
Adenylate Kinase 4—A Key Regulator of Proliferation and Metabolic Shift in Human Pulmonary Arterial Smooth Muscle Cells via Akt and HIF-1α Signaling Pathways

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
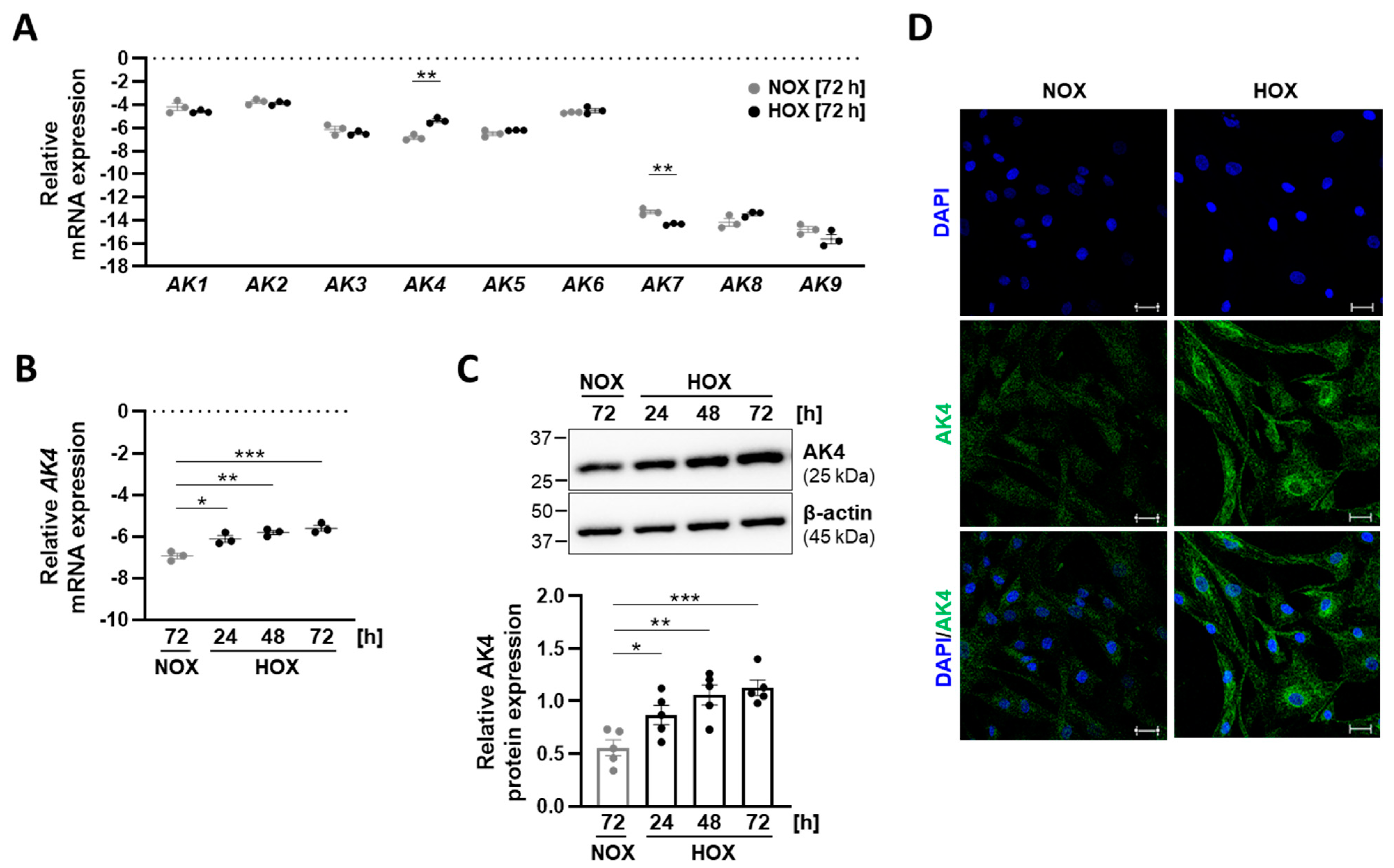
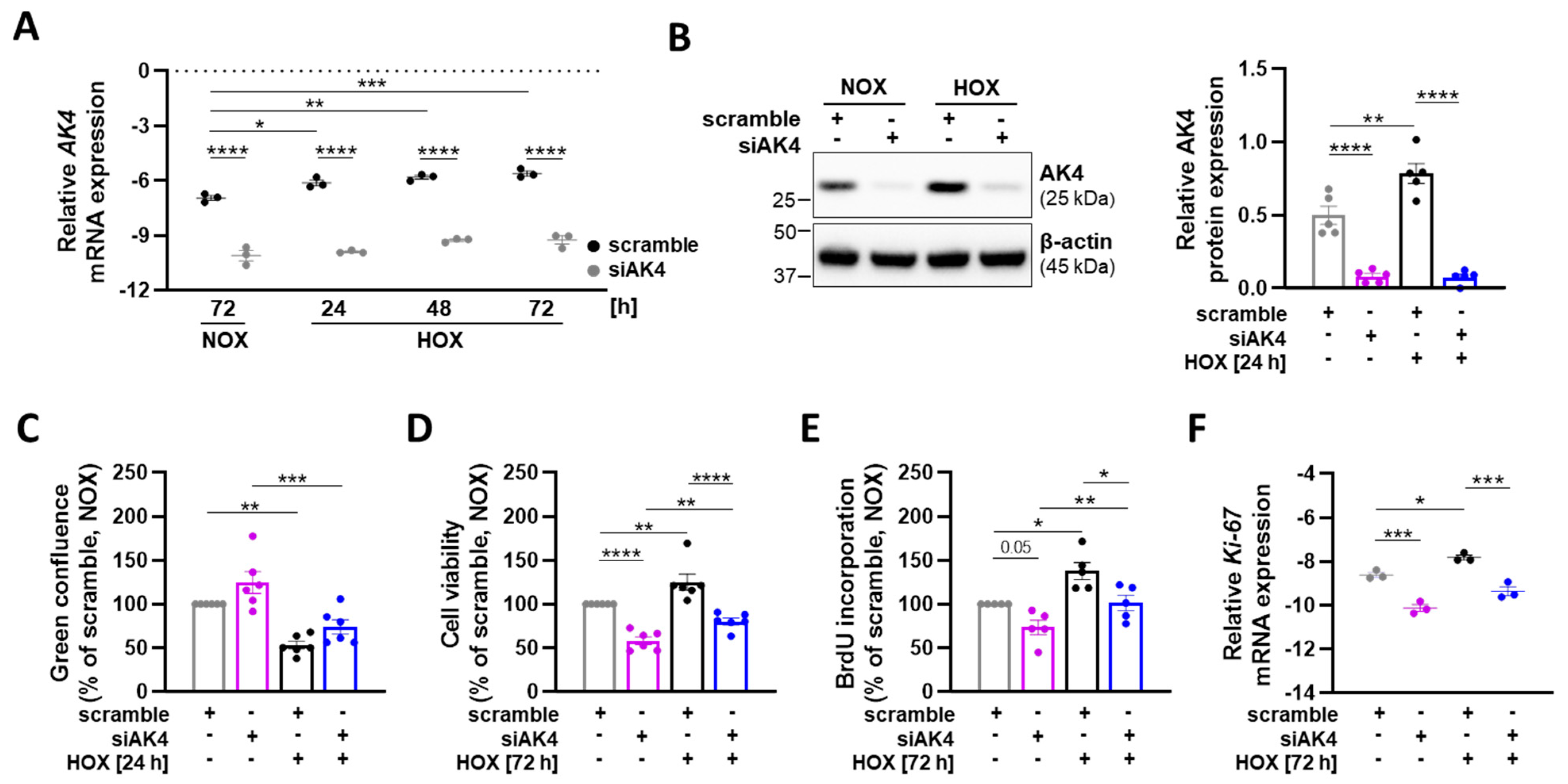
2.1. Hypoxia Upregulates AK4 in Primary Human Pulmonary Arterial Smooth Muscle Cells (PASMCs)
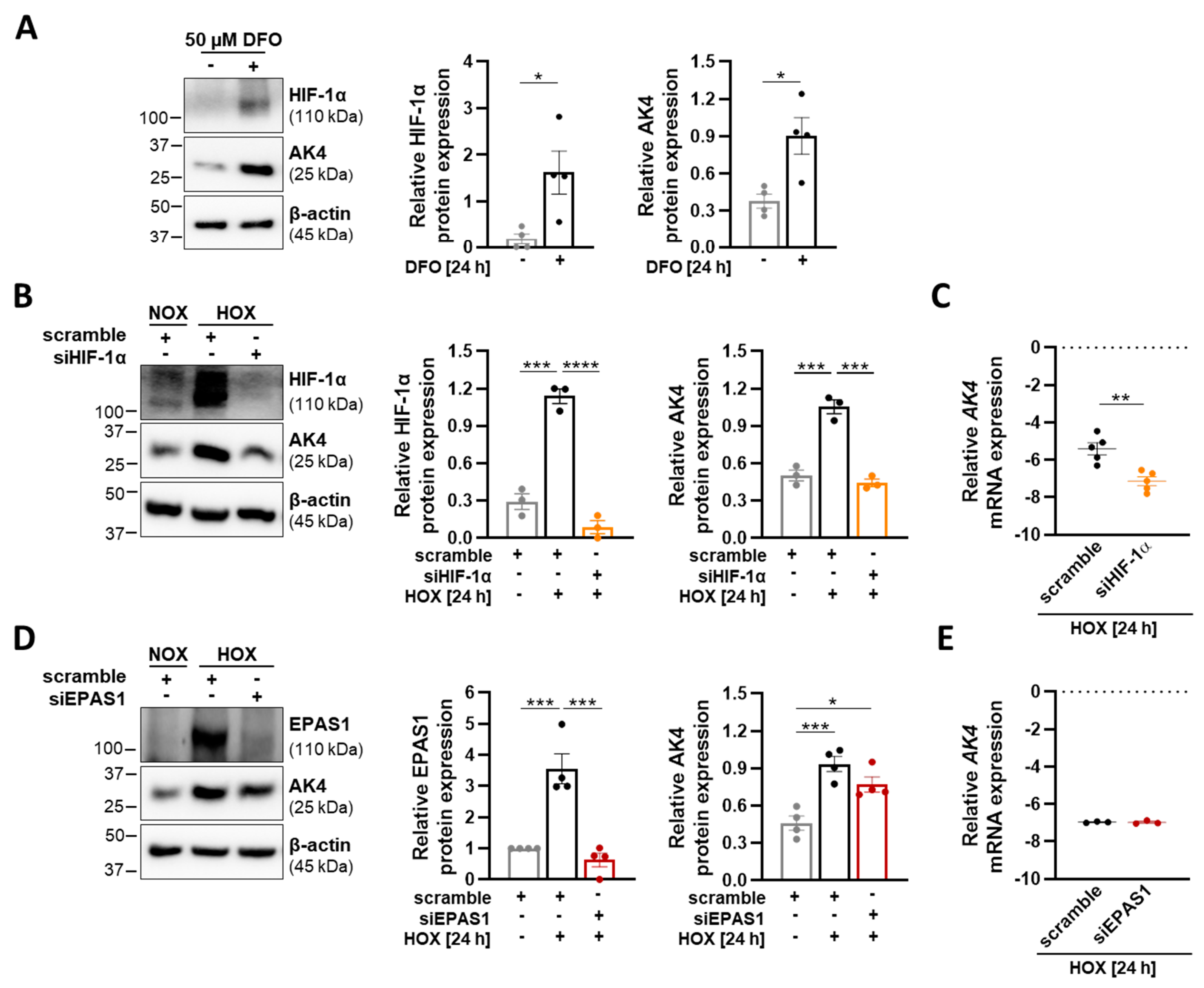
2.2. AK4 Is Upregulated in Hypoxic PASMCs in a HIF-1α-Dependent Manner
2.3. AK4 Regulates the Viability and Proliferation of PASMCs under Both Normoxia and Hypoxia
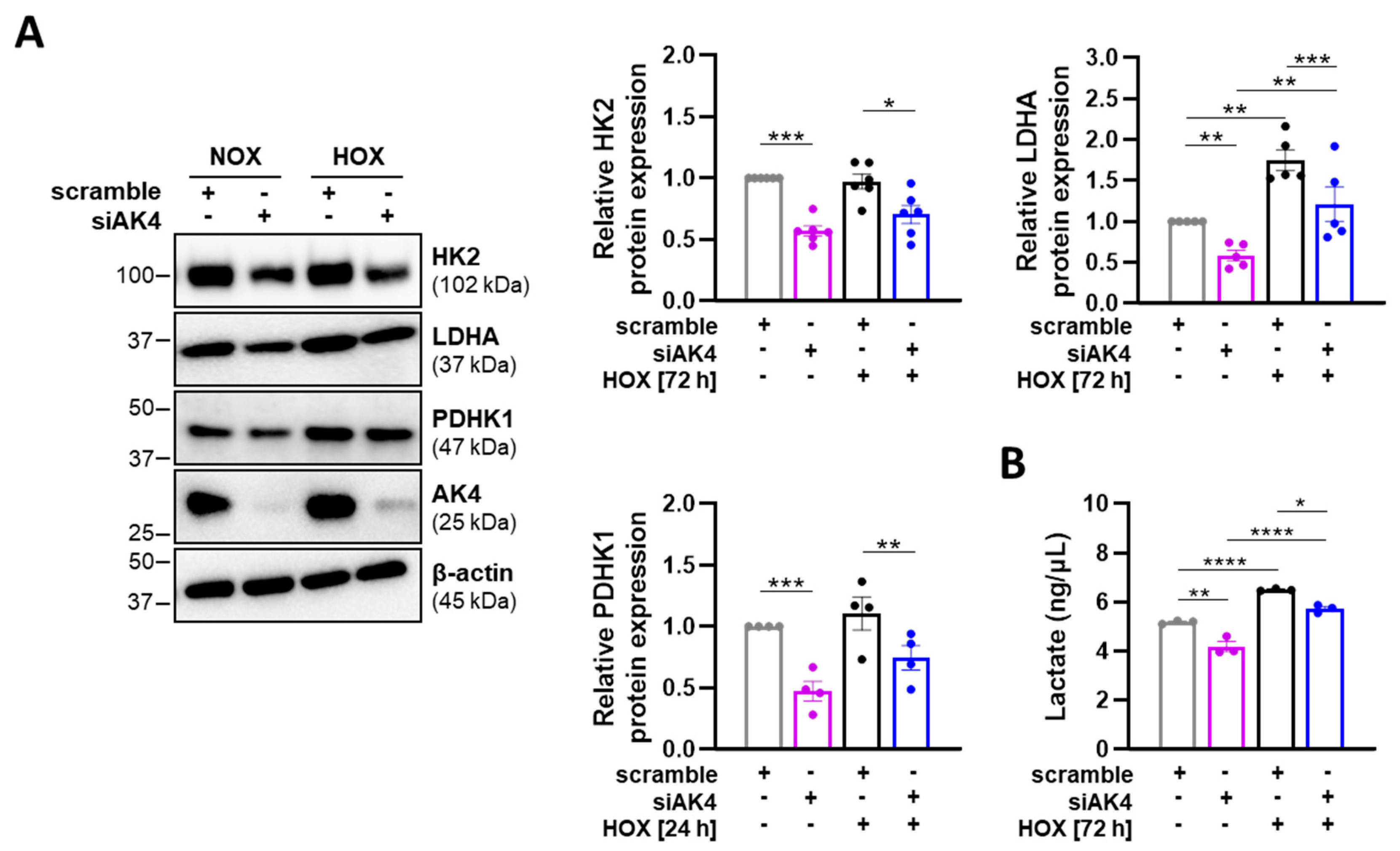
2.4. Downregulation of AK4 Augments the Mitochondrial Respiratory Function and Reduces the Glycolytic Metabolism in PASMCs
2.5. AK4 Interacts with HIF-1α and Akt Signaling Pathways in PASMCs
2.6. AK4 Is Increased in the Lungs of IPAH Patients and Regulates the Glycolytic Metabolism of IPAH-PASMCs
3. Discussion
4. Materials and Methods
4.1. Human Lung Samples
4.2. Cell Culture and Hypoxia Exposure of PASMCs
4.3. RNA Interference by Synthetic siRNA
4.4. Cell Viability Assay
4.5. Apoptosis Assay
4.6. Cell Proliferation Assay
4.7. Lactate Production Assay
4.8. Measurement of PASMC Mitochondrial Respiration Using High-Resolution Respirometry
4.9. Immunocytochemistry
4.10. Immunohistochemistry
4.11. RNA Isolation and RT-qPCR
4.12. Western Blot
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AK4 | adenylate kinase 4 |
| Akt | protein kinase B |
| B2M | beta-2-microglobulin |
| BrdU | bromodeoxyuridine |
| DFO | deferoxamine |
| EPAS1 | endothelial PAS domain-containing protein 1 (HIF-2α) |
| FCCP | carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone |
| HIF | hypoxia inducible factor |
| HK2 | hexokinase 2 |
| HOX | hypoxia |
| IPAH | idiopathic pulmonary arterial hypertension |
| LDHA | lactate dehydrogenase A |
| Ki-67 | marker of proliferation Ki-67 |
| NOX | normoxia |
| OCR | oxygen consumption rate |
| OXPHOS | oxidative phosphorylation |
| PASMCs | pulmonary arterial smooth muscle cells |
| PBS | phosphate-buffered saline |
| PDHK1 | pyruvate dehydrogenase kinase 1 |
| PH | pulmonary hypertension |
| RT | room temperature |
| RT-qPCR | reverse transcription quantitative polymerase chain reaction |
| scramble | scrambled siRNA (control siRNA) |
| TCA | tricarboxylic acid |
| VSMCs | vascular smooth muscle cells |
References
- Young, J.M.; Williams, D.R.; Thompson, A.A.R. Thin Air, Thick Vessels: Historical and Current Perspectives on Hypoxic Pulmonary Hypertension. Front. Med. 2019, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Pullamsetti, S.S.; Mamazhakypov, A.; Weissmann, N.; Seeger, W.; Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Invest. 2020, 130, 5638–5651. [Google Scholar] [CrossRef]
- Lee, J.W.; Ko, J.; Ju, C.; Eltzschig, H.K. Hypoxia signaling in human diseases and therapeutic targets. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Semenza, G.L. O2-regulated gene expression: Transcriptional control of cardiorespiratory physiology by HIF-1. J. Appl. Physiol. 2004, 96, 1173–1177. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, K.R.; Fagan, K.A.; Frid, M.G. Hypoxia-Induced Pulmonary Vascular Remodeling. Circ. Res. 2006, 99, 675–691. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Ball, M.K.; Waypa, G.B.; Mungai, P.T.; Nielsen, J.M.; Czech, L.; Dudley, V.J.; Beussink, L.; Dettman, R.W.; Berkelhamer, S.K.; Steinhorn, R.H.; et al. Regulation of Hypoxia-induced Pulmonary Hypertension by Vascular Smooth Muscle Hypoxia-Inducible Factor-1α. Am. J. Respir. Crit. Care Med. 2014, 189, 314–324. [Google Scholar] [CrossRef]
- Tajsic, T.; Morrell, N.W. Smooth Muscle Cell Hypertrophy, Proliferation, Migration and Apoptosis in Pulmonary Hypertension. Compr. Physiol. 2010, 1, 295–317. [Google Scholar] [CrossRef]
- Veith, C.; Schermuly, R.T.; Brandes, R.P.; Weissmann, N. Molecular mechanisms of hypoxia-inducible factor-induced pulmonary arterial smooth muscle cell alterations in pulmonary hypertension. J. Physiol. 2016, 594, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Culley, M.K.; Chan, S.Y. Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Investig. 2018, 128, 3704–3715. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Savai, R.; Seeger, W.; Goncharova, E.A. From Cancer Biology to New Pulmonary Arterial Hypertension Therapeutics. Targeting Cell Growth and Proliferation Signaling Hubs. Am. J. Respir. Crit. Care Med. 2017, 195, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. 2013, 22, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Belaiba, R.S.; Bonello, S.; Zähringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Gö, A. Hypoxia Up-Regulates Hypoxia-Inducible Factor-1 Transcription by Involving Phosphatidylinositol 3-Kinase and Nuclear Factor B in Pulmonary Artery Smooth Muscle Cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karger, S.; Qin, X.; Xiao, Y.; Peng, H.; Hong, C.; Chen, Z.; Deng, X.; Wang, A.; Yang, F.; Yang, L.; et al. PDGF Promotes the Warburg Effect in Pulmonary Arterial Smooth Muscle Cells via Activation of the PI3K/AKT/ mTOR/HIF-1α Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 1603–1613. [Google Scholar] [CrossRef]
- Liu, P.; Gu, Y.; Luo, J.; Ye, P.; Zheng, Y.; Yu, W.; Chen, S. Inhibition of Src activation reverses pulmonary vascular remodeling in experimental pulmonary arterial hypertension via Akt/mTOR/HIF-1<alpha> signaling pathway. Exp. Cell Res. 2019, 380, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Zhou, Q.; Chen, J.; Rexius-Hall, M.L.; Rehman, J.; Zhou, G. Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nat. Commun. 2018, 9, 3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Nemutlu, E.; Terzic, A.; Dzeja, P. Adenylate Kinase Isoform Network: A Major Hub in Cell Energetics and Metabolic Signaling. In Systems Biology of Metabolic and Signaling Networks. Energy, Mass and Information Transfer; Springer: Berlin/Heidelberg, Germany, 2014; pp. 145–162. [Google Scholar]
- Dzeja, P.; Terzic, A. Adenylate Kinase and AMP Signaling Networks: Metabolic Monitoring, Signal Communication and Body Energy Sensing. Int. J. Mol. Sci. 2009, 10, 1729–1772. [Google Scholar] [CrossRef] [Green Version]
- Panayiotou, C.; Solaroli, N.; Karlsson, A. The many isoforms of human adenylate kinases. Int. J. Biochem. Cell Biol. 2014, 49, 75–83. [Google Scholar] [CrossRef]
- Ionescu, M.I. Adenylate Kinase: A Ubiquitous Enzyme Correlated with Medical Conditions. Protein J. 2019, 38, 120–133. [Google Scholar] [CrossRef]
- Dzeja, P.P.; Chung, S.; Faustino, R.S.; Behfar, A.; Terzic, A. Developmental Enhancement of Adenylate Kinase-AMPK Metabolic Signaling Axis Supports Stem Cell Cardiac Differentiation. PLoS ONE 2011, 6, e19300. [Google Scholar] [CrossRef]
- Oshima, K.; Saiki, N.; Tanaka, M.; Imamura, H.; Niwa, A.; Tanimura, A.; Nagahashi, A.; Hirayama, A.; Okita, K.; Hotta, A.; et al. Human AK2 links intracellular bioenergetic redistribution to the fate of hematopoietic progenitors. Biochem. Biophys. Res. Commun. 2018, 497, 719–725. [Google Scholar] [CrossRef]
- Lorès, P.; Coutton, C.; El Khouri, E.; Stouvenel, L.; Givelet, M.; Thomas, L.; Rode, B.; Schmitt, A.; Louis, B.; Sakheli, Z.; et al. Homozygous missense mutation L673P in adenylate kinase 7 (AK7) leads to primary male infertility and multiple morphological anomalies of the flagella but not to primary ciliary dyskinesia. Hum. Mol. Genet. 2018, 27, 1196–1211. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, C.; Tang, Z.; Yang, Y.; Tian, Y.; Yao, H.; Zhu, X.; Zhang, Z.; Ji, J.; Zheng, X. Adenylate kinase hCINAP determines self-renewal of colorectal cancer stem cells by facilitating LDHA phosphorylation. Nat. Commun. 2017, 8, 15308. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Pu, Y.; Amina, Q.; Wang, Q.; Zhang, M.; Song, J.; Guo, J.; Mardan, M. Prognostic and therapeutic potential of Adenylate kinase 2 in lung adenocarcinoma. Sci. Rep. 2019, 9, 17757. [Google Scholar] [CrossRef] [Green Version]
- Jan, Y.-H.; Tsai, H.-Y.; Yang, C.-J.; Huang, M.-S.; Yang, Y.-F.; Lai, T.-C.; Lee, C.-H.; Jeng, Y.-M.; Huang, C.-Y.; Su, J.-L.; et al. Adenylate Kinase-4 Is a Marker of Poor Clinical Outcomes That Promotes Metastasis of Lung Cancer by Downregulating the Transcription Factor ATF3. Cancer Res. 2012, 72, 5119–5129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Qin, X.; Wang, Y.; Mao, F. Identification of AK4 as a novel therapeutic target for serous ovarian cancer. Oncol. Lett. 2020, 20, 346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yin, Y.-T.; Wu, C.-H.; Qiu, R.-L.; Jiang, W.-J.; Deng, X.-G.; Li, Z.-X. AK4 Promotes the Progression of HER2-Positive Breast Cancer by Facilitating Cell Proliferation and Invasion. Dis. Markers 2019, 2019, 8186091. [Google Scholar] [CrossRef]
- Fujisawa, K.; Terai, S.; Takami, T.; Yamamoto, N.; Yamasaki, T.; Matsumoto, T.; Yamaguchi, K.; Owada, Y.; Nishina, H.; Noma, T.; et al. Modulation of anti-cancer drug sensitivity through the regulation of mitochondrial activity by adenylate kinase 4. J. Exp. Clin. Cancer Res. 2016, 35, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanning, N.J.; Looyenga, B.D.; Kauffman, A.L.; Niemi, N.M.; Sudderth, J.; DeBerardinis, R.J.; MacKeigan, J.P. A Mitochondrial RNAi Screen Defines Cellular Bioenergetic Determinants and Identifies an Adenylate Kinase as a Key Regulator of ATP Levels. Cell Rep. 2014, 7, 907–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.; Binas, B.; Moon, J.H.; Kang, S.S.; Kim, H.J. Differential expression of adenylate kinase 4 in the context of disparate stress response strategies of HEK293 and HepG2 cells. Arch. Biochem. Biophys. 2013, 533, 11–17. [Google Scholar] [CrossRef]
- Liu, R.; Ström, A.-L.; Zhai, J.; Gal, J.; Bao, S.; Gong, W.; Zhu, H. Enzymatically inactive adenylate kinase 4 interacts with mitochondrial ADP/ATP translocase. Int. J. Biochem. Cell Biol. 2009, 41, 1371–1380. [Google Scholar] [CrossRef] [Green Version]
- Panayiotou, C.; Solaroli, N.; Johansson, M.; Karlsson, A. Evidence of an intact N-terminal translocation sequence of human mitochondrial adenylate kinase 4. Int. J. Biochem. Cell Biol. 2010, 42, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Jan, Y.H.; Lai, T.C.; Yang, C.J.; Lin, Y.F.; Huang, M.S.; Hsiao, M. Adenylate kinase 4 modulates oxidative stress and stabilizes HIF-1α to drive lung adenocarcinoma metastasis. J. Hematol. Oncol. 2019, 12, 12. [Google Scholar] [CrossRef]
- Klepinin, A.; Zhang, S.; Klepinina, L.; Rebane-Klemm, E.; Terzic, A.; Kaambre, T.; Dzeja, P. Adenylate Kinase and Metabolic Signaling in Cancer Cells. Front. Oncol. 2020, 10, 660. [Google Scholar] [CrossRef] [PubMed]
- Xin, F.; Yao, D.W.; Fan, L.; Liu, J.H.; Liu, X.D. Adenylate kinase 4 promotes bladder cancer cell proliferation and invasion. Clin. Exp. Med. 2019, 19, 525–534. [Google Scholar] [CrossRef]
- Chin, W.-Y.; He, C.-Y.; Chow, T.W.; Yu, Q.-Y.; Lai, L.-C.; Miaw, S.-C. Adenylate Kinase 4 Promotes Inflammatory Gene Expression via Hif1α and AMPK in Macrophages. Front. Immunol. 2021, 12, 630318. [Google Scholar] [CrossRef]
- Hu, C.-J.; Iyer, S.; Sataur, A.; Covello, K.L.; Chodosh, L.A.; Simon, M.C. Differential Regulation of the Transcriptional Activities of Hypoxia-Inducible Factor 1 Alpha (HIF-1α) and HIF-2α in Stem Cells. Mol. Cell. Biol. 2006, 26, 3514–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greijer, A.E.; van der Groep, P.; Kemming, D.; Shvarts, A.; Semenza, G.L.; Meijer, G.A.; van de Wiel, M.A.; Belien, J.A.M.; van Diest, P.J.; van der Wall, E. Up-regualtion of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor I (HIF-I). J. Pathol. 2005, 206, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Harvey, L.; Chan, S. Emerging Metabolic Therapies in Pulmonary Arterial Hypertension. J. Clin. Med. 2017, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Cool, C.D.; Kuebler, W.M.; Bogaard, H.J.; Spiekerkoetter, E.; Nicolls, M.R.; Voelkel, N.F. The hallmarks of severe pulmonary arterial hypertension: The cancer hypothesis—ten years later. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L1115–L1130. [Google Scholar] [CrossRef]
- Ma, Z.; Xiang, X.; Li, S.; Xie, P.; Gong, Q.; Goh, B.-C.; Wang, L. Targeting hypoxia-inducible factor-1, for cancer treatment: Recent advances in developing small-molecule inhibitors from natural compounds. Semin. Cancer Biol. 2020, S1044-579X(20)30202–9. [Google Scholar] [CrossRef]
- Soto-Heredero, G.; Gómez de las Heras, M.M.; Gabandé-Rodríguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Chan, X.Y.; Volkova, E.; Eoh, J.; Black, R.; Fang, L.; Gorashi, R.; Song, J.; Wang, J.; Elliott, M.B.; Barreto-Ortiz, S.F.; et al. HIF2A gain-of-function mutation modulates the stiffness of smooth muscle cells and compromises vascular mechanics. iScience 2021, 24, 102246. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Frid, M.G.; Mouradian, G.; Li, M.; et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur. Respir. J. 2019, 54, 1900378. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L.; Ludovica Fracanzani, A.; Gatti, S.; Cairo, G.; Fargion, S. Iron Depletion by Deferoxamine Up-Regulates Glucose Uptake and Insulin Signaling in Hepatoma Cells and in Rat Liver. Am. J. Pathol. 2008, 172, 738–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djafarzadeh, S.; Jakob, S.M. High-resolution Respirometry to Assess Mitochondrial Function in Permeabilized and Intact Cells. J. Vis. Exp. 2017, 2017, 54985. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fink, T.; Ebbesen, P.; Zachar, V. Temporal transcriptome of mouse ATDC5 chondroprogenitors differentiating under hypoxic conditions. Exp. Cell Res. 2006, 312, 1727–1744. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Madamanchi, N.R.; Hakim, Z.S.; Rojas, M.; Runge, M.S. Thrombin and NAD(P)H Oxidase–Mediated Regulation of CD44 and BMP4-Id Pathway in VSMC, Restenosis, and Atherosclerosis. Circ. Res. 2006, 98, 1254–1263. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Kikkawa, R.; Yamada, H.; Horii, I. Investigation of proteomic biomarkers in in vivo hepatotoxicity study of rat liver: Toxicity differentiation in hepatotoxicants. J. Toxicol. Sci. 2006, 31, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veith, C.; Marsh, L.M.; Wygrecka, M.; Rutschmann, K.; Seeger, W.; Weissmann, N.; Kwapiszewska, G. Paxillin Regulates Pulmonary Arterial Smooth Muscle Cell Function in Pulmonary Hypertension. Am. J. Pathol. 2012, 181, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Seimetz, M.; Sommer, N.; Bednorz, M.; Pak, O.; Veith, C.; Hadzic, S.; Gredic, M.; Parajuli, N.; Kojonazarov, B.; Kraut, S.; et al. NADPH oxidase subunit NOXO1 is a target for emphysema treatment in COPD. Nat. Metab. 2020, 2, 532–546. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wujak, M.; Veith, C.; Wu, C.-Y.; Wilke, T.; Kanbagli, Z.I.; Novoyatleva, T.; Guenther, A.; Seeger, W.; Grimminger, F.; Sommer, N.; et al. Adenylate Kinase 4—A Key Regulator of Proliferation and Metabolic Shift in Human Pulmonary Arterial Smooth Muscle Cells via Akt and HIF-1α Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 10371. https://doi.org/10.3390/ijms221910371
Wujak M, Veith C, Wu C-Y, Wilke T, Kanbagli ZI, Novoyatleva T, Guenther A, Seeger W, Grimminger F, Sommer N, et al. Adenylate Kinase 4—A Key Regulator of Proliferation and Metabolic Shift in Human Pulmonary Arterial Smooth Muscle Cells via Akt and HIF-1α Signaling Pathways. International Journal of Molecular Sciences. 2021; 22(19):10371. https://doi.org/10.3390/ijms221910371
Chicago/Turabian StyleWujak, Magdalena, Christine Veith, Cheng-Yu Wu, Tessa Wilke, Zeki Ilker Kanbagli, Tatyana Novoyatleva, Andreas Guenther, Werner Seeger, Friedrich Grimminger, Natascha Sommer, and et al. 2021. "Adenylate Kinase 4—A Key Regulator of Proliferation and Metabolic Shift in Human Pulmonary Arterial Smooth Muscle Cells via Akt and HIF-1α Signaling Pathways" International Journal of Molecular Sciences 22, no. 19: 10371. https://doi.org/10.3390/ijms221910371
APA StyleWujak, M., Veith, C., Wu, C.-Y., Wilke, T., Kanbagli, Z. I., Novoyatleva, T., Guenther, A., Seeger, W., Grimminger, F., Sommer, N., Schermuly, R. T., & Weissmann, N. (2021). Adenylate Kinase 4—A Key Regulator of Proliferation and Metabolic Shift in Human Pulmonary Arterial Smooth Muscle Cells via Akt and HIF-1α Signaling Pathways. International Journal of Molecular Sciences, 22(19), 10371. https://doi.org/10.3390/ijms221910371