liver diseases such as modulation of the NO-cGMP pathway for treatment of portal hypertension [33], reversal of fibrosis/cirrhosis, therapy of hepatic encephalopathy using PDE-5-inhibitors, and determination of serum/plasma cGMP levels as an indicator of portal hypertension.
2. Current Research Directions in Components of the NO-cGMP Pathway in Experimental Liver Damage
NO and eNOS: In a healthy liver, eNOS is constitutively expressed. A number of studies showed reduced activity of eNOS in a cirrhotic liver. However, it is not clear whether this reflects reduced expression or disturbed post-translational modification of the enzyme [34,35,36,37,38,39,40]. Several more recent studies [41,42,43] found increased eNOS expression in a cirrhotic liver, however, this may depend on the stage or type of liver damage. Schaffner et al. (2018) [42] demonstrated upregulation of eNOS in TAA-induced rat liver fibrosis and cirrhosis and high expression of PDE-5. The same findings were reported by Uschner et al. (2020) [43]—eNOS was upregulated in both BDL-induced and CCl4-induced liver cirrhosis, accompanied by PDE-5 upregulation. In hepatic arteries of human cirrhotics, however, eNOS was upregulated and PDE-5 was downregulated, while in the aortic wall of cirrhotic rats, eNOS was upregulated, and PDE-5 was downregulated.
sGC: Using immunohistochemistry, Theilig et al. (2001) [44] detected sGC in HSCs and found decreasing amounts of this enzyme from the periphery of the hepatic lobule towards the central vein as an indicator of metabolic zonation of the NO-cGMP pathway. According to Davies et al. (2006) [45], sGC activity was lowered in BDL-induced rat liver cirrhosis. In contrast, Loureiro-Silva et al. (2006) [46], Lee et al. (2010) [47], Schwabl et al. (2018, 2020) [41,48], and Schaffner et al. (2018) [42] described overexpression of sGC in different types and stages of experimental liver damage. Hall et al. (2019) [49] described sGC in HSCs and myofibroblasts in a rat model of non-alcoholic steatohepatitis (NASH), however, it was not present in hepatocytes. In CCl4-induced rat liver cirrhosis, sGC was overexpressed and was also found in large amounts in fibrotic tissue within a cirrhotic liver. These findings were confirmed in human NASH. The sGC stimulator praliciguat markedly increased hepatic cGMP levels in experimental liver cirrhosis and reduced fibrosis and inflammation in a damaged liver [49]. Schwabl et al. (2018) described upregulation of the β1-subunit of sGC in BDL-induced liver cirrhosis and to a lesser extent in CCl4-induced liver cirrhosis, whereas the α1-subunit remained unchanged [41]. In a healthy rat liver, sGC was primarily detected in HSCs and hepatocytes, as well as in LSECs. After bile duct ligation, sGC expression was observed in HSCs, hepatocytes, and Kupffer cells. Stimulation of sGC by Riociguat decreased portal pressure and improved fibrosis, as demonstrated by histology and measurement of αSMA expression. Inflammatory markers were reduced in both types of experimental liver damage [48].
PDE-5: Perri et al. (2006) [50] described a defective cGMP-PKG pathway in activated HSCs contributing to an impaired NO-dependent response. Activity or expression of PDE-5 was not considered in this study. Loureiro-Silva et al. (2006) [46] found a decreased vasodilatory response to NO in experimental liver cirrhosis as a consequence of PDE-5 overexpression. Markedly increased protein expression of PDE-5 and a slight overexpression of sGCα1β1 were reported by Lee et al. (2010) [47] in BDL-induced liver cirrhosis. They observed that after a one-week administration of Sildenafil, sGC was further upregulated and PDE-5 was reduced. This effect was accompanied by a reduction of portal venous pressure. Schaffner et al. (2018) [42], on the other hand, found that PDE-5 is overexpressed in different stages of TAA-induced liver fibrosis and cirrhosis. This may override the overexpression of eNOS demonstrated by the same group. The description of a zonation of PDE-5 yielded a new aspect in the regulation of cGMP levels inside the liver—high sGC in the periphery of the liver lobule [44] stimulated cGMP production, whereas high PDE-5 in perivenular regions converted cGMP to inactive 5′-GMP before entering systemic circulation. In TAA-induced liver damage, this zonation was lost and apart from increased expression of PDE-5 in perisinusoidal cells, high amounts of this enzyme were detected in fibrotic liver tissue [42]. Uschner et al. (2020) [43] confirmed upregulation of eNOS and PDE-5 in BDL- or CCl4-induced rat liver cirrhosis. Consequently, intrahepatic cGMP was reduced. In contrast, in aortic wall tissue, PDE-5 was reduced and eNOS was upregulated in cirrhosis models. Corresponding results were obtained in cirrhotic livers of human transplant recipients: PDE-5 and eNOS were upregulated. However, in hepatic arteries, PDE-5 mRNA was reduced and eNOS mRNA was increased in comparison to those of healthy liver donors. Moreover, it was reported that eNOS was markedly expressed in LSECs, whereas PDE-5 was mainly found in HSCs. Udenafil treatment increased or normalized cGMP levels in a cirrhotic rat liver, inducing dilation of sinusoids and reduction of portal pressure, however, cGMP was further increased in aortic tissue of cirrhotic rats. The combination of udenafil and propranolol blunted the effect of udenafil alone on cGMP in aortic tissue, however, the dilating effect inside the liver was additive. Furthermore, eNOS and PDE-5 were reduced in aortic tissue after combined application of udenafil and propranolol. These data yielded the rationale for a combination therapy of cirrhotic portal hypertension using a PDE-5-inhibitor (dilation of sinusoids) and propranolol (constriction of splanchnic blood vessels by blockade of beta-2 adrenoreceptors). In addition, these data showed that peripheral vasodilation can be explained by increased cGMP due to peripheral PDE-5 downregulation. eNOS was not the main player in this circulatory disturbance.
Another study by Brusilovskaya et al. (2020) investigated the effects of tadalafil (a long-acting inhibitor of PDE-5), riociguat (a sGC-stimulator), and cinaciguat (a sGC-activator) in BDL-induced liver cirrhosis [48]. Tadalafil and riociguat decreased portal pressure by about 20% and 10%, respectively, and decreased intrahepatic vascular resistance with a minor effect on systemic circulatory parameters. Both tadalafil and riociguat reduced the extent of liver fibrosis in comparison to that in control and decreased serum levels of aminotransferases, suggesting a beneficial effect on liver damage. Cinaciguat did not show the same beneficial effects. After tadalafil and riociguat administration, hepatic smooth muscle actin alpha protein concentration decreased and expression of phosphorylated moensin was reduced. BDL did not lead to a decrease of intrahepatic cGMP content. However, both tadalafil and riociguat induced a marked increase of intrahepatic cGMP by 240% and 32%, respectively. This was the first study which compared the effects of targeting sGC or PDE-5. The results do not clearly favor any of the two.
cGMP: Niederberger et al. (1995) [51] confirmed high cGMP in aortic walls in experimental cirrhosis and hypothesized an increase in NO synthesis caused by eNOS. The authors demonstrated that a correction of increased NO production normalized hyperdynamic circulatory disturbance [52]. Tahseldar et al. (2009) [53] investigated activities of PDEs in kidney and liver of rats with cirrhosis. They found an increase of cGMP hydrolyzing activity by 24% in cirrhosis (mainly PDE-5). It was observed that while PDE-5 was increased in mesenteric arteries, it remained unchanged in the aorta of cirrhotic animals. The study conducted by Uschner et al. (2020) [43] clarified some contrasting data about the disturbance of the NO-cGMP pathway in peripheral blood vessels in cirrhosis—PDE-5 was found to be downregulated in the aortic wall in experimental cirrhosis and in hepatic arteries in human cirrhosis. These data are consistent with the increase of serum cGMP irrespective of eNOS activity.
3. Role of the NO-cGMP Pathway in PH Pathophysiology
Considering these recent findings, we propose explanations for these controversial findings and thereby offer an alternative perspective in understanding the pathophysiology of PH. We suggest a paradigm highlighting how the NO-cGMP pathway is altered in liver cirrhosis. Modulation of sGC and/or PDE-5 regulates sinusoidal cGMP availability, thus influencing sinusoidal tone, portal pressure, and portal venous blood flow (Figure 1).
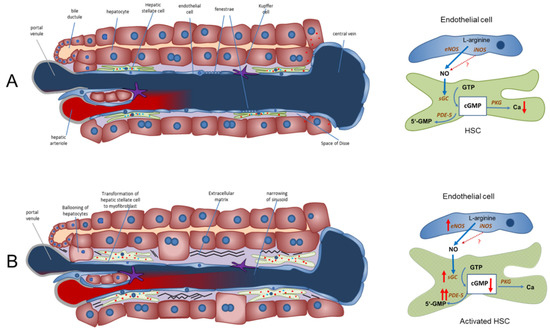
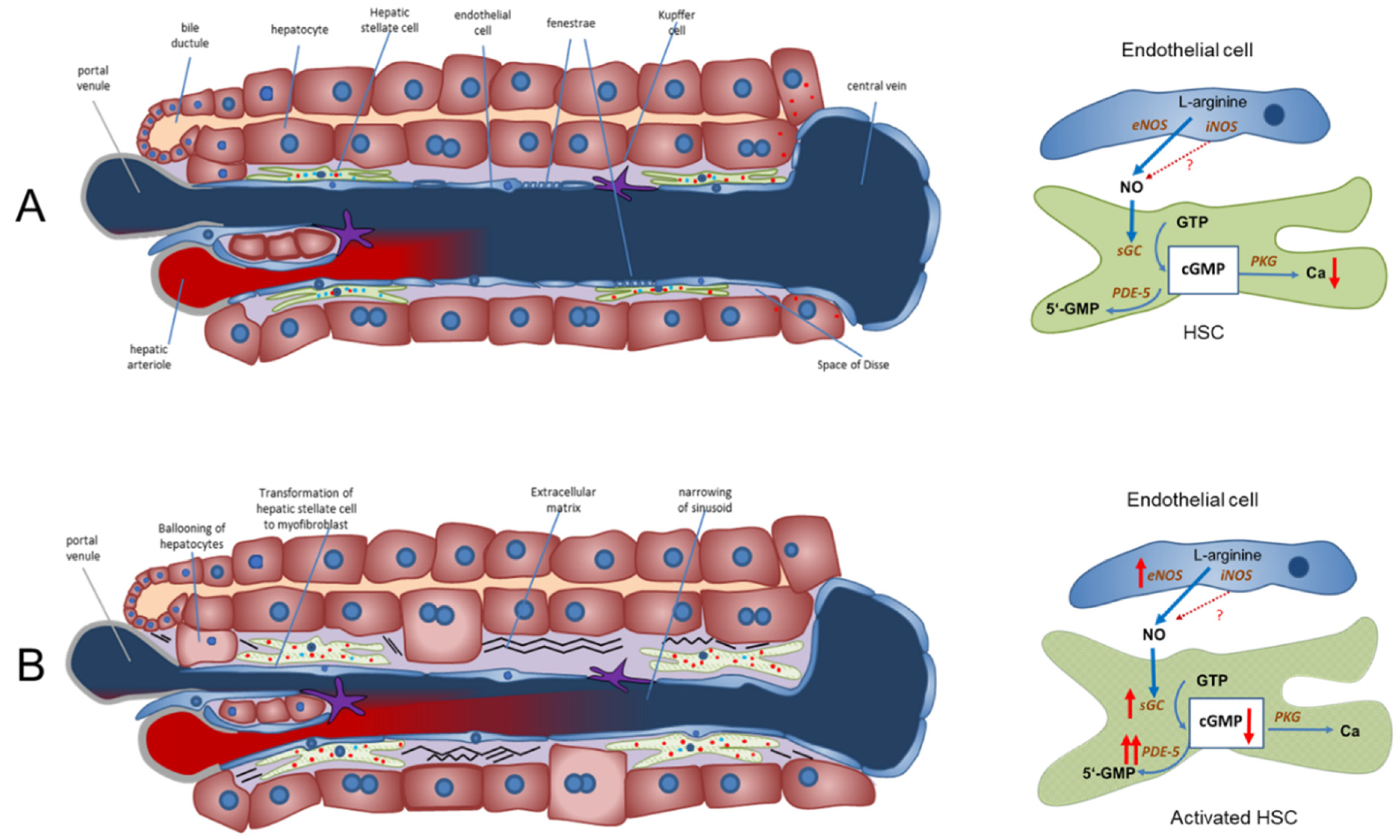
Figure 1.
The NO-cGMP pathway as the main regulator of sinusoidal and vascular tone in a healthy and cirrhotic liver (modified from Schaffner et al. (2018) [42] and Kreisel and Schaffner et al. (2020) [33]). (A) Regulation of sinusoidal tone in a healthy liver. In a healthy liver, an opposing zonation of sGC and PDE-5 may lead to high cGMP production in the peripheral parts of the hepatic lobule (high sGC), in which cGMP may exert its physiological function inside the sinusoids. Excess cGMP is possibly degraded by a high presence of PDE-5 (zone 3 hepatocytes) before entering extrahepatic vasculature. (B) Disturbed regulation of sinusoidal tone in liver cirrhosis. Altered expression of key enzymes in the NO-cGMP pathway (overexpression of eNOS and sGC and marked overexpression of PDE-5 in perisinusoidal cell, mainly activated HSCs) leads to reduced cGMP concentrations and sinusoidal constriction, increasing portal pressure. ↑ increased expression; ↑↑ markedly increased expression; cGMP ↓ decreased concentration. Red dots: PDE-5. Blue dots: sGC.
NO is generated by eNOS in sinusoidal endothelial cells and diffuses into the neighboring hepatic stellate cells, where it binds to and activates sGC. This enzyme catalyzes the conversion of GTP to cGMP. This intracellular second messenger, in turn, triggers distinct downstream signaling effects (e.g., lowering of intracellular Ca ions) which eventually exert vasodilation. As a negative feedback mechanism, rising cGMP concentrations initiate the activation of PDE-5, which mediates cGMP inactivation. The opposing zonation of sGC and PDE-5 may suggest regulation of sinusoidal cGMP. It is synthesized in the periphery of the sinusoids (high sGC concentration), exerts its physiological function along the sinusoids and is inactivated before it enters systemic circulation (high PDE-5 concentration in zone 3 hepatocytes). Figure 1, panel A.
In liver cirrhosis, both eNOS and sGC are overexpressed. Their activity is overridden by the marked overexpression of PDE-5 primarily in perisinusoidal cells and the loss of the physiological zonation of PDE-5. The final dilator cGMP is converted to inactive 5′-GMP. Sinusoids persist in a constricted state. Figure 1, panel B.
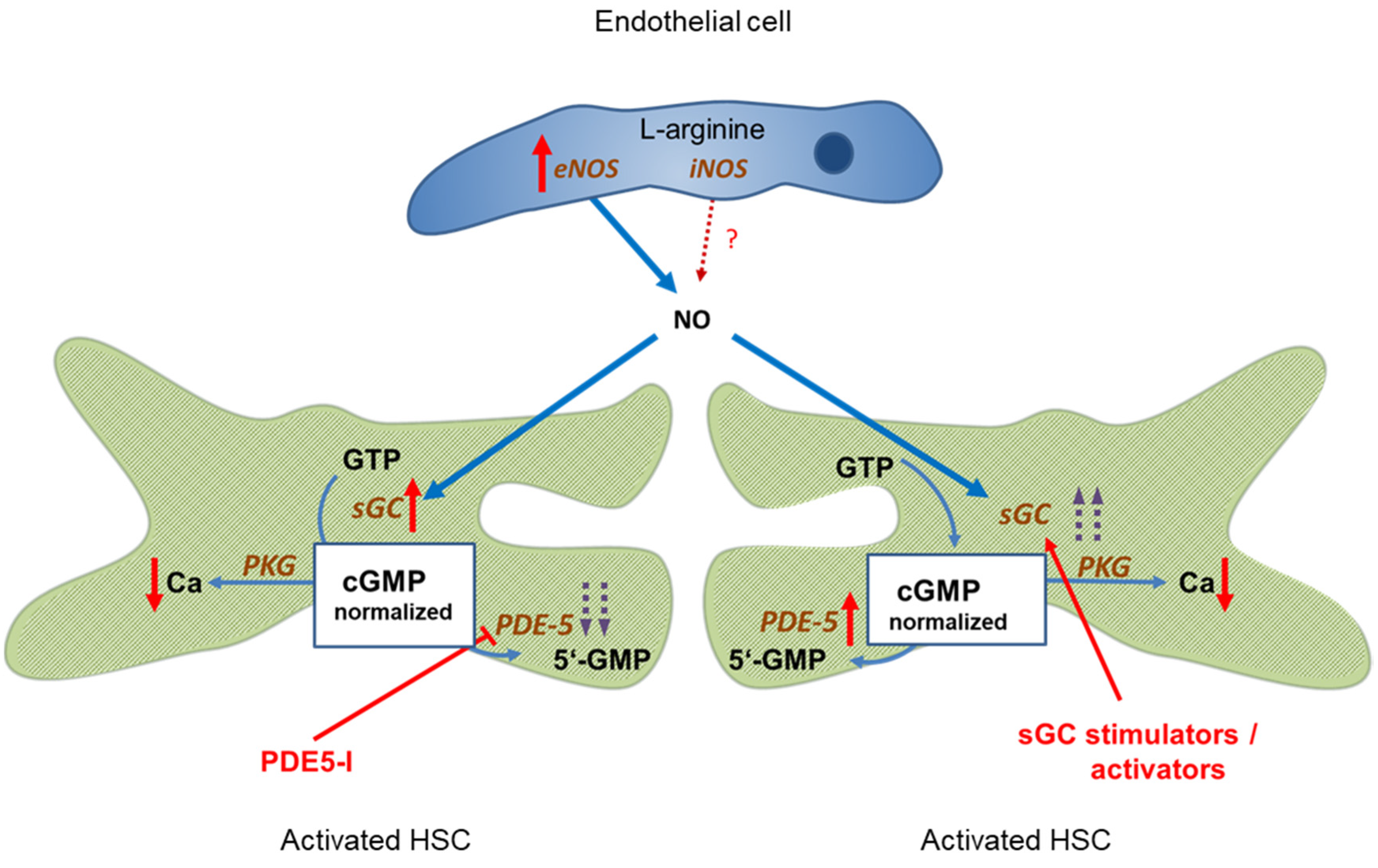
PDE-5 is markedly overexpressed in liver cirrhosis, however, its activity can be blocked by PDE-5 inhibitors. This leads to a normalization of cGMP concentrations and, consequently, sinusoidal dilation. Figure 2, left side.
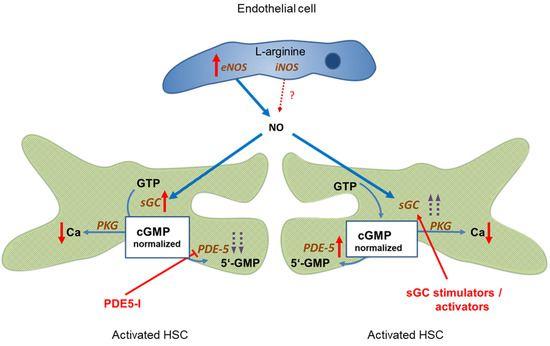
Figure 2.
Targeting the NO-cGMP pathway for therapy of portal hypertension. Left side: Administration of PDE-5 inhibitors in liver cirrhosis leads to normalization of cGMP concentration and sinusoidal dilation. Portal pressure decreases and portal blood flow increases. Right side: Enhancing sGC activity in liver cirrhosis using an sGC stimulator or activator leads to increased cGMP levels and decreased sinusoidal tone. Portal pressure decreases and portal blood flow increases. ↑ increased expression; ↑↑ markedly increased expression; Ca ↓ decreased concentration;  inhibition;
inhibition;  stimulation/activation.
stimulation/activation.
inhibition; stimulation/activation.
Comparable effects are achieved if sGC activity is further enhanced by either an sGC stimulator or activator. This overrides the overexpression of PDE-5, leading to normalization of cGMP levels and sinusoidal dilation. Figure 2, right side.
Further studies should show whether the etiology of liver damage has an effect on these changes in the NO-cGMP pathway.
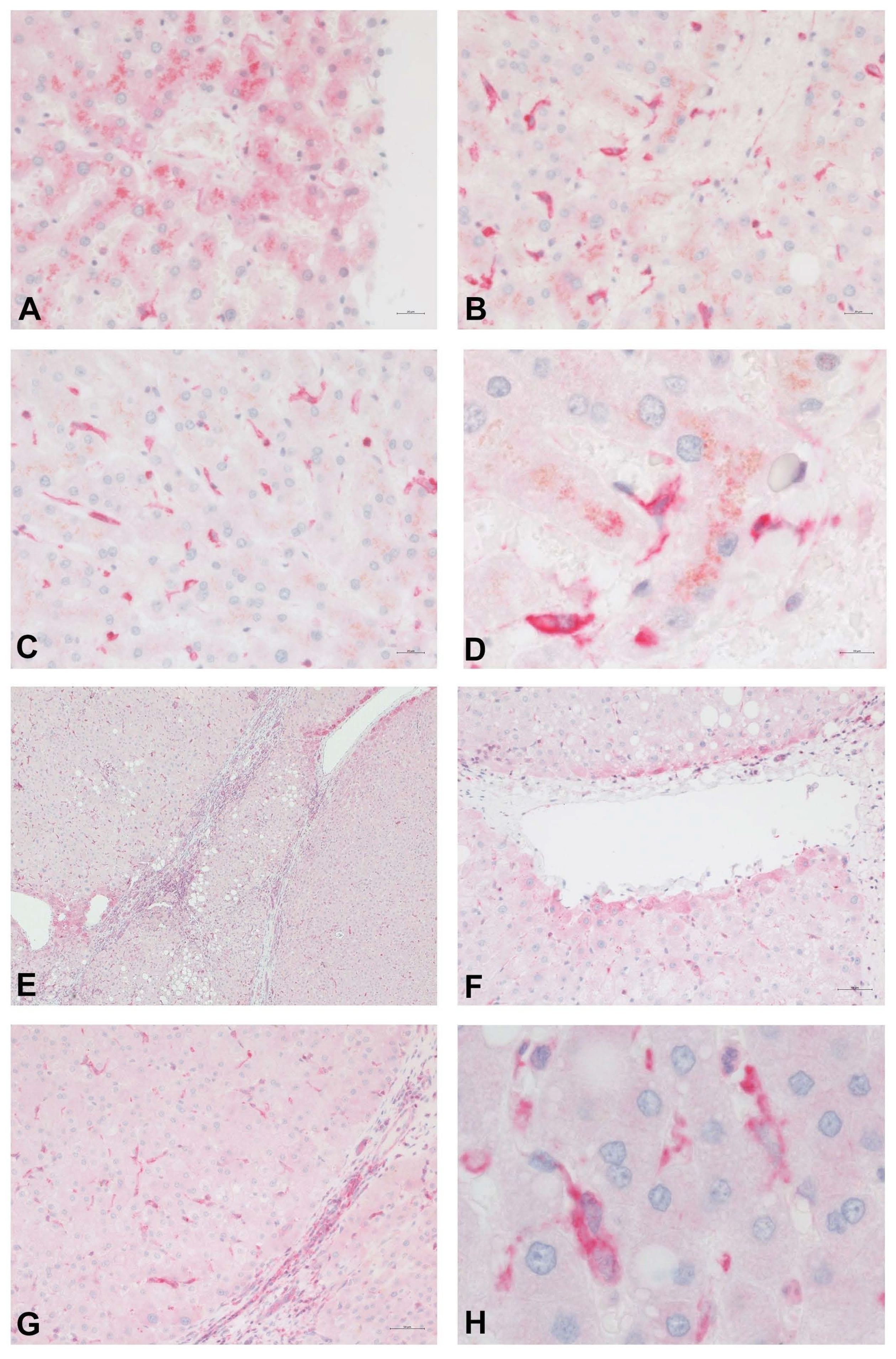
Figure 3 shows the distribution of PDE-5 in perisinusoidal cells and the expression in zone 3 hepatocytes in a healthy human liver. Immunolabeling of cirrhotic liver tissue highlights the presence of PDE-5 in fibrous septa, in hepatocytes adjacent to veins, and in perisinusoidal cells scattered throughout the parenchymal nodules.
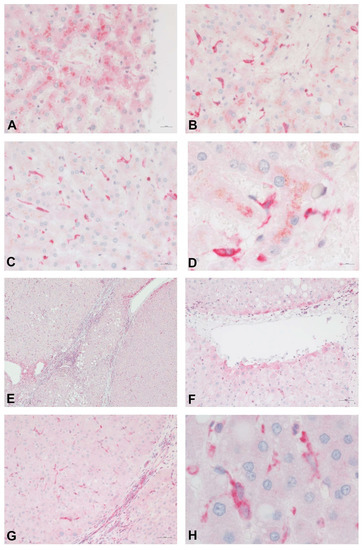
Figure 3.
Immunostain for PDE-5 (red) of normal (A–D) and cirrhotic (E–H) human liver tissue. In normal control liver samples, PDE-5 is weakly expressed in hepatocytes around terminal hepatic venules (A). A higher intensity of staining is noted in perisinusoidal cells, especially in the perivenular region (B–D). Immunolabeling of cirrhotic liver tissue highlights the presence of PDE-5 in fibrous septa (E,C), in hepatocytes adjacent to veins (E,F), and in perisinusoidal cells scattered throughout the parenchyma.
Targeting the components of the NO-cGMP pathway in cirrhotic liver management using PDE-5 inhibitors or sGC modulators could lower portal pressure and increase portal perfusion. These effects might have clinical consequences.
- These drugs are potential therapeutic alternatives for portal hypertension.
- PDE-5 inhibitors or sGC modulators may be useful for reversal of liver fibrosis/cirrhosis, as shown in animal studies.
- PDE-5 inhibitors are a promising therapy of hepatic encephalopathy in liver cirrhosis.
- Serum levels of cGMP can be used as a simple non-invasive marker of clinically significant portal hypertension.
4. Modulation of the NO-cGMP Pathway in a Healthy and Cirrhotic Liver
4.1. Effect in Portal Hypertension
The effects of PDE-5 inhibitors on portal hemodynamics in experimental models of liver damage were investigated by several authors [41,42,43,47,48,54,55]. Consistently, a decrease of portal pressure was demonstrated, as well as an increase of sinusoidal flow or portal blood flow. PDE-5 inhibitors were also tested in the clinical setting by several teams of researchers [56,57,58,59,60,61,62]. A decrease in HVPG of >10% in acute testing, indicating a beneficial clinical effect, was proven in the majority of studies [54,56,57,59,61]. For details of the studies, see Table 1.

Table 1.
Effect of PDE-5 inhibitors and sGC modulators on portal pressure, Kreisel and Schaffner et al. (2020) [33].
Preclinical and clinical data provide evidence that targeting sCG (stimulators or activators) or PDE-5 inhibitors in liver cirrhosis lowers portal pressure and intrahepatic resistance, thereby increasing sinusoidal blood flow. Thus, PDE-5-inhibitors and sGC stimulators/activators can be used as a novel therapy in treating portal hypertension. The data also suggest that although the effects of sGC and PDE-5 inhibitors are dependent on NO synthesis, eNOS activity it not the limiting factor. The main regulator of sinusoidal tone, cGMP, can be modulated by targeting sGC and/or PDE-5.
The results of preclinical and clinical studies are summarized in Table 1. For further details, we refer to Kreisel and Schaffner et al. (2020) [33].
4.2. Targeting the NO-cGMP Pathway May Contribute to Reversal of Liver Fibrosis/Cirrhosis
Recent clinical and experimental studies suggested that chronic liver disease and resulting portal hypertension may be at least partially reversible [64,65,66,67]. Zoubek et al. (2017) [68] summarized the aims of antifibrotic therapy as follows: “The fundaments of fibrosis resolution rely on four cardinal points: discontinuation of the primary cause of chronic hepatic injury; regression of myofibroblasts from an activated to a deactivated status and/or their elimination; degradation of excessive matrix; switch of proinflammatory milieu to a restorative environment”.
For example, regression of fibrosis/cirrhosis has been observed in alcoholic liver disease after cessation of alcohol consumption [69,70], in chronic HBV infection [71,72,73] after long-term antiviral therapy, in chronic HCV infection after elimination of the virus with direct antiviral agents [65,66,74,75,76], and in non-alcoholic fatty liver disease after bariatric surgery [77,78]. Influencing the gut microbiome may play an important role in the treatment of NAFLD [79].
The cellular key players in the development of liver fibrosis are HSCs, which differentiate into myofibroblasts. The discovery of novel pathways and mediators revealed the plasticity and complexity of HSC activation but also yielded information about potential approaches to reverse HSC activation [80]. Reversal of fibrosis is accompanied by apoptosis of myofibroblasts or redifferentiation into inactivated HSCs. Elimination of HSCs is achieved via apoptosis or senescence [81,82]. According to Kisseleva et al. (2021) [83], several drugs are under consideration for therapy of liver fibrosis that interfere with HSC activation, such as PPARy agonists, LPAR1 agonists, NOX inhibitor, and hedgehog inhibitors, among others. Thus, it is worth investigating whether drugs which modulate the NO-cGMP pathway, influencing the function of HSCs, may be capable of reversing liver damage. Selicean et al. (2021) [67] reviewed pharmacological approaches to address portal pressure in the regression of chronic liver disease, such as ACE-inhibitors or ARBs, statins, the FXR agonist obeticholic acid, the Rho-kinase inhibitor fasudil, endothelin receptor antagonists, and the multikinase inhibitor sorafenib. The authors mentioned therapy with inhibitors of PDE-5 as a further possibility.
Furthermore, Knorr et al. (2011) [84] demonstrated that the sGC activator BAY 60-2770 ameliorated fibrosis in the rat model of BDL-induced cirrhosis independent of NO. Choi et al. (2009) [55] showed that the PDE-5 inhibitor udenafil decreased portal pressure by about 30%, reduced the degree of fibrosis, and suppressed the expression of procollagen type I and alpha-smooth muscle actin mRNA as an indicator for reduced HSC activity. According to Xie et al. (2012) [85], the sGC activator BAY 60-2770 accelerated the complete reversal of capillarization (restored differentiation of LSECs) in TAA-induced liver damage. Restoration of differentiation to LSECs led to quiescence of HSCs and regression of fibrosis even in the absence of further exposure to the sGC activator.
In a NASH model investigated by Flores-Costa et al. (2018) [86], sCG and cGMP were both lowered. These were restored to normal levels by the sGC stimulator IW-1973. This had a protective effect on hepatic steatosis, inflammation, and fibrosis. Similarly, as demonstrated by Hall et al. (2019) [49] in CCl4-induced liver cirrhosis, the sGC stimulator praliciguat markedly increased hepatic cGMP levels and reduced fibrosis and inflammation in the damaged liver. Schwabl et al. (2018) [41] reported that stimulation of sGC by riociguat decreased portal pressure and also improved fibrosis as demonstrated by histology and measurement of α-smooth muscle actin expression in BDL-induced and CCl4-induced liver cirrhosis. Inflammatory markers were reduced in both types of experimental liver damage. In the study of Brusilovskaya et al. (2020) [48], the long-acting PDE-5 inhibitor tadalafil and the sGC stimulator riociguat decreased portal pressure and intrahepatic vascular resistance and diminished the extent of fibrosis in BDL-induced cirrhosis.
Preclinical data suggest that modulation of the NO-cGMP pathway may induce or accelerate reversal of fibrosis/cirrhosis.
4.3. Role of Inhibitors of PDE-5 or Modulators of sGC in Hepatic Encephalopathy
The pathogenesis of hepatic encephalopathy (HE) is a complex process involving a disturbed interplay of the gut, liver, kidneys, skeletal muscles, and brain [87,88,89,90,91,92,93]. The common denominator is hyperammonemia. Therapy of HE [94,95,96,97,98] aims to correct underlying and precipitating factors—treatment of viral infections, cessation of offending drugs or alcohol, management of bleeding episodes, prevention of malnutrition or electrolyte imbalance, correction of intestinal dysbiosis, therapy of portosystemic shunts which bypass portal blood from the liver, and improvement of liver perfusion. The most promising therapeutic approaches target hyperammonemia [88,91,93,97,99,100].
Ammonia is produced mainly from protein digestion in the gastrointestinal tract and, in part, from glutamine via glutaminase in the kidney [101]. In healthy individuals, the ammonia level is stable and can be modified by urea synthesis in periportal hepatocytes and/or synthesis of glutamine in perivenous hepatocytes in the liver. Although hyperammonemia has been considered the main factor responsible for neurological changes, other factors like inflammation, inflammatory cytokines, disturbed cerebral blood flow, hyperthermia, hyponatremia, oxidative stress, catabolic state in liver cirrhosis, and increased levels of bile acids and lactate may also be involved. Blood ammonia levels do not always correlate with the severity of encephalopathy. On the other hand, biochemical changes observed in HE can be reproduced by increasing blood or brain ammonia levels in experimental animals. In cultured astrocytes, exposure to ammonium salts reproduces the morphological and biochemical findings observed in HE [102,103,104,105]. Hyperammonemia leads to an increase of glutamine in astrocytes that causes hypertonicity, cytotoxic astrocyte swelling, and brain edema [105,106,107]. These astrocytic functional changes impair the glutamate-glutamine metabolism, leading to excitotoxicity and neuronal dysfunction. Damage to astrocyte-neuronal communication as well as the impairment of glutamatergic and GABAergic neurotransmission further compromise cerebral function and may explain neurological deficits of HE.
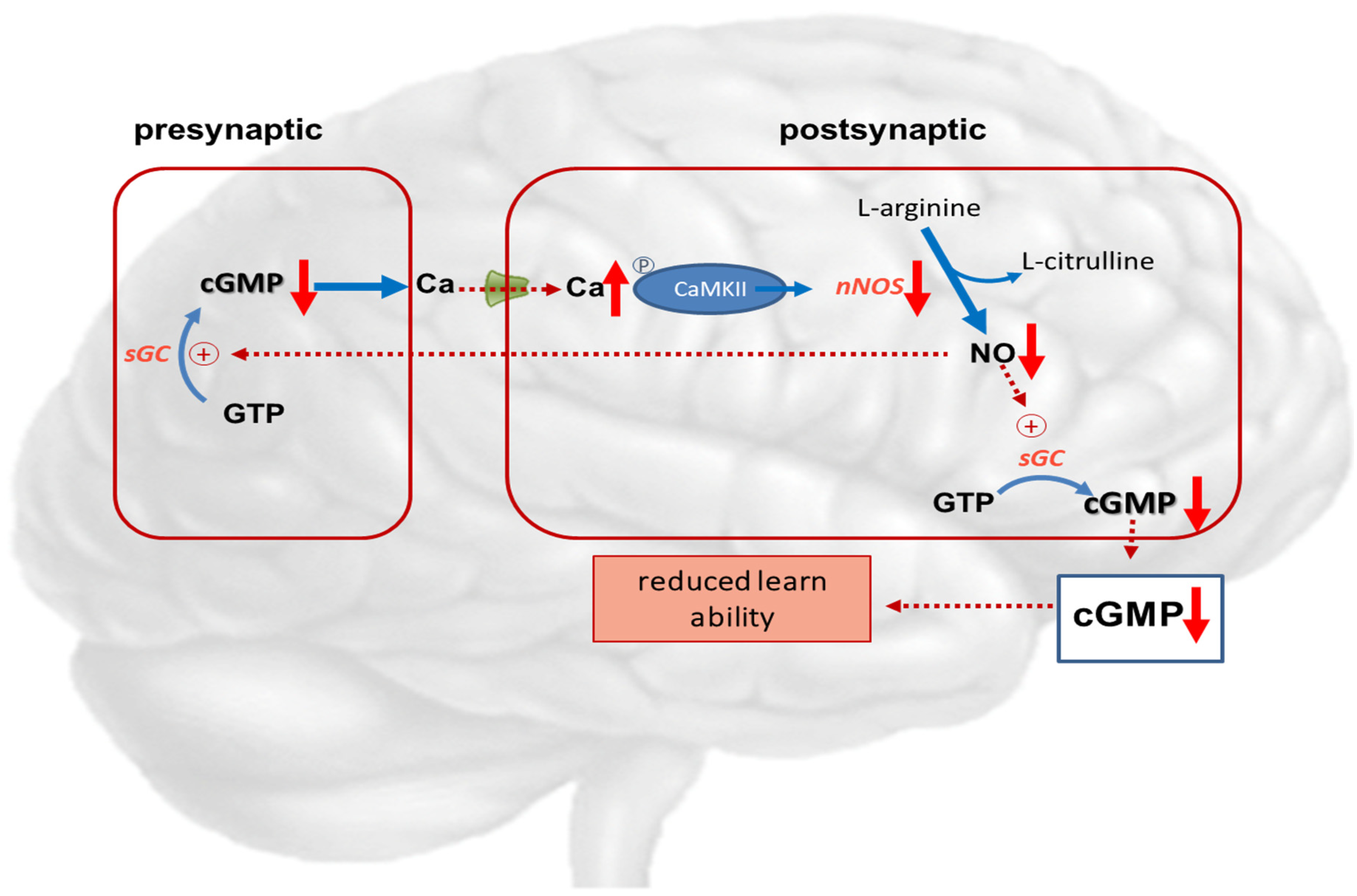
The glutamate-NO-cGMP pathway, including N-methyl-D-aspartate (NMDA) receptors, plays important roles in function and intercellular communication. Several investigators demonstrated modifiable learning ability depending on extracellular cGMP levels in rats [102,103,104].
Hyperammonemic rats show impaired function of the glutamate-NO-cGMP pathway, reduced levels of extracellular cGMP in the cerebellum, and reduced ability for spatial learning and memory [108]. Treatments that increase cGMP in the cerebellum were observed to restore learning in rats with hyperammonemia or hepatic encephalopathy [102,103,104,105].
These studies suggest that in pathological situations wherein reduced extracellular cGMP concentration is associated with reduced function of the glutamate-NO-cGMP pathway, it is possible to improve the mechanisms of such a pathway and enhance learning ability using treatments that increase cGMP levels. This may be achieved by administering phosphodiesterase inhibitors such as zaprinast or sildenafil [102,106], anti-inflammatory drugs like ibuprofen, inhibitors of MAP-kinase p38 [107,109], or compounds reducing GABAA receptor activation [108,110,111]. Continuous intracerebral administration of zaprinast, an inhibitor of phosphodiesterase, increased extracellular cGMP and restored learning ability in hyperammonemic rats. In rats with porto-caval anastomosis or hyperammonemia, sildenafil normalized the function of the glutamate-NO-cGMP pathway and extracellular cGMP in the brain in vivo [112]. Other research groups showed that administration of sildenafil or vardenafil improved the memory performance of hyperammonemic rats in an object recognition task [111], and sildenafil improved long-term retention of an inhibitory avoidance response in mice [113].
The memory-enhancing effects of PDE-5 inhibition in experimental studies may possibly be related to increased blood flow and, consequently, increased glucose metabolism, as PDE-5 inhibitors are known vasodilators [112]. Biochemical and immunohistochemical studies support the effect of PDE-5 inhibition on the glutamate-NO-cGMP pathway as the mechanism of action. Additionally, sildenafil was shown to increase axon and myelin density and modulate gene expression in rats with multiple microinfarction, as well as decrease inflammatory responses in aged rats [114]. This suggests that the mechanisms involved go beyond a mainly vasomotor mechanism of action. Figure 4 describes the disturbance of the NO—cGMP pathways in hepatic encephalopathy.
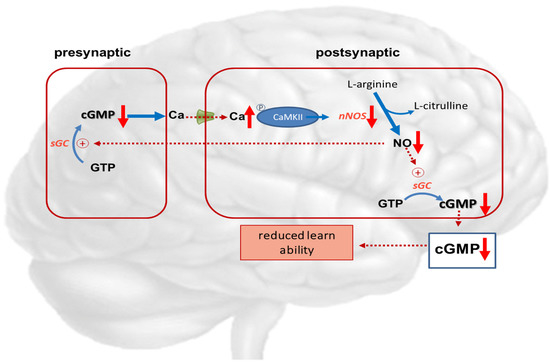
Figure 4.
Potential role of cGMP in hepatic encephalopathy. Hyperammonemia reduces the function of the glutamate-nitric oxide-cGMP pathway. Activation of NMDA receptors leads to increased intracellular calcium in the postsynaptic neuron. Calcium binds to calmodulin, which in turn activates function of calcium/calmodulin-dependent protein kinase II (CaMKII) by phosphorylation. CaMKII itself phosphorylates neuronal NO-synthase, reducing its activity and NO formation. This results in reduced synthesis of cGMP, leading to reduced learning ability.  NMDA-receptor; ↑ increased expression; ↑↑ markedly increased expression; ↑ increased concentration; ↓ decreased concentration.
NMDA-receptor; ↑ increased expression; ↑↑ markedly increased expression; ↑ increased concentration; ↓ decreased concentration.
NMDA-receptor; ↑ increased expression; ↑↑ markedly increased expression; ↑ increased concentration; ↓ decreased concentration.
The influence of PDE-5 inhibitors on cerebral function in HE is complex. In addition to modifying the glutamate-NO-cGMP pathway, changes in cerebral blood flow may also play a role. Influencing inflammation via intracellular cGMP concentration is also conceivable. The increase in sinusoidal perfusion, decrease of portal pressure, and reduction of shunt volume might lead to an improvement in the detoxification capacity of the liver. Further clinical studies are needed to prove the effect of PDE-5 inhibitors on hepatic encephalopathy.
4.4. Plasma/Serum cGMP as a Potential Marker of Clinically Significant Portal Hypertension (CSPH)
Numerous blood tests have been established to recognize or differentiate various liver diseases [115,116,117,118]. Several biochemical parameters in liver cirrhosis indicate hepatocyte damage or cholestasis, while others indicate stimulated synthesis of extracellular matrix proteins (laminin, hyaluronic acid, procollagen-III-peptide, Mac-2-binding protein glycosylation isomer) by stimulated HSCs (myofibroblasts), thus reflecting the degree of liver fibrosis. Various serum markers are suitable tools to detect clinically significant portal hypertension [119,120,121,122,123].
Serum/plasma cGMP could be a potential indicator of clinically significant portal hypertension in which the pathophysiological background is based on the opposed disturbance of the NO-cGMP pathway inside (low cGMP) compared to outside the cirrhotic liver (high cGMP). Inside the cirrhotic liver, cGMP is converted into biologically inactive 5′-cGMP by overexpressed PDE-5. This results in low cGMP in sinusoids and in liver outflow [42]. In contrast, in peripheral arteries, PDE-5 is underexpressed in liver cirrhosis [43]. This leads to high cGMP levels in the peripheral circulatory system. The disturbance of the NO—cGMP pathway in the peripheral circulation is shown in Figure 5.
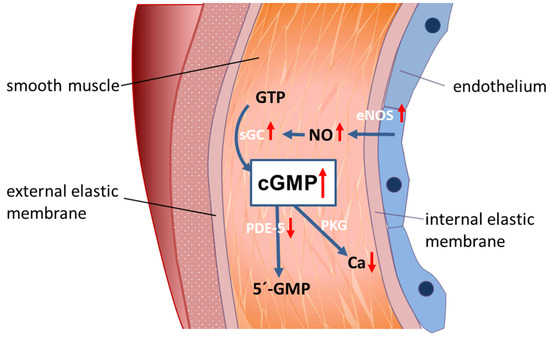
Figure 5.
A model for peripheral vasodilation in progressed liver cirrhosis. In liver cirrhosis, NO is overexpressed in peripheral arterial blood vessels, while PDE-5 is decreased. This leads to high cGMP concentration and peripheral vasodilation.
In patients with progressed liver cirrhosis, similar findings were reported. Kirstetter et al. (1997) [124] described elevated plasma cGMP levels in patients with progressed cirrhosis. Levels of ANP (a stimulator of membrane bound guanylate cyclase) were unchanged, while sGC was not measured. Montoliu et al. (2005) [125] described increased levels of plasma cGMP, as well as nitrates and ANP, in human liver cirrhosis. To further explain these data, increased stimulation of sGC by NO and membrane-bound GC by ANP as well as enhanced release of intracellular cGMP were discussed. The same authors (2010) demonstrated increased plasma cGMP levels in patients with HE [126] in a separate study. Similarly, Siqueira et al. (2008) [127] described significantly increased plasma levels of cGMP in patients with alcoholic liver cirrhosis. cGMP levels were negatively correlated with the MELD score, suggesting that progression of cirrhosis leads to higher plasma cGMP levels. According to Felipo et al. (2014) [128], patients with cirrhosis and minimal HE displayed increased blood ammonia, cGMP, IL-6, IL-18, and 3-nitrotyrosine. Sturm et al. (2021) [129] also published preliminary data that plasma cGMP was found to be significantly elevated in patients with clinically significant portal hypertension (CSPH), whereas in cirrhotics without CSPH, cGMP remained normal. These data suggest that plasma/serum cGMP might be an indicator of CSPH.
5. Conclusions
The “NO-paradox” states that NO availability inside the liver is reduced and NO production in the peripheral systemic circulation is increased, leading to vascular disturbance in liver cirrhosis. However, recent data suggest that altered availability of cGMP better elucidates the contrasting findings of intrahepatic vasoconstriction and peripheral systemic vasodilation. We suggest a focus shift from NO to cGMP and propose the alternative “cGMP-paradox”. The enzymes sGC and PDE-5 play a crucial role in the regulation of local cGMP levels. Hence, targeting these enzymes by PDE-5 inhibitors or sGC stimulators/activators could induce dilation of hepatic sinusoids, decrease intrahepatic resistance, increase portal venous blood flow, and consequently, lower portal pressure in cirrhotic portal hypertension. These effects suggest further clinical applications in liver cirrhosis. Inhibitors of PDE-5 or stimulators/activators of sGC may contribute to reversal of liver fibrosis or cirrhosis. Inhibitors of PDE-5 may have therapeutic potential for hepatic encephalopathy. Finally, plasma/serum levels of cGMP can be used as non-invasive marker of clinically significant portal hypertension.
Funding
The article processing charge was funded by the German Research Foundation (DFG) and the Albert Ludwigs University Freiburg in the funding programme Open Access Publishing.
Institutional Review Board Statement
Ethical review and approval were waived for this manuscript. Only existing data are reviewed.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original data are available in the cited references.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| NO | nitric oxide |
| GTP | guanosine triphosphate |
| cGMP | cyclic guanosine monophosphate |
| PKG | protein kinase G |
| CaMKII | calcium/calmodulin-dependent protein kinase II |
| eNOS | endothelial NO synthase |
| nNOS | neuronal NO synthase |
| sGC | soluble guanylate cyclase |
| PDE-5 | phosphodiesterase-5 |
| PDE-5-I | phosphodiesterase-5-inhibitor |
| CSPH | clinically significant portal hypertension |
| ANP | atria natriuretic factor |
| MELD | Model End Stage Liver Disease |
| HE | hepatic encephalopathy |
| PVP | portal venous pressure |
| MAP | mean arterial pressure |
| PPHTN | porto-pulmonary hypertension |
References
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Turco, L.; Garcia-Tsao, G. Portal Hypertension: Pathogenesis and Diagnosis. Clin. Liver Dis. 2019, 23, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Berzigotti, A.; Seijo, S.; Reverter, E.; Bosch, J. Assessing portal hypertension in liver diseases. Expert Rev. Gastroenterol. Hepatol. 2013, 7, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Pizcueta, P.; Feu, F.; Fernández, M.; Garcia-Pagan, J.C. Pathophysiology of portal hypertension. Gastroenterol. Clin. N. Am. 1992, 21, 1–14. [Google Scholar] [CrossRef]
- Bosch, J. Vascular deterioration in cirrhosis: The big picture. J. Clin. Gastroenterol. 2007, 41 (Suppl. S3), S247–S253. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Gonzalez-Abraldes, J.; Berzigotti, A.; Garcia-Pagan, J.C. The clinical use of HVPG measurements in chronic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 573–582. [Google Scholar] [CrossRef]
- Bosch, J.; Groszmann, R.J.; Shah, V.H. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J. Hepatol. 2015, 62, S121–S130. [Google Scholar] [CrossRef] [Green Version]
- Wongcharatrawee, S.; Groszmann, R.J. Diagnosing portal hypertension. Baillieres. Best Pract. Res. Clin. Gastroenterol. 2000, 14, 881–894. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Maeso-Díaz, R.; Bosch, J. Pathophysiology and a Rational Basis of Therapy. Dig. Dis. 2015, 33, 508–514. [Google Scholar] [CrossRef]
- De Franchis, R.; Dell’Era, A.; Primignani, M. Diagnosis and monitoring of portal hypertension. Dig. Liver Dis. 2008, 40, 312–317. [Google Scholar] [CrossRef]
- De Franchis, R.; Baveno, V.I. Faculty. Expanding consensus in portal hypertension: Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J. Hepatol. 2015, 63, 743–752. [Google Scholar] [CrossRef] [Green Version]
- La Mura, V.; Abraldes, J.G.; Raffa, S.; Retto, O.; Berzigotti, A.; García-Pagán, J.C.; Bosch, J. Prognostic value of acute hemodynamic response to i.v. propranolol in patients with cirrhosis and portal hypertension. J. Hepatol. 2009, 51, 279–287. [Google Scholar] [CrossRef]
- Villanueva, C.; Aracil, C.; Colomo, A.; Hernández-Gea, V.; López-Balaguer, J.M.; Alvarez-Urturi, C.; Torras, X.; Balanzó, J.; Guarner, C. Acute hemodynamic response to beta-blockers and prediction of long-term outcome in primary prophylaxis of variceal bleeding. Gastroenterology 2009, 137, 119–128. [Google Scholar] [CrossRef]
- Alvarado Tapias, E.; Ardevol, A.; Garcia-Guix, M.; Montañés, R.; Pavel, O.; Cuyas, B.; Graupera, I.; Brujats, A.; Vilades, D.; Colomo, A.; et al. Short-term hemodynamic effects of β-blockers influence survival of patients with decompensated cirrhosis. J. Hepatol. 2020, 73, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M. Molecular pathophysiology of portal hypertension. Hepatology 2015, 61, 1406–1415. [Google Scholar] [CrossRef]
- Mccuskey, R.S. The Hepatic Microvascular System in Health and Its Response to Toxicants. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2008, 291, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrød, B. Liver Sinusoidal Endothelial Cells. Compr. Physiol. 2015, 5, 1751–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakiri, Y.; Groszmann, R.J. Vascular endothelial dysfunction in cirrhosis. J. Hepatol. 2007, 46, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y. Pathophysiology of Portal Hypertension. Clin. Liver Dis. 2014, 18, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.; Iwakiri, Y. The portal hypertension syndrome: Etiology, classification, relevance, and animal models. Hepatol. Int. 2018, 12, 1–10. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Trebicka, J. Portal hypertension in cirrhosis: Pathophysiological mechanisms and therapy. JHEP Rep. 2021, 3, 100316. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.; Di Pascoli, M.; Verardo, A.; Gatta, A. Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis. World J. Gastroenterol. 2014, 20, 2555–2563. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Moncada, S. Hyperdynamic circulation in cirrhosis: A role for nitric oxide? Lancet 1991, 337, 776–778. [Google Scholar] [CrossRef]
- Iwakiri, Y. Nitric oxide in liver fibrosis: The role of inducible nitric oxide synthase. Clin. Mol. Hepatol. 2015, 21, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.D.; Francis, S.H. Cyclic GMP Phosphodiesterase-5: Target of Sildenafil. J. Biol. Chem. 1999, 274, 13729–13732. [Google Scholar] [CrossRef] [Green Version]
- Rybalkin, S.D.; Yan, C.; Bornfeldt, K.E.; Beavo, J.A. Cyclic GMP Phosphodiesterases and Regulation of Smooth Muscle Function. Circ. Res. 2003, 93, 280–291. [Google Scholar] [CrossRef]
- Wall, M.E.; Francis, S.H.; Corbin, J.D.; Grimes, K.; Richie-Jannetta, R.; Kotera, J.; Macdonald, B.A.; Gibson, R.R.; Trewhella, J. Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 2380–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Clark, J.W.; Bryan, R.M.; Robertson, C.S. Mathematical modeling of the nitric oxide/cGMP pathway in the vascular smooth muscle cell. Am. J. Physiol. Circ. Physiol. 2005, 289, H886–H897. [Google Scholar] [CrossRef]
- Shah, V.; Lyford, G.; Gores, G.; Farrugia, G. Nitric oxide in gastrointestinal health and disease. Gastroenterology 2004, 126, 903–913. [Google Scholar] [CrossRef]
- Rockey, D.C.; Shah, V. Nitric oxide biology and the liver: Report of an AASLD research workshop. Hepatology 2004, 39, 250–257. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Grisham, M.; Shah, V. Vascular biology and pathobiology of the liver: Report of a single-topic symposium. Hepatology 2008, 47, 1754–1763. [Google Scholar] [CrossRef]
- Langer, D.A.; Shah, V.H. Nitric oxide and portal hypertension: Interface of vasoreactivity and angiogenesis. J. Hepatol. 2005, 44, 209–216. [Google Scholar] [CrossRef]
- Kreisel, W.; Schaffner, D.; Lazaro, A.; Trebicka, J.; Merfort, I.; Schmitt-Graeff, A.; Deibert, P. Phosphodiesterases in the Liver as Potential Therapeutic Targets of Cirrhotic Portal Hypertension. Int. J. Mol. Sci. 2020, 21, 6223. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Kim, M.Y. Nitric oxide in liver diseases. Trends Pharmacol. Sci. 2015, 36, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Trebicka, J.; Hennenberg, M.; Odenthal, M.; Shir, K.; Klein, S.; Granzow, M.; Vogt, A.; Dienes, H.-P.; Lammert, F.; Reichen, J.; et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J. Hepatol. 2010, 53, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.S.; George, J.; Wang, J.H. Current concepts on the role of nitric oxide in portal hypertension. World J. Gastroenterol. 2013, 19, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Wang, S.-S.; Chan, C.-Y.; Chen, Y.-C.; Lee, F.-Y.; Chang, F.-Y.; Chu, C.-J.; Lin, H.-C.; Lu, R.-H.; Lee, S.-D. Role of Hepatic Nitric Oxide Synthases in Rats with Thioacetamide-induced Acute Liver Failure and Encephalopathy. J. Chin. Med. Assoc. 2007, 70, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Leung, T.-M.; Tipoe, G.L.; Liong, E.C.; Lau, T.Y.; Fung, M.-L.; Nanji, A.A. Endothelial nitric oxide synthase is a critical factor in experimental liver fibrosis. Int. J. Exp. Pathol. 2008, 89, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Trebicka, J.; Hennenberg, M.; Laleman, W.; Shelest, N.; Biecker, E.; Schepke, M.; Nevens, F.; Sauerbruch, T.; Heller, J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 2007, 46, 242–253. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, L.; Puttagunta, L.; Martinez-Cuesta, M.A.; Kneteman, N.; Mayers, I.; Moqbel, R.; Hamid, Q.; Radomski, M.W. Distribution of nitric oxide synthase in normal and cirrhotic human liver. Proc. Natl. Acad. Sci. USA 2002, 99, 17161–17166. [Google Scholar] [CrossRef] [Green Version]
- Schwabl, P.; Brusilovskaya, K.; Supper, P.; Bauer, D.; Königshofer, P.; Riedl, F.; Hayden, H.; Fuchs, C.; Stift, J.; Oberhuber, G.; et al. The soluble guanylate cyclase stimulator riociguat reduces fibrogenesis and portal pressure in cirrhotic rats. Sci. Rep. 2018, 8, 9372. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, D.; Lazaro, A.; Deibert, P.; Hasselblatt, P.; Stoll, P.; Fauth, L.; Baumstark, M.W.; Merfort, I.; Schmitt-Graeff, A.; Kreisel, W. Analysis of the nitric oxide-cyclic guanosine monophosphate pathway in experimental liver cirrhosis suggests phosphodiesterase-5 as potential target to treat portal hypertension. World J. Gastroenterol. 2018, 24, 4356–4368. [Google Scholar] [CrossRef]
- Uschner, F.E.; Glückert, K.; Paternostro, R.; Gnad, T.; Schierwagen, R.; Mandorfer, M.; Magdaleno, F.; Ortiz, C.; Schwarzkopf, K.; Kamath, P.S.; et al. Combination of phosphodiesterase-5-inhibitors and beta blockers improves experimental portal hypertension and erectile dysfunction. Liver Int. 2020, 40, 2228–2241. [Google Scholar] [CrossRef] [PubMed]
- Theilig, F.; Bostanjoglo, M.; Pavenstädt, H.; Grupp, C.; Holland, G.; Slosarek, I.; Gressner, A.M.; Russwurm, M.; Koesling, D.; Bachmann, S. Cellular distribution and function of soluble guanylyl cyclase in rat kidney and liver. J. Am. Soc. Nephrol. 2001, 12, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.A.; Hodges, S.J.; Pitsillides, A.A.; Mookerjee, R.; Jalan, R.; Mehdizadeh, S. Hepatic guanylate cyclase activity is decreased in a model of cirrhosis: A quantitative cytochemistry study. FEBS Lett. 2006, 580, 2123–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro-Silva, M.R.; Iwakiri, Y.; Abraldes, J.G.; Haq, O.; Groszmann, R.J. Increased phosphodiesterase-5 expression is involved in the decreased vasodilator response to nitric oxide in cirrhotic rat livers. J. Hepatol. 2006, 44, 886–893. [Google Scholar] [CrossRef]
- Lee, K.-C.; Yang, Y.-Y.; Huang, Y.-T.; Lee, F.-Y.; Hou, M.-C.; Lin, H.-C.; Lee, S.-D. Administration of a low dose of sildenafil for 1 week decreases intrahepatic resistance in rats with biliary cirrhosis: The role of NO bioavailability. Clin. Sci. 2010, 119, 45–55. [Google Scholar] [CrossRef]
- Brusilovskaya, K.; Königshofer, P.; Lampach, D.; Szodl, A.; Supper, P.; Bauer, D.; Beer, A.; Stift, J.; Timelthaler, G.; Oberhuber, G.; et al. Soluble guanylyl cyclase stimulation and phosphodiesterase-5 inhibition improve portal hypertension and reduce liver fibrosis in bile duct–ligated rats. United Eur. Gastroenterol. J. 2020, 8, 1174–1185. [Google Scholar] [CrossRef]
- Hall, K.C.; Bernier, S.G.; Jacobson, S.; Liu, G.; Zhang, P.Y.; Sarno, R.; Catanzano, V.; Currie, M.G.; Masferrer, J.L. sGC stimulator praliciguat suppresses stellate cell fibrotic transformation and inhibits fibrosis and inflammation in models of NASH. Proc. Natl. Acad. Sci. USA 2019, 116, 11057–11062. [Google Scholar] [CrossRef] [Green Version]
- Perri, R.E.; Langer, D.A.; Chatterjee, S.; Gibbons, S.J.; Gadgil, J.; Cao, S.; Farrugia, G.; Shah, V.H. Defects in cGMP-PKG pathway contribute to impaired NO-dependent responses in hepatic stellate cells upon activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G535–G542. [Google Scholar] [CrossRef]
- Niederberger, M.; Ginès, P.; Tsai, P.; Martin, P.-Y.; Morris, K.; Weigert, A.; McMurtry, I.; Schrier, R.W. Increased aortic cyclic guanosine monophosphate concentration in experimental cirrhosis in rats: Evidence for a role of nitric oxide in the pathogenesis of arterial vasodilation in cirrhosis. Hepatology 1995, 21, 1625–1631. [Google Scholar] [CrossRef]
- Niederberger, M.; Martin, P.-Y.; Ginès, P.; Morris, K.; Tsai, P.; Xu, D.-L.; McMurtry, I.; Schrier, R.W. Normalization of nitric oxide production corrects arterial vasodilation and hyperdynamic circulation in cirrhotic rats. Gastroenterology 1995, 109, 1624–1630. [Google Scholar] [CrossRef]
- Tahseldar-Roumieh, R.; Keravis, T.; Maarouf, S.; Justiniano, H.; Sabra, R.; Lugnier, C. PDEs1-5 activity and expression in tissues of cirrhotic rats reveal a role for aortic PDE3 in NO desensitization. Int. J. Exp. Pathol. 2009, 90, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Halverscheid, L.; Deibert, P.; Schmidt, R.; Blum, H.E.; Dunkern, T.; Pannen, B.H.J.; Kreisel, W. Phosphodiesterase-5 inhibitors have distinct effects on the hemodynamics of the liver. BMC Gastroenterol. 2009, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.-M.; Shin, J.-H.; Kim, J.-M.; Lee, C.-H.; Kang, K.-K.; Ahn, B.-O.; Yoo, M. Effect of Udenafil on Portal Venous Pressure and Hepatic Fibrosis in Rats. Arzneimittelforschung 2009, 59, 641–646. [Google Scholar] [CrossRef]
- Deibert, P.; Schumacher, Y.-O.; Ruecker, G.; Opitz, O.G.; Blum, H.E.; Rossle, M.; Kreisel, W. Effect of vardenafil, an inhibitor of phosphodiesterase-5, on portal haemodynamics in normal and cirrhotic liver—Results of a pilot study. Aliment. Pharmacol. Ther. 2006, 23, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bremer, H.C.; Kreisel, W.; Roecker, K.; Dreher, M.; Koenig, D.; Kurz-Schmieg, A.K.; Blum, H.E.; Roessle, M.; Deibert, P. Phosphodiesterase 5 inhibitors lower both portal and pulmonary pressure in portopulmonary hypertension: A case report. J. Med. Case Rep. 2007, 1, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-C.; Yang, Y.-Y.; Wang, Y.-W.; Hou, M.-C.; Lee, F.-Y.; Lin, H.-C.; Lee, S.-D. Acute administration of sildenafil enhances hepatic cyclic guanosine monophosphate production and reduces hepatic sinusoid resistance in cirrhotic patients. Hepatol. Res. 2008, 38, 1186–1193. [Google Scholar] [CrossRef]
- Clemmesen, J.O.; Giraldi, A.; Ott, P.; Dalhoff, K.; Hansen, B.A.; Larsen, F.S. Sildenafil does not influence hepatic venous pressure gradient in patients with cirrhosis. World J. Gastroenterol. 2008, 14, 6208–6212. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Inayat, I.; Tal, M.; Spector, M.; Shea, M.; Groszmann, R.J.; Garcia–Tsao, G. Sildenafil Has No Effect on Portal Pressure but Lowers Arterial Pressure in Patients with Compensated Cirrhosis. Clin. Gastroenterol. Hepatol. 2010, 8, 546–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreisel, W.; Deibert, P.; Kupcinskas, L.; Sumskiene, J.; Appenrodt, B.; Roth, S.; Neagu, M.; Rössle, M.; Zipprich, A.; Caca, K.; et al. The phosphodiesterase-5-inhibitor udenafil lowers portal pressure in compensated preascitic liver cirrhosis. A dose-finding phase-II-study. Dig. Liver Dis. 2015, 47, 144–150. [Google Scholar] [CrossRef]
- Deibert, P.; Lazaro, A.; Stankovic, Z.; Schaffner, D.; Rössle, M.; Kreisel, W. Beneficial long term effect of a phosphodiesterase-5-inhibitor in cirrhotic portal hypertension: A case report with 8 years follow-up. World J. Gastroenterol. 2018, 24, 438–444. [Google Scholar] [CrossRef]
- Colle, I.; De Vriese, A.; Van Vlierberghe, H.; Lameire, N.H.; DeVos, M. Systemic and splanchnic haemodynamic effects of sildenafil in an in vivo animal model of cirrhosis support for a risk in cirrhotic patients. Liver Int. 2004, 24, 63–68. [Google Scholar] [CrossRef]
- Jung, Y.K.; Yim, H.J. Reversal of liver cirrhosis: Current evidence and expectations. Korean J. Intern. Med. 2017, 32, 213–228. [Google Scholar] [CrossRef] [Green Version]
- Rockey, D.C. Fibrosis reversal after hepatitis C virus elimination. Curr. Opin. Gastroenterol. 2019, 35, 137–144. [Google Scholar] [CrossRef]
- Rockey, D.C.; Friedman, S.L. Fibrosis Regression After Eradication of Hepatitis C Virus: From Bench to Bedside. Gastroenterology 2021, 160, 1502–1520.e1. [Google Scholar] [CrossRef]
- Selicean, S.; Wang, C.; Guixé-Muntet, S.; Stefanescu, H.; Kawada, N.; Gracia-Sancho, J. Regression of portal hypertension: Underlying mechanisms and therapeutic strategies. Hepatol. Int. 2021, 15, 36–50. [Google Scholar] [CrossRef]
- Zoubek, M.E.; Trautwein, C.; Strnad, P. Reversal of liver fibrosis: From fiction to reality. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 129–141. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [Green Version]
- Su, T.-H.; Hu, T.-H.; Chen, C.-Y.; Huang, Y.-W.; Chuang, W.-L.; Lin, C.-C.; Wang, C.-C.; Su, W.-W.; Chen, M.-Y.; Peng, C.-Y.; et al. Four-year entecavir therapy reduces hepatocellular carcinoma, cirrhotic events and mortality in chronic hepatitis B patients. Liver Int. 2016, 36, 1755–1764. [Google Scholar] [CrossRef]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Schall, R.A.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Lampertico, P.; Invernizzi, F.; Viganò, M.; Loglio, A.; Mangia, G.; Facchetti, F.; Primignani, M.; Jovani, M.; Iavarone, M.; Fraquelli, M.; et al. The long-term benefits of nucleos(t)ide analogs in compensated HBV cirrhotic patients with no or small esophageal varices: A 12-year prospective cohort study. J. Hepatol. 2015, 63, 1118–1125. [Google Scholar] [CrossRef]
- Mandorfer, M.; Kozbial, K.; Schwabl, P.; Freissmuth, C.; Schwarzer, R.; Stern, R.; Chromy, D.; Stättermayer, A.F.; Reiberger, T.; Beinhardt, S.; et al. Sustained virologic response to interferon-free therapies ameliorates HCV-induced portal hypertension. J. Hepatol. 2016, 65, 692–699. [Google Scholar] [CrossRef]
- Schwabl, P.; Mandorfer, M.; Steiner, S.; Scheiner, B.; Chromy, D.; Herac, M.; Bucsics, T.; Hayden, H.; Grabmeier-Pfistershammer, K.; Ferlitsch, A.; et al. Interferon-free regimens improve portal hypertension and histological necroinflammation in HIV/HCV patients with advanced liver disease. Aliment. Pharmacol. Ther. 2016, 45, 139–149. [Google Scholar] [CrossRef]
- Lens, S.; Alvarado-Tapias, E.; Mariño, Z.; Londoño, M.-C.; LLop, E.; Martinez, J.; Fortea, J.I.; Ibañez, L.; Ariza, X.; Baiges, A.; et al. Effects of all-oral anti-viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus-associated cirrhosis. Gastroenterology 2017, 153, 1273–1283.e1. [Google Scholar] [CrossRef] [Green Version]
- Laursen, T.L.; Hagemann, C.A.; Wei, C.; Kazankov, K.; Thomsen, K.L.; Knop, F.K.; Grønbæk, H. Bariatric surgery in patients with non-alcoholic fatty liver disease—From pathophysiology to clinical effects. World J. Hepatol. 2019, 11, 138–149. [Google Scholar] [CrossRef]
- Seymour, K.A.; Abdelmalek, M.F. The role of bariatric surgery in the management of nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2021, 37, 208–215. [Google Scholar] [CrossRef]
- Lee, N.; Suk, K. The Role of the Gut Microbiome in Liver Cirrhosis Treatment. Int. J. Mol. Sci. 2020, 22, 199. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Tacke, F.; Trautwein, C. Mechanisms of liver fibrosis resolution. J. Hepatol. 2015, 63, 1038–1039. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Resolution of Liver Fibrosis: Basic Mechanisms and Clinical Relevance. Semin. Liver Dis. 2015, 35, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Knorr, A.; Hirth-Dietrich, C.; Alonso-Alija, C.; Härter, M.; Hahn, M.; Keim, Y.; Wunder, F.; Stasch, J.-P. Nitric Oxide-independent Activation of Soluble Guanylate Cyclase by BAY 60-2770 in Experimental Liver Fibrosis. Arzneimittelforschung 2008, 58, 71–80. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; DeLeve, L.D. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 2012, 142, 918–927.e6. [Google Scholar] [CrossRef] [Green Version]
- Flores-Costa, R.; Alcaraz-Quiles, J.; Titos, E.; López-Vicario, C.; Casulleras, M.; Duran-Güell, M.; Rius, B.; Diaz, A.; Hall, K.; Shea, C.; et al. The soluble guanylate cyclase stimulator IW-1973 prevents inflammation and fibrosis in experimental non-alcoholic steatohepatitis. Br. J. Pharmacol. 2018, 175, 953–967. [Google Scholar] [CrossRef]
- Jayakumar, A.; Norenberg, M.D. Hyperammonemia in Hepatic Encephalopathy. J. Clin. Exp. Hepatol. 2018, 8, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Hadjihambi, A.; Khetan, V.; Jalan, R. Pharmacotherapy for hyperammonemia. Expert Opin. Pharmacother. 2014, 15, 1685–1695. [Google Scholar] [CrossRef]
- Ochoa-Sanchez, R.; Rose, C.F. Pathogenesis of Hepatic Encephalopathy in Chronic Liver Disease. J. Clin. Exp. Hepatol. 2018, 8, 262–271. [Google Scholar] [CrossRef]
- DeMorrow, S.; Cudalbu, C.; Davies, N.; Jayakumar, A.R.; Rose, C.F. 2021 ISHEN guidelines on animal models of hepatic encephalopathy. Liver Int. 2021, 41, 1474–1488. [Google Scholar] [CrossRef]
- Rudler, M.; Weiss, N.; Bouzbib, C.; Thabut, D. Diagnosis and Management of Hepatic Encephalopathy. Clin. Liver Dis. 2021, 25, 393–417. [Google Scholar] [CrossRef]
- Alsahhar, J.S.; Rahimi, R. Updates on the pathophysiology and therapeutic targets for hepatic encephalopathy. Curr. Opin. Gastroenterol. 2019, 35, 145–154. [Google Scholar] [CrossRef]
- Butterworth, R.F. Hepatic Encephalopathy in Cirrhosis: Pathology and Pathophysiology. Drugs 2019, 79, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver: Vilstrup et al. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef]
- Angeli, P.; Bernardi, M.; Villanueva, C.; Francoz, C.; Mookerjee, R.; Trebicka, J.; Krag, A.; Laleman, W.; Gines, P. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460. [Google Scholar] [CrossRef] [Green Version]
- Ferenci, P. Hepatic encephalopathy. Gastroenterol. Rep. 2017, 5, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.F.; Amodio, P.; Bajaj, J.S.; Dhiman, R.K.; Montagnese, S.; Taylor-Robinson, S.D.; Vilstrup, H.; Jalan, R. Hepatic encephalopathy: Novel insights into classification, pathophysiology and therapy. J. Hepatol. 2020, 73, 1526–1547. [Google Scholar] [CrossRef]
- Bajaj, J.S. Diagnosis and Treatment of Hepatic Encephalopathy. Gastroenterol. Hepatol. 2019, 15, 434–436. [Google Scholar]
- Kircheis, G.; Lüth, S. Pharmacokinetic and Pharmacodynamic Properties of l-Ornithine l-Aspartate (LOLA) in Hepatic Encephalopathy. Drugs 2019, 79, 23–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterworth, R.F. Ammonia Removal by Metabolic Scavengers for the Prevention and Treatment of Hepatic Encephalopathy in Cirrhosis. Drugs R&D 2021, 21, 123–132. [Google Scholar] [CrossRef]
- Levitt, D.G.; Levitt, M.D. A model of blood-ammonia homeostasis based on a quantitative analysis of nitrogen metabolism in the multiple organs involved in the production, catabolism, and excretion of ammonia in humans. Clin. Exp. Gastroenterol. 2018, 11, 193–215. [Google Scholar] [CrossRef] [Green Version]
- Erceg, S.; Monfort, P.; Hernandez-Viadel, M.; Llansola, M.; Montoliu, C.; Felipo, V. Restoration of learning ability in hyperammonemic rats by increasing extracellular cGMP in brain. Brain Res. 2005, 1036, 115–121. [Google Scholar] [CrossRef]
- Cabrera-Pastor, A.; Hernandez-Rabaza, V.; Taoro-Gonzalez, L.; Balzano, T.; Llansola, M.; Felipo, V. In vivo administration of extracellular cGMP normalizes TNF-α and membrane expression of AMPA receptors in hippocampus and spatial reference memory but not IL-1β, NMDA receptors in membrane and working memory in hyperammonemic rats. Brain Behav. Immun. 2016, 57, 360–370. [Google Scholar] [CrossRef]
- Rodrigo, R.; Monfort, P.; Cauli, O.; Erceg, S.; Felipo, V. Pharmacological manipulation of cyclic GMP levels in brain restores learning ability in animal models of hepatic encephalopathy: Therapeutic implications. Neuropsychiatr. Dis. Treat. 2006, 2, 53–63. [Google Scholar]
- Felipo, V. Hepatic encephalopathy: Effects of liver failure on brain function. Nat. Rev. Neurosci. 2013, 14, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Erceg, S.; Monfort, P.; Hernández-Viadel, M.; Rodrigo, R.; Montoliu, C.; Felipo, V. Oral administration of sildenafil restores learning ability in rats with hyperammonemia and with portacaval shunts. Hepatology 2005, 41, 299–306. [Google Scholar] [CrossRef]
- Cauli, O.; Rodrigo, R.; Piedrafita, B.; Boix, J.; Felipo, V. Inflammation and hepatic encephalopathy: Ibuprofen restores learning ability in rats with portacaval shunts. Hepatology 2007, 46, 514–519. [Google Scholar] [CrossRef]
- Gonzalez-Usano, A.; Cauli, O.; Agusti, A.; Felipo, V. Pregnenolone sulfate restores the glutamate-nitric-oxide-cGMP pathway and extracellular GABA in cerebellum and learning and motor coordination in hyperammonemic rats. ACS Chem. Neurosci. 2014, 5, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agusti, A.; Cauli, O.; Rodrigo, R.; Llansola, M.; Hernández-Rabaza, V.; Felipo, V. p38 MAP kinase is a therapeutic target for hepatic encephalopathy in rats with portacaval shunts. Gut 2011, 60, 1572–1579. [Google Scholar] [CrossRef]
- Cauli, O.; Mansouri, M.T.; Agusti, A.; Felipo, V. Hyperammonemia Increases GABAergic Tone in the Cerebellum but Decreases It in the Rat Cortex. Gastroenterology 2009, 136, 1359–1367.e2. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Agusti, A.; Llansola, M.; Montoliu, C.; Strömberg, J.; Malinina, E.; Ragagnin, G.; Doverskog, M.; Backstrom, T.; Felipo, V. GR3027 antagonizes GABAA receptor-potentiating neurosteroids and restores spatial learning and motor coordination in rats with chronic hyperammonemia and hepatic encephalopathy. Am. J. Physiol. Liver Physiol. 2015, 309, G400–G409. [Google Scholar] [CrossRef] [Green Version]
- Prickaerts, J.; van Staveren, W.; Şik, A.; Ittersum, M.M.-V.; Niewöhner, U.; van der Staay, F.; Blokland, A.; de Vente, J. Effects of two selective phosphodiesterase type 5 inhibitors, sildenafil and vardenafil, on object recognition memory and hippocampal cyclic GMP levels in the rat. Neuroscience 2002, 113, 351–361. [Google Scholar] [CrossRef]
- Baratti, C.M.; Boccia, M.M. Effects of sildenafil on long-term retention of an inhibitory avoidance response in mice. Behav. Pharmacol. 1999, 10, 731–737. [Google Scholar] [CrossRef]
- Venkat, P.; Chopp, M.; Zacharek, A.; Cui, C.; Landschoot-Ward, J.; Qian, Y.; Chen, Z.; Chen, J. Sildenafil treatment of vascular dementia in aged rats. Neurochem. Int. 2019, 127, 103–112. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; LaVine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2017, 67, 328–357. [Google Scholar] [CrossRef]
- Lala, V.; Goyal, A.; Bansal, P.; Minter, D.A. Liver Function Tests; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK482489/ (accessed on 22 July 2021).
- Tripodi, A.; Mannucci, P.M. The Coagulopathy of Chronic Liver Disease. N. Engl. J. Med. 2011, 365, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Kwo, P.Y.; Cohen, S.M.; Lim, J.K. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. Am. J. Gastroenterol. 2017, 112, 18–35. [Google Scholar] [CrossRef]
- Nababan, S.; Kalista, K.; Jasirwan, C.; Kurniawan, J.; Lesmana, C.; Sulaiman, A.; Hasan, I.; Gani, R. Mac-2 Binding Protein Glycosylation Isomer for Screening High-Risk Esophageal Varices in Liver Cirrhotic Patient. Livers 2021, 1, 6. [Google Scholar] [CrossRef]
- Karatzas, A.; Konstantakis, C.; Aggeletopoulou, I.; Kalogeropoulou, C.; Thomopoulos, K.; Triantos, C. Νon-invasive screening for esophageal varices in patients with liver cirrhosis. Ann. Gastroenterol. 2018, 31, 305–314. [Google Scholar] [CrossRef]
- Pons, M.; Augustin, S.; Scheiner, B.; Guillaume, M.; Rosselli, M.; Rodrigues, S.G.; Stefanescu, H.; Ma, M.M.; Mandorfer, M.; Mergeay-Fabre, M.; et al. Noninvasive Diagnosis of Portal Hypertension in Patients with Compensated Advanced Chronic Liver Disease. Am. J. Gastroenterol. 2021, 116, 723–732. [Google Scholar] [CrossRef]
- Procopet, B.; Berzigotti, A. Diagnosis of cirrhosis and portal hypertension: Imaging, non-invasive markers of fibrosis and liver biopsy. Gastroenterol. Rep. 2017, 5, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Mandorfer, M.; Hernández-Gea, V.; García-Pagán, J.C.; Reiberger, T. Noninvasive Diagnostics for Portal Hypertension: A Comprehensive Review. Semin. Liver Dis. 2020, 40, 240–255. [Google Scholar] [CrossRef]
- Kirstetter, P.; Moreau, R.; Vachiery, F.; Gadano, A.; Soupison, T.; Pilette, C.; Pussard, E.; Cailmail, S.; Takahashi, H.; Lebrec, D. Plasma concentrations of cyclic 3′,5′-guanosine monophosphate in patients with cirrhosis: Relationship with atrial natriuretic peptide and haemodynamics. J. Gastroenterol. Hepatol. 1997, 12, 233–236. [Google Scholar] [CrossRef]
- Montoliu, C.; Kosenko, E.; Del Olmo, J.A.; Serra, M.A.; Rodrigo, J.M.; Felipo, V. Correlation of nitric oxide and atrial natriuretic peptide changes with altered cGMP homeostasis in liver cirrhosis. Liver Int. 2005, 25, 787–795. [Google Scholar] [CrossRef]
- Montoliu, C.; Rodrigo, R.; Monfort, P.; Llansola, M.; Cauli, O.; Boix, J.; Elmlili, N.; Agusti, A.; Felipo, V. Cyclic GMP pathways in hepatic encephalopathy. Neurological and therapeutic implications. Metab. Brain Dis. 2010, 25, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, C.; de Moura, M.C.; Pedro, A.J.; Rocha, P. Elevated nitric oxide and 3′,5′ cyclic guanosine monophosphate levels in patients with alcoholic cirrhosis. World J. Gastroenterol. 2008, 14, 236–242. [Google Scholar] [CrossRef]
- Felipo, V.; Urios, A.; Giménez-Garzó, C.; Cauli, O.; Andrés-Costa, M.-J.; González, O.; Serra, M.A.; Sánchez-González, J.; Aliaga, R.; Giner-Durán, R.; et al. Non invasive blood flow measurement in cerebellum detects minimal hepatic encephalopathy earlier than psychometric tests. World J. Gastroenterol. 2014, 20, 11815–11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, L.; Roth, L.; Bettinger, D.; Zoldan, K.; Boettler, T.; Huber, J.; Kaeser, R.; Kreisel, W.; Thimme, R.; Schultheiss, M. Blood cyclic guanosine monophosphate levels as potential marker of portal hypertension in patients with liver cirrhosis. Z. Gastroenterol. 2021, 59, 2–31. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).