
Targeting Histone Deacetylases in Idiopathic Pulmonary Fibrosis: A Future Therapeutic Option


Abstract
:1. Introduction
1.1. Pathomechanisms of Idiopathic Pulmonary Fibrosis
1.1.1. Genetic Factors Affecting IPF-Epithelial Cells
1.1.2. The Core in IPF: Disturbed AECII–Mesenchymal Communication and AECII/Fibroblast Apoptosis Imbalance
1.1.3. Key Fibrotic Pathways behind IPF
1.1.4. Deregulation of microRNAs in IPF
1.2. Treatment of Idiopathic Pulmonary Fibrosis
1.2.1. Established Therapies for IPF
1.2.2. Therapeutic Targets Proposed for IPF Treatment
1.2.3. Senotherapies for IPF
1.2.4. Current Therapies for IPF in Development
1.2.5. Past Treatment Strategies Not Recommended Anymore
1.3. Similarities between IPF and Cancer: Histone Deacetylases as Novel Therapeutic Targets in IPF
1.4. The HDAC Family/HDAC Classes and Their Function—Lessons from Cancer Research
1.5. Histone Deacetylase Inhibitors
2. Imbalanced Histone Deacetylase (HDAC) Activities in Idiopathic Pulmonary Fibrosis: Effects and Therapeutic Correction
2.1. Class I Histone Deacetylases in IPF: Expression Profile, Function and Preclinical Studies
2.1.1. Class I HDAC Inhibitors in Preclinical Studies of Lung Fibrosis
2.1.2. Class I Isoform-Selective Inhibitors in Preclinical Models of Lung Fibrosis
2.1.3. Loss of Sin3-HDAC1/HDAC2 Repressor Complex Activity in AECII Results in Alveolar Senescence
2.2. Class IIA Histone Deacetylases in IPF: Expression Profile, Function, and Preclinical Studies
2.3. Class IIB Histone Deacetylases in IPF: Expression Profile, Function and Preclinical Studies
2.3.1. Peculiar Role of HDAC6 in IPF Epithelial Cells?
2.3.2. HDAC6 Selective Inhibitors in Preclinical Models of Lung Fibrosis
3. Discussion of HDAC Inhibitors as Therapeutic Option for IPF
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [PubMed]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Cerri, S.; Monari, M.; Guerrieri, A.; Donatelli, P.; Bassi, I.; Garuti, M.; Luppi, F.; Betti, S.; Bandelli, G.; Carpano, M.; et al. Real-life comparison of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis: A 24-month assessment. Respir. Med. 2019, 159, 105803. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell Signal. 2020, 66, 109482. [Google Scholar]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Murer, B.; Lestani, M.; Cancellieri, A.; Montagna, L.; Piccoli, P.; Cangi, G.; Semenzato, G.; Doglioni, C. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: The role of deltaN-p63. Lab. Investig. 2002, 82, 1335–1345. [Google Scholar] [CrossRef]
- Plantier, L.; Crestani, B.; Wert, S.E.; Dehoux, M.; Zweytick, B.; Guenther, A.; Whitsett, J.A. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011, 66, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.L.; Rostami, M.R.; LeBlanc, M.; Kaner, R.J.; O’Beirne, S.L.; Mezey, J.G.; Leopold, P.L.; Quast, K.; Visvanathan, S.; Fine, J.S.; et al. Dysregulation of club cell biology in idiopathic pulmonary fibrosis. PLoS ONE 2020, 15, e0237529. [Google Scholar] [CrossRef]
- Chilosi, M.; Zamo, A.; Doglioni, C.; Reghellin, D.; Lestani, M.; Montagna, L.; Pedron, S.; Ennas, M.G.; Cancellieri, A.; Murer, B.; et al. Migratory marker expression in fibroblast foci of idiopathic pulmonary fibrosis. Respir. Res. 2006, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Myers, J.L.; Katzenstein, A.L. Epithelial necrosis and alveolar collapse in the pathogenesis of usual interstitial pneumonia. Chest 1988, 94, 1309–1311. [Google Scholar]
- Uhal, B.D.; Joshi, I.; Hughes, W.F.; Ramos, C.; Pardo, A.; Selman, M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am. J. Physiol. 1998, 275, L1192–L1199. [Google Scholar]
- Barbas-Filho, J.V.; Ferreira, M.A.; Sesso, A.; Kairalla, R.A.; Carvalho, C.R.; Capelozzi, V.L. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J. Clin. Pathol. 2001, 54, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Kuwano, K.; Kunitake, R.; Kawasaki, M.; Nomoto, Y.; Hagimoto, N.; Nakanishi, Y.; Hara, N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1996, 154 (2 Pt 1), 477–483. [Google Scholar] [CrossRef]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [Green Version]
- Cha, S.I.; Ryerson, C.J.; Lee, J.S.; Kukreja, J.; Barry, S.S.; Jones, K.D.; Elicker, B.M.; Kim, D.S.; Papa, F.R.; Collard, H.R.; et al. Cleaved cytokeratin-18 is a mechanistically informative biomarker in idiopathic pulmonary fibrosis. Respir. Res. 2012, 13, 105. [Google Scholar] [CrossRef] [Green Version]
- Klymenko, O.; Huehn, M.; Wilhelm, J.; Wasnick, R.; Shalashova, I.; Ruppert, C.; Henneke, I.; Hezel, S.; Guenther, K.; Mahavadi, P.; et al. Regulation and role of the ER stress transcription factor CHOP in alveolar epithelial type-II cells. J. Mol. Med. 2019, 97, 973–990. [Google Scholar] [PubMed] [Green Version]
- Thomas, A.Q.; Lane, K.; Phillips, J., 3rd; Prince, M.; Markin, C.; Speer, M.; Schwartz, D.A.; Gaddipati, R.; Marney, A.; Johnson, J.; et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 2002, 165, 1322–1328. [Google Scholar] [PubMed]
- Mulugeta, S.; Nguyen, V.; Russo, S.J.; Muniswamy, M.; Beers, M.F. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am. J. Respir. Cell Mol. Biol. 2005, 32, 521–530. [Google Scholar] [PubMed]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar]
- Maitra, M.; Wang, Y.; Gerard, R.D.; Mendelson, C.R.; Garcia, C.K. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J. Biol. Chem. 2010, 285, 22103–22113. [Google Scholar] [CrossRef] [Green Version]
- Katzen, J.; Wagner, B.D.; Venosa, A.; Kopp, M.; Tomer, Y.; Russo, S.J.; Headen, A.C.; Basil, M.C.; Stark, J.M.; Mulugeta, S.; et al. An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight 2019, 4, e126125. [Google Scholar] [CrossRef] [Green Version]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Kropski, J.A.; Mitchell, D.B.; Markin, C.; Polosukhin, V.V.; Choi, L.; Johnson, J.E.; Lawson, W.E.; Phillips, J.A., 3rd; Cogan, J.D.; Blackwell, T.S.; et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest 2014, 146, e1–e7. [Google Scholar]
- Fukuhara, A.; Tanino, Y.; Ishii, T.; Inokoshi, Y.; Saito, K.; Fukuhara, N.; Sato, S.; Saito, J.; Ishida, T.; Yamaguchi, H.; et al. Pulmonary fibrosis in dyskeratosis congenita with TINF2 gene mutation. Eur. Respir. J. 2013, 42, 1757–1759. [Google Scholar]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [Green Version]
- Kropski, J.A.; Lawson, W.E.; Young, L.R.; Blackwell, T.S. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis. Model. Mech. 2013, 6, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Courtwright, A.M.; El-Chemaly, S. Telomeres in Interstitial Lung Disease: The Short and the Long of It. Ann. Am. Thorac. Soc. 2019, 16, 175–181. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef]
- Hancock, L.A.; Hennessy, C.E.; Solomon, G.M.; Dobrinskikh, E.; Estrella, A.; Hara, N.; Hill, D.B.; Kissner, W.J.; Markovetz, M.R.; Grove Villalon, D.E.; et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat. Commun. 2018, 9, 5363. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar]
- Bouros, E.; Filidou, E.; Arvanitidis, K.; Mikroulis, D.; Steiropoulos, P.; Bamias, G.; Bouros, D.; Kolios, G. Lung fibrosis-associated soluble mediators and bronchoalveolar lavage from idiopathic pulmonary fibrosis patients promote the expression of fibrogenic factors in subepithelial lung myofibroblasts. Pulm. Pharmacol. Ther. 2017, 46, 78–87. [Google Scholar]
- Yang, J.; Velikoff, M.; Canalis, E.; Horowitz, J.C.; Kim, K.K. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L786–L796. [Google Scholar]
- Osterholzer, J.J.; Christensen, P.J.; Lama, V.; Horowitz, J.C.; Hattori, N.; Subbotina, N.; Cunningham, A.; Lin, Y.; Murdock, B.J.; Morey, R.E.; et al. PAI-1 promotes the accumulation of exudate macrophages and worsens pulmonary fibrosis following type II alveolar epithelial cell injury. J. Pathol. 2012, 228, 170–180. [Google Scholar]
- Kim, K.K.; Dotson, M.R.; Agarwal, M.; Yang, J.; Bradley, P.B.; Subbotina, N.; Osterholzer, J.J.; Sisson, T.H. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis. 2018, 9, 1056. [Google Scholar]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharmacol. 2014, 4, 173. [Google Scholar]
- Waghray, M.; Cui, Z.; Horowitz, J.C.; Subramanian, I.M.; Martinez, F.J.; Toews, G.B.; Thannickal, V.J. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005, 19, 854–856. [Google Scholar]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar]
- Sisson, T.H.; Maher, T.M.; Ajayi, I.O.; King, J.E.; Higgins, P.D.; Booth, A.J.; Sagana, R.L.; Huang, S.K.; White, E.S.; Moore, B.B.; et al. Increased survivin expression contributes to apoptosis-resistance in IPF fibroblasts. Adv. Biosci. Biotechnol. 2012, 3, 657–664. [Google Scholar]
- Predescu, S.A.; Zhang, J.; Bardita, C.; Patel, M.; Godbole, V.; Predescu, D.N. Mouse Lung Fibroblast Resistance to Fas-Mediated Apoptosis Is Dependent on the Baculoviral Inhibitor of Apoptosis Protein 4 and the Cellular FLICE-Inhibitory Protein. Front. Physiol. 2017, 8, 128. [Google Scholar]
- Golan-Gerstl, R.; Wallach-Dayan, S.B.; Zisman, P.; Cardoso, W.V.; Goldstein, R.H.; Breuer, R. Cellular FLICE-like inhibitory protein deviates myofibroblast fas-induced apoptosis toward proliferation during lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, E.; Gili, E.; Fruciano, M.; Korfei, M.; Fagone, E.; Iemmolo, M.; Lo Furno, D.; Giuffrida, R.; Crimi, N.; Guenther, A.; et al. PI3K p110gamma overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: In vitro effects of its inhibition. Lab. Investig. 2013, 93, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Wei, K.; Jacobs, S.S.; Upadhyay, D.; Weill, D.; Rosen, G.D. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J. Biol. Chem. 2010, 285, 8196–8206. [Google Scholar] [PubMed] [Green Version]
- Ajayi, I.O.; Sisson, T.H.; Higgins, P.D.; Booth, A.J.; Sagana, R.L.; Huang, S.K.; White, E.S.; King, J.E.; Moore, B.B.; Horowitz, J.C. X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 86–95. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Quintanilla, M.; Bernabeu, C. TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin. Sci. 2011, 121, 233–251. [Google Scholar] [CrossRef] [Green Version]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004, 114, 1308–1316. [Google Scholar] [CrossRef]
- Zhang, Y.; Dees, C.; Beyer, C.; Lin, N.Y.; Distler, A.; Zerr, P.; Palumbo, K.; Susok, L.; Kreuter, A.; Distler, O.; et al. Inhibition of casein kinase II reduces TGFbeta induced fibroblast activation and ameliorates experimental fibrosis. Ann. Rheum. Dis. 2015, 74, 936–943. [Google Scholar] [CrossRef]
- Milara, J.; Ballester, B.; Morell, A.; Ortiz, J.L.; Escriva, J.; Fernandez, E.; Perez-Vizcaino, F.; Cogolludo, A.; Pastor, E.; Artigues, E.; et al. JAK2 mediates lung fibrosis, pulmonary vascular remodelling and hypertension in idiopathic pulmonary fibrosis: An experimental study. Thorax 2018, 73, 519–529. [Google Scholar]
- Wang, J.; Hu, K.; Cai, X.; Yang, B.; He, Q.; Wang, J.; Weng, Q. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharm. Sin. B 2022, 12, 18–32. [Google Scholar] [CrossRef]
- Hu, Y.; Peng, J.; Feng, D.; Chu, L.; Li, X.; Jin, Z.; Lin, Z.; Zeng, Q. Role of extracellular signal-regulated kinase, p38 kinase, and activator protein-1 in transforming growth factor-beta1-induced alpha smooth muscle actin expression in human fetal lung fibroblasts in vitro. Lung 2006, 184, 33–42. [Google Scholar]
- Xia, H.; Khalil, W.; Kahm, J.; Jessurun, J.; Kleidon, J.; Henke, C.A. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am. J. Pathol. 2010, 176, 2626–2637. [Google Scholar] [CrossRef]
- Conte, E.; Fruciano, M.; Fagone, E.; Gili, E.; Caraci, F.; Iemmolo, M.; Crimi, N.; Vancheri, C. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: The role of class I P110 isoforms. PLoS ONE 2011, 6, e24663. [Google Scholar]
- Zhang, X.L.; Xing, R.G.; Chen, L.; Liu, C.R.; Miao, Z.G. PI3K/Akt signaling is involved in the pathogenesis of bleomycin-induced pulmonary fibrosis via regulation of epithelial-mesenchymal transition. Mol. Med. Rep. 2016, 14, 5699–5706. [Google Scholar] [CrossRef]
- Wei, X.; Han, J.; Chen, Z.Z.; Qi, B.W.; Wang, G.C.; Ma, Y.H.; Zheng, H.; Luo, Y.F.; Wei, Y.Q.; Chen, L.J. A phosphoinositide 3-kinase-gamma inhibitor, AS605240 prevents bleomycin-induced pulmonary fibrosis in rats. Biochem. Biophys. Res. Commun. 2010, 397, 311–317. [Google Scholar] [CrossRef]
- Guerreiro, A.S.; Fattet, S.; Kulesza, D.W.; Atamer, A.; Elsing, A.N.; Shalaby, T.; Jackson, S.P.; Schoenwaelder, S.M.; Grotzer, M.A.; Delattre, O.; et al. A sensitized RNA interference screen identifies a novel role for the PI3K p110gamma isoform in medulloblastoma cell proliferation and chemoresistance. Mol. Cancer Res. 2011, 9, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Azad, N.; Wang, L.; Iyer, A.K.; Castranova, V.; Jiang, B.H.; Rojanasakul, Y. Phosphatidylinositol-3-kinase/Akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am. J. Respir. Cell Mol. Biol. 2010, 42, 432–441. [Google Scholar] [CrossRef]
- Spassov, S.G.; Donus, R.; Ihle, P.M.; Engelstaedter, H.; Hoetzel, A.; Faller, S. Hydrogen Sulfide Prevents Formation of Reactive Oxygen Species through PI3K/Akt Signaling and Limits Ventilator-Induced Lung Injury. Oxid. Med. Cell. Longev. 2017, 2017, 3715037. [Google Scholar] [CrossRef]
- O’Donoghue, R.J.; Knight, D.A.; Richards, C.D.; Prele, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- Pedroza, M.; Le, T.T.; Lewis, K.; Karmouty-Quintana, H.; To, S.; George, A.T.; Blackburn, M.R.; Tweardy, D.J.; Agarwal, S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016, 30, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Luan, J.; Gao, S.; Li, S.; Jiang, Q.; Liu, R.; Liang, Q.; Zhang, R.; Zhang, F.; Li, X.; et al. Fedratinib Attenuates Bleomycin-Induced Pulmonary Fibrosis via the JAK2/STAT3 and TGF-beta1 Signaling Pathway. Molecules 2021, 26, 4491. [Google Scholar] [CrossRef]
- Chuang, Y.F.; Huang, S.W.; Hsu, Y.F.; Yu, M.C.; Ou, G.; Huang, W.J.; Hsu, M.J. WMJ-8-B, a novel hydroxamate derivative, induces MDA-MB-231 breast cancer cell death via the SHP-1-STAT3-survivin cascade. Br. J. Pharmacol. 2017, 174, 2941–2961. [Google Scholar] [CrossRef] [Green Version]
- Prele, C.M.; Yao, E.; O’Donoghue, R.J.; Mutsaers, S.E.; Knight, D.A. STAT3: A central mediator of pulmonary fibrosis? Proc. Am. Thorac. Soc. 2012, 9, 177–182. [Google Scholar] [CrossRef]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escriva, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef] [Green Version]
- Chambers, R.C. Procoagulant signalling mechanisms in lung inflammation and fibrosis: Novel opportunities for pharmacological intervention? Br. J. Pharmacol. 2008, 153, S367–S378. [Google Scholar] [CrossRef] [Green Version]
- Howell, D.C.; Laurent, G.J.; Chambers, R.C. Role of thrombin and its major cellular receptor, protease-activated receptor-1, in pulmonary fibrosis. Biochem. Soc. Trans. 2002, 30, 211–216. [Google Scholar] [CrossRef]
- Scotton, C.J.; Krupiczojc, M.A.; Konigshoff, M.; Mercer, P.F.; Lee, Y.C.; Kaminski, N.; Morser, J.; Post, J.M.; Maher, T.M.; Nicholson, A.G.; et al. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J. Clin. Investig. 2009, 119, 2550–2563. [Google Scholar] [CrossRef] [Green Version]
- Bai, K.J.; Chen, B.C.; Pai, H.C.; Weng, C.M.; Yu, C.C.; Hsu, M.J.; Yu, M.C.; Ma, H.P.; Wu, C.H.; Hong, C.Y.; et al. Thrombin-induced CCN2 expression in human lung fibroblasts requires the c-Src/JAK2/STAT3 pathway. J. Leukoc. Biol. 2013, 93, 101–112. [Google Scholar] [CrossRef]
- Atanelishvili, I.; Liang, J.; Akter, T.; Spyropoulos, D.D.; Silver, R.M.; Bogatkevich, G.S. Thrombin increases lung fibroblast survival while promoting alveolar epithelial cell apoptosis via the endoplasmic reticulum stress marker, CCAAT enhancer-binding homologous protein. Am. J. Respir. Cell Mol. Biol. 2014, 50, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, X.; Shan, H.; Liang, H. MicroRNAs in idiopathic pulmonary fibrosis: Involvement in pathogenesis and potential use in diagnosis and therapeutics. Acta Pharm. Sin. B 2016, 6, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Yang, L.; Wang, W.; Wang, J.; Wang, J.; Xu, Z. Discovery and validation of extracellular/circulating microRNAs during idiopathic pulmonary fibrosis disease progression. Gene 2015, 562, 138–144. [Google Scholar] [CrossRef]
- Yang, S.; Banerjee, S.; de Freitas, A.; Sanders, Y.Y.; Ding, Q.; Matalon, S.; Thannickal, V.J.; Abraham, E.; Liu, G. Participation of miR-200 in pulmonary fibrosis. Am. J. Pathol. 2012, 180, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Liu, L.; Qian, H. Up-regulation of miR-21 promotes cell proliferation and collagen synthesis in pulmonary fibroblasts. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin. J. Cell. Mol. Immunol. 2015, 31, 918–922. [Google Scholar]
- Lino Cardenas, C.L.; Henaoui, I.S.; Courcot, E.; Roderburg, C.; Cauffiez, C.; Aubert, S.; Copin, M.C.; Wallaert, B.; Glowacki, F.; Dewaeles, E.; et al. miR-199a-5p is upregulated during fibrogenic response to tissue injury and mediates TGFbeta-induced lung fibroblast activation by targeting caveolin-1. PLoS Genet. 2013, 9, e1003291. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Xu, C.; Pan, Z.; Zhang, Y.; Xu, Z.; Chen, Y.; Li, T.; Li, X.; Liu, Y.; Huangfu, L.; et al. The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol. Ther. 2014, 22, 1122–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierro-Fernandez, M.; Busnadiego, O.; Sandoval, P.; Espinosa-Diez, C.; Blanco-Ruiz, E.; Rodriguez, M.; Pian, H.; Ramos, R.; Lopez-Cabrera, M.; Garcia-Bermejo, M.L.; et al. miR-9-5p suppresses pro-fibrogenic transformation of fibroblasts and prevents organ fibrosis by targeting NOX4 and TGFBR2. EMBO Rep. 2015, 16, 1358–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, J.; Beisang, D.J.; Peterson, M.; Forster, C.; Gilbertsen, A.; Benyumov, A.; Smith, K.; Korenczuk, C.E.; Barocas, V.H.; Guenther, K.; et al. Dicer1 Deficiency in the Idiopathic Pulmonary Fibrosis Fibroblastic Focus Promotes Fibrosis by Suppressing MicroRNA Biogenesis. Am. J. Respir. Crit. Care Med. 2018, 198, 486–496. [Google Scholar] [CrossRef]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Moimas, S.; Salton, F.; Kosmider, B.; Ring, N.; Volpe, M.C.; Bahmed, K.; Braga, L.; Rehman, M.; Vodret, S.; Graziani, M.L.; et al. miR-200 family members reduce senescence and restore idiopathic pulmonary fibrosis type II alveolar epithelial cell transdifferentiation. ERJ Open Res. 2019, 5, 00138–02019. [Google Scholar] [CrossRef]
- Ge, L.; Habiel, D.M.; Hansbro, P.M.; Kim, R.Y.; Gharib, S.A.; Edelman, J.D.; Konigshoff, M.; Parimon, T.; Brauer, R.; Huang, Y.; et al. miR-323a-3p regulates lung fibrosis by targeting multiple profibrotic pathways. JCI Insight 2016, 1, e90301. [Google Scholar] [CrossRef]
- Hayton, C.; Chaudhuri, N. Managing Idiopathic Pulmonary Fibrosis: Which Drug for Which Patient? Drugs Aging 2017, 34, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Krauss, E.; Tello, S.; Wilhelm, J.; Schmidt, J.; Stoehr, M.; Seeger, W.; Dartsch, R.C.; Crestani, B.; Guenther, A. Assessing the Effectiveness of Pirfenidone in Idiopathic Pulmonary Fibrosis: Long-Term, Real-World Data from European IPF Registry (eurIPFreg). J. Clin. Med. 2020, 9, 3763. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 291, 367–373. [Google Scholar] [PubMed]
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 2008, 590, 400–408. [Google Scholar] [CrossRef]
- Gurujeyalakshmi, G.; Hollinger, M.A.; Giri, S.N. Pirfenidone inhibits PDGF isoforms in bleomycin hamster model of lung fibrosis at the translational level. Am. J. Physiol. 1999, 276, L311–L318. [Google Scholar] [CrossRef]
- Grattendick, K.J.; Nakashima, J.M.; Feng, L.; Giri, S.N.; Margolin, S.B. Effects of three anti-TNF-alpha drugs: Etanercept, infliximab and pirfenidone on release of TNF-alpha in medium and TNF-alpha associated with the cell in vitro. Int. Immunopharmacol. 2008, 8, 679–687. [Google Scholar] [CrossRef]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-beta-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef]
- Lehmann, M.; Buhl, L.; Alsafadi, H.N.; Klee, S.; Hermann, S.; Mutze, K.; Ota, C.; Lindner, M.; Behr, J.; Hilgendorff, A.; et al. Differential effects of Nintedanib and Pirfenidone on lung alveolar epithelial cell function in ex vivo murine and human lung tissue cultures of pulmonary fibrosis. Respir. Res. 2018, 19, 175. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, F.; Kang, L.; Wang, Z.; Wang, Y. Pirfenidone attenuates bleomycin-induced pulmonary fibrosis in mice by regulating Nrf2/Bach1 equilibrium. BMC Pulm. Med. 2017, 17, 63. [Google Scholar] [CrossRef] [Green Version]
- Pourgholamhossein, F.; Rasooli, R.; Pournamdari, M.; Pourgholi, L.; Samareh-Fekri, M.; Ghazi-Khansari, M.; Iranpour, M.; Poursalehi, H.R.; Heidari, M.R.; Mandegary, A. Pirfenidone protects against paraquat-induced lung injury and fibrosis in mice by modulation of inflammation, oxidative stress, and gene expression. Food Chem. Toxicol. 2018, 112, 39–46. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Crestani, B.; Huggins, J.T.; Kaye, M.; Costabel, U.; Glaspole, I.; Ogura, T.; Song, J.W.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: Results from the open-label extension study, INPULSIS-ON. Lancet Respir. Med. 2019, 7, 60–68. [Google Scholar] [CrossRef]
- Vancheri, C.; Kreuter, M.; Richeldi, L.; Ryerson, C.J.; Valeyre, D.; Grutters, J.C.; Wiebe, S.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Nintedanib with Add-on Pirfenidone in Idiopathic Pulmonary Fibrosis. Results of the INJOURNEY Trial. Am. J. Respir. Crit. Care Med. 2018, 197, 356–363. [Google Scholar] [CrossRef]
- Koli, K.; Myllarniemi, M.; Keski-Oja, J.; Kinnula, V.L. Transforming growth factor-beta activation in the lung: Focus on fibrosis and reactive oxygen species. Antioxid. Redox Signal. 2008, 10, 333–342. [Google Scholar] [CrossRef]
- Saini, G.; Porte, J.; Weinreb, P.H.; Violette, S.M.; Wallace, W.A.; McKeever, T.M.; Jenkins, G. αvβ6 integrin may be a potential prognostic biomarker in interstitial lung disease. Eur. Respir. J. 2015, 46, 486–494. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, T.; Saigal, A.; Johnson, J.; Morrisson, J.; Tabrizifard, S.; Hollingsworth, S.A.; Eddins, M.J.; Mao, W.; O’Neill, K.; et al. Discovery of a new class of integrin antibodies for fibrosis. Sci. Rep. 2021, 11, 2118. [Google Scholar] [CrossRef]
- John, A.E.; Graves, R.H.; Pun, K.T.; Vitulli, G.; Forty, E.J.; Mercer, P.F.; Morrell, J.L.; Barrett, J.W.; Rogers, R.F.; Hafeji, M.; et al. Translational pharmacology of an inhaled small molecule alphavbeta6 integrin inhibitor for idiopathic pulmonary fibrosis. Nat. Commun. 2020, 11, 4659. [Google Scholar] [CrossRef]
- Mullally, A.; Hood, J.; Harrison, C.; Mesa, R. Fedratinib in myelofibrosis. Blood Adv. 2020, 4, 1792–1800. [Google Scholar]
- D’Alessandro, M.; Perillo, F.; Metella Refini, R.; Bergantini, L.; Bellisai, F.; Selvi, E.; Cameli, P.; Manganelli, S.; Conticini, E.; Cantarini, L.; et al. Efficacy of baricitinib in treating rheumatoid arthritis: Modulatory effects on fibrotic and inflammatory biomarkers in a real-life setting. Int. Immunopharmacol. 2020, 86, 106748. [Google Scholar] [CrossRef]
- Strand, V.; van Vollenhoven, R.F.; Lee, E.B.; Fleischmann, R.; Zwillich, S.H.; Gruben, D.; Koncz, T.; Wilkinson, B.; Wallenstein, G. Tofacitinib or adalimumab versus placebo: Patient-reported outcomes from a phase 3 study of active rheumatoid arthritis. Rheumatology 2016, 55, 1031–1041. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liang, R.; Chen, C.W.; Mallano, T.; Dees, C.; Distler, A.; Reich, A.; Bergmann, C.; Ramming, A.; Gelse, K.; et al. JAK1-dependent transphosphorylation of JAK2 limits the antifibrotic effects of selective JAK2 inhibitors on long-term treatment. Ann. Rheum. Dis. 2017, 76, 1467–1475. [Google Scholar] [CrossRef]
- Emery, P.; Keystone, E.; Tony, H.P.; Cantagrel, A.; van Vollenhoven, R.; Sanchez, A.; Alecock, E.; Lee, J.; Kremer, J. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: Results from a 24-week multicentre randomised placebo-controlled trial. Ann. Rheum. Dis. 2008, 67, 1516–1523. [Google Scholar] [CrossRef]
- Abidi, E.; El Nekidy, W.S.; Alefishat, E.; Rahman, N.; Petroianu, G.A.; El-Lababidi, R.; Mallat, J. Tocilizumab and COVID-19: Timing of Administration and Efficacy. Front. Pharmacol. 2022, 13, 825749. [Google Scholar] [CrossRef]
- Coltro, G.; Vannucchi, A.M. The safety of JAK kinase inhibitors for the treatment of myelofibrosis. Expert Opin. Drug Saf. 2021, 20, 139–154. [Google Scholar] [CrossRef]
- Merkt, W.; Bueno, M.; Mora, A.L.; Lagares, D. Senotherapeutics: Targeting senescence in idiopathic pulmonary fibrosis. Semin. Cell Dev. Biol. 2020, 101, 104–110. [Google Scholar] [CrossRef]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50, 1602367. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Luo, S.; Kan, J.; Zhang, J.; Ye, P.; Wang, D.; Jiang, X.; Li, M.; Zhu, L.; Gu, Y. Bioactive Compounds from Coptidis Rhizoma Alleviate Pulmonary Arterial Hypertension by Inhibiting Pulmonary Artery Smooth Muscle Cells’ Proliferation and Migration. J. Cardiovasc. Pharmacol. 2021, 78, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.A., Jr.; Hogaboam, C.M. Quercetin Enhances Ligand-induced Apoptosis in Senescent Idiopathic Pulmonary Fibrosis Fibroblasts and Reduces Lung Fibrosis In Vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, J.M. Senolytic therapies for healthy longevity. Science 2019, 364, 636–637. [Google Scholar] [CrossRef]
- Ozgur Yurttas, N.; Eskazan, A.E. Dasatinib-induced pulmonary arterial hypertension. Br. J. Clin. Pharmacol. 2018, 84, 835–845. [Google Scholar] [CrossRef]
- Shea, B.S.; Tager, A.M. Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Ninou, I.; Kaffe, E.; Muller, S.; Budd, D.C.; Stevenson, C.S.; Ullmer, C.; Aidinis, V. Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2018, 52, 32–40. [Google Scholar] [CrossRef]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef]
- Gill, M.W.; Sivaraman, L.; Cheng, P.T.W.; Murphy, B.J.; Chadwick, K.; Lehman-McKeeman, L.; Graziano, M.; Bristol-Myers Squibb. BMS-986278, an LPA1 receptor antagonist for idiopathic pulmonary fibrosis: Preclinical assessments of potential hepatobiliary toxicity [abstract]. Am. J. Respir. Crit. Care Med. 2019, 199, 8755. [Google Scholar]
- Corte, T.J.; Lancaster, L.; Swigris, J.J.; Maher, T.M.; Goldin, J.G.; Palmer, S.M.; Suda, T.; Ogura, T.; Minnich, A.; Zhan, X.; et al. Phase 2 trial design of BMS-986278, a lysophosphatidic acid receptor 1 (LPA1) antagonist, in patients with idiopathic pulmonary fibrosis (IPF) or progressive fibrotic interstitial lung disease (PF-ILD). BMJ Open Respir. Res. 2021, 8, e001026. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Fernandez Perez, E.R.; Costabel, U.; Albera, C.; Lederer, D.J.; Flaherty, K.R.; Ettinger, N.; Perez, R.; Scholand, M.B.; Goldin, J.; et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2020, 8, 25–33. [Google Scholar] [CrossRef]
- Pilling, D.; Galvis-Carvajal, E.; Karhadkar, T.R.; Cox, N.; Gomer, R.H. Monocyte differentiation and macrophage priming are regulated differentially by pentraxins and their ligands. BMC Immunol. 2017, 18, 30. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.A.; Chen, Q.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Peng, X.; Gulati, M.; Homer, R.J.; Russell, T.; van Rooijen, N.; et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int. J. Biochem. Cell Biol. 2011, 43, 154–162. [Google Scholar] [CrossRef]
- Korfei, M.; von der Beck, D.; Henneke, I.; Markart, P.; Ruppert, C.; Mahavadi, P.; Ghanim, B.; Klepetko, W.; Fink, L.; Meiners, S.; et al. Comparative proteome analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF), non-specific interstitial pneumonia (NSIP) and organ donors. J. Proteom. 2013, 85, 109–128. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Effect of Recombinant Human Pentraxin 2 vs Placebo on Change in Forced Vital Capacity in Patients with Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef]
- Jia, W.; Wang, Z.; Gao, C.; Wu, J.; Wu, Q. Trajectory modeling of endothelial-to-mesenchymal transition reveals galectin-3 as a mediator in pulmonary fibrosis. Cell Death Dis. 2021, 12, 327. [Google Scholar] [CrossRef]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Hirani, N.; MacKinnon, A.C.; Nicol, L.; Ford, P.; Schambye, H.; Pedersen, A.; Nilsson, U.J.; Leffler, H.; Sethi, T.; Tantawi, S.; et al. Target inhibition of galectin-3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2021, 57, 2002559. [Google Scholar] [CrossRef]
- Noth, I.; Anstrom, K.J.; Calvert, S.B.; de Andrade, J.; Flaherty, K.R.; Glazer, C.; Kaner, R.J.; Olman, M.A.; Idiopathic Pulmonary Fibrosis Clinical Research Network. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar]
- Behr, J.; Bendstrup, E.; Crestani, B.; Gunther, A.; Olschewski, H.; Skold, C.M.; Wells, A.; Wuyts, W.; Koschel, D.; Kreuter, M.; et al. Safety and tolerability of acetylcysteine and pirfenidone combination therapy in idiopathic pulmonary fibrosis: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2016, 4, 445–453. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Brown, K.K.; Raghu, G.; du Bois, R.M.; Lynch, D.A.; Martinez, F.; Valeyre, D.; Leconte, I.; Morganti, A.; Roux, S.; et al. BUILD-3: A randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 92–99. [Google Scholar] [CrossRef]
- Raghu, G.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Nathan, S.D.; Wells, A.U.; Collard, H.R.; et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann. Intern Med. 2013, 158, 641–649. [Google Scholar] [CrossRef]
- Kolb, M.; Raghu, G.; Wells, A.U.; Behr, J.; Richeldi, L.; Schinzel, B.; Quaresma, M.; Stowasser, S.; Martinez, F.J.; INSTAGE Investigators. Nintedanib plus Sildenafil in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 379, 1722–1731. [Google Scholar] [CrossRef]
- Daniels, C.E.; Lasky, J.A.; Limper, A.H.; Mieras, K.; Gabor, E.; Schroeder, D.R.; Imatinib-IPF Study Investigators. Imatinib treatment for idiopathic pulmonary fibrosis: Randomized placebo-controlled trial results. Am. J. Respir. Crit. Care Med. 2010, 181, 604–610. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryu, J.H.; Douglas, W.W.; Schwarz, M.I.; Curran-Everett, D.; King, T.E., Jr.; Brown, K.K. Combined corticosteroid and cyclophosphamide therapy does not alter survival in idiopathic pulmonary fibrosis. Chest 2004, 125, 2169–2174. [Google Scholar] [CrossRef] [Green Version]
- King, T.E., Jr.; Albera, C.; Bradford, W.Z.; Costabel, U.; Hormel, P.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; Szwarcberg, J.; Thomeer, M.; et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): A multicentre, randomised, placebo-controlled trial. Lancet 2009, 374, 222–228. [Google Scholar] [CrossRef]
- Raghu, G.; Brown, K.K.; Collard, H.R.; Cottin, V.; Gibson, K.F.; Kaner, R.J.; Lederer, D.J.; Martinez, F.J.; Noble, P.W.; Song, J.W.; et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: A randomised, double-blind, controlled, phase 2 trial. Lancet Respir. Med. 2017, 5, 22–32. [Google Scholar] [CrossRef]
- Wheaton, A.K.; Velikoff, M.; Agarwal, M.; Loo, T.T.; Horowitz, J.C.; Sisson, T.H.; Kim, K.K. The vitronectin RGD motif regulates TGF-beta-induced alveolar epithelial cell apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L1206–L1217. [Google Scholar] [CrossRef] [Green Version]
- Distler, J.H.W.; Gyorfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Konigshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [Green Version]
- White, E.S.; Thannickal, V.J.; Carskadon, S.L.; Dickie, E.G.; Livant, D.L.; Markwart, S.; Toews, G.B.; Arenberg, D.A. Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: A role for phosphatase and tensin homologue deleted on chromosome 10. Am. J. Respir. Crit. Care Med. 2003, 168, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [Green Version]
- Vancheri, C. Idiopathic pulmonary fibrosis and cancer: Do they really look similar? BMC Med. 2015, 13, 220. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Huang, Y.; Pang, L.; Gu, W.; Wang, N.; Hu, J.; Cui, X.; Zhang, J.; Zhao, J.; Liu, C.; et al. Prognostic value of the MicroRNA-29 family in multiple human cancers: A meta-analysis and systematic review. Clin. Exp. Pharmacol. Physiol. 2017, 44, 441–454. [Google Scholar] [CrossRef]
- Babaei, G.; Raei, N.; Toofani Milani, A.; Gholizadeh-Ghaleh Aziz, S.; Pourjabbar, N.; Geravand, F. The emerging role of miR-200 family in metastasis: Focus on EMT, CSCs, angiogenesis, and anoikis. Mol. Biol. Rep. 2021, 48, 6935–6947. [Google Scholar] [CrossRef]
- De Santis, C.; Gotte, M. The Role of microRNA Let-7d in Female Malignancies and Diseases of the Female Reproductive Tract. Int. J. Mol. Sci. 2021, 22, 7359. [Google Scholar] [CrossRef]
- Lettieri, S.; Oggionni, T.; Lancia, A.; Bortolotto, C.; Stella, G.M. Immune Stroma in Lung Cancer and Idiopathic Pulmonary Fibrosis: A Common Biologic Landscape? Int. J. Mol. Sci. 2021, 22, 2882. [Google Scholar] [CrossRef]
- Singh, A.; Singh, A.K.; Giri, R.; Kumar, D.; Sharma, R.; Valis, M.; Kuca, K.; Garg, N. The role of microRNA-21 in the onset and progression of cancer. Future Med. Chem. 2021, 13, 1885–1906. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Watts, K.; Feghali-Bostwick, C.A.; Knox, A.; Pang, L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol. Cell. Biol. 2009, 29, 4325–4339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.K.; Scruggs, A.M.; Donaghy, J.; Horowitz, J.C.; Zaslona, Z.; Przybranowski, S.; White, E.S.; Peters-Golden, M. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 2013, 4, e621. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Hagood, J.S.; Liu, H.; Zhang, W.; Ambalavanan, N.; Thannickal, V.J. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur. Respir. J. 2014, 43, 1448–1458. [Google Scholar] [CrossRef]
- Korfei, M.; Skwarna, S.; Henneke, I.; MacKenzie, B.; Klymenko, O.; Saito, S.; Ruppert, C.; von der Beck, D.; Mahavadi, P.; Klepetko, W.; et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 2015, 70, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone deacetylases: A saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 2014, 62, 11–33. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, H.; Hock, T.; Thannickal, V.J.; Sanders, Y.Y. Histone deacetylase inhibition downregulates collagen 3A1 in fibrotic lung fibroblasts. Int. J. Mol. Sci. 2013, 14, 19605–19617. [Google Scholar] [CrossRef]
- Ota, C.; Yamada, M.; Fujino, N.; Motohashi, H.; Tando, Y.; Takei, Y.; Suzuki, T.; Takahashi, T.; Kamata, S.; Makiguchi, T.; et al. Histone deacetylase inhibitor restores surfactant protein-C expression in alveolar-epithelial type II cells and attenuates bleomycin-induced pulmonary fibrosis in vivo. Exp. Lung Res. 2015, 41, 422–434. [Google Scholar] [CrossRef]
- Conforti, F.; Davies, E.R.; Calderwood, C.J.; Thatcher, T.H.; Jones, M.G.; Smart, D.E.; Mahajan, S.; Alzetani, A.; Havelock, T.; Maher, T.M.; et al. The histone deacetylase inhibitor, romidepsin, as a potential treatment for pulmonary fibrosis. Oncotarget 2017, 8, 48737–48754. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Alam, A.; Pac-Soo, A.; Chen, Q.; Shang, Y.; Zhao, H.; Yao, S.; Ma, D. Pretreatment with valproic acid alleviates pulmonary fibrosis through epithelial-mesenchymal transition inhibition in vitro and in vivo. Lab. Investig. 2021, 101, 1166–1175. [Google Scholar] [CrossRef]
- Saito, S.; Zhuang, Y.; Suzuki, T.; Ota, Y.; Bateman, M.E.; Alkhatib, A.L.; Morris, G.F.; Lasky, J.A. HDAC8 inhibition ameliorates pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L175–L186. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Burgess, A.; Fairlie, D.P.; Leonard, H.; Parsons, P.G.; Gabrielli, B.G. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef] [Green Version]
- Campiani, G.; Cavella, C.; Osko, J.D.; Brindisi, M.; Relitti, N.; Brogi, S.; Saraswati, A.P.; Federico, S.; Chemi, G.; Maramai, S.; et al. Harnessing the Role of HDAC6 in Idiopathic Pulmonary Fibrosis: Design, Synthesis, Structural Analysis, and Biological Evaluation of Potent Inhibitors. J. Med. Chem. 2021, 64, 9960–9988. [Google Scholar] [CrossRef]
- Prasse, A.; Binder, H.; Schupp, J.C.; Kayser, G.; Bargagli, E.; Jaeger, B.; Hess, M.; Rittinghausen, S.; Vuga, L.; Lynn, H.; et al. BAL Cell Gene Expression Is Indicative of Outcome and Airway Basal Cell Involvement in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 622–630. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Eberharter, A.; Becker, P.B. Histone acetylation: A switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002, 3, 224–229. [Google Scholar] [CrossRef]
- Kanno, T.; Kanno, Y.; Siegel, R.M.; Jang, M.K.; Lenardo, M.J.; Ozato, K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell 2004, 13, 33–43. [Google Scholar] [CrossRef]
- Spange, S.; Wagner, T.; Heinzel, T.; Kramer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Buchwald, M.; Kramer, O.H.; Heinzel, T. HDACi—Targets beyond chromatin. Cancer Lett. 2009, 280, 160–167. [Google Scholar] [CrossRef]
- Kramer, O.H. HDAC2: A critical factor in health and disease. Trends Pharm. Sci 2009, 30, 647–655. [Google Scholar] [CrossRef]
- Fritsche, P.; Seidler, B.; Schuler, S.; Schnieke, A.; Gottlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [Green Version]
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef]
- Liu, P.; Xiao, J.; Wang, Y.; Song, X.; Huang, L.; Ren, Z.; Kitazato, K. Posttranslational modification and beyond: Interplay between histone deacetylase 6 and heat-shock protein 90. Mol. Med. 2021, 27, 110. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Yeon, S.K.; Kim, G.W.; Lee, D.H.; Jeon, Y.H.; Yoo, J.; Kim, S.Y.; Kwon, S.H. HDAC6-Selective Inhibitor Overcomes Bortezomib Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 1341. [Google Scholar] [CrossRef]
- Shan, B.; Yao, T.P.; Nguyen, H.T.; Zhuo, Y.; Levy, D.R.; Klingsberg, R.C.; Tao, H.; Palmer, M.L.; Holder, K.N.; Lasky, J.A. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008, 283, 21065–21073. [Google Scholar] [CrossRef] [Green Version]
- Deskin, B.; Lasky, J.; Zhuang, Y.; Shan, B. Requirement of HDAC6 for activation of Notch1 by TGF-beta1. Sci. Rep. 2016, 6, 31086. [Google Scholar] [CrossRef] [PubMed]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. HDAC6-an Emerging Target Against Chronic Myeloid Leukemia? Cancers 2020, 12, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafon-Hughes, L.; Di Tomaso, M.V.; Mendez-Acuna, L.; Martinez-Lopez, W. Chromatin-remodelling mechanisms in cancer. Mutat. Res. 2008, 658, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shang, Y.P.; Chen, H.Y.; Li, J. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol. Res. 2017, 47, 149–159. [Google Scholar] [CrossRef]
- Harms, K.L.; Chen, X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007, 67, 3145–3152. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Bae, H.J.; Xie, H.J.; Chang, Y.G.; Kim, M.G.; Park, H.; Lee, J.Y.; et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J. Cell. Biochem. 2012, 113, 2167–2177. [Google Scholar] [CrossRef]
- Von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361–371.e5. [Google Scholar] [CrossRef]
- Mariadason, J.M. Dissecting HDAC3-mediated tumor progression. Cancer Biol. Ther. 2008, 7, 1581–1583. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, W. Histone deacetylase 3 (HDAC3) as an important epigenetic regulator of kidney diseases. J. Mol. Med. 2022, 100, 43–51. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byun, D.S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.F.; Wei, A.M.; Kou, Q.; Zhu, Q.Y.; Zhang, L. Histone deacetylase 4 increases progressive epithelial ovarian cancer cells via repression of p21 on fibrillar collagen matrices. Oncol. Rep. 2016, 35, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Spaety, M.E.; Gries, A.; Badie, A.; Venkatasamy, A.; Romain, B.; Orvain, C.; Yanagihara, K.; Okamoto, K.; Jung, A.C.; Mellitzer, G.; et al. HDAC4 Levels Control Sensibility toward Cisplatin in Gastric Cancer via the p53-p73/BIK Pathway. Cancers 2019, 11, 1747. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qi, Z.; Yin, H.; Yang, G. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1alpha-mediated regulation of apoptosis and autophagy. Theranostics 2019, 9, 1096–1114. [Google Scholar] [CrossRef]
- Ye, M.; Fang, Z.; Gu, H.; Song, R.; Ye, J.; Li, H.; Wu, Z.; Zhou, S.; Li, P.; Cai, X.; et al. Histone deacetylase 5 promotes the migration and invasion of hepatocellular carcinoma via increasing the transcription of hypoxia-inducible factor-1alpha under hypoxia condition. Tumor Biol. 2017, 39, 1010428317705034. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Sun, S.; Yao, S.; Han, X.; Gu, M.; Shi, J. Histone deacetylase 5 promotes the proliferation and invasion of lung cancer cells. Oncol. Rep. 2018, 40, 2224–2232. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; D’Mello, S.R. Neuroprotection by histone deacetylase-7 (HDAC7) occurs by inhibition of c-Jun expression through a deacetylase-independent mechanism. J. Biol. Chem. 2011, 286, 4819–4828. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Chen, Q.; Xie, Z.; Ai, J.; Tong, L.; Ding, J.; Geng, M. The role of histone deacetylase 7 (HDAC7) in cancer cell proliferation: Regulation on c-Myc. J. Mol. Med. 2011, 89, 279–289. [Google Scholar] [CrossRef]
- Sang, Y.; Sun, L.; Wu, Y.; Yuan, W.; Liu, Y.; Li, S.W. Histone deacetylase 7 inhibits plakoglobin expression to promote lung cancer cell growth and metastasis. Int. J. Oncol. 2019, 54, 1112–1122. [Google Scholar] [CrossRef] [Green Version]
- Cutano, V.; Di Giorgio, E.; Minisini, M.; Picco, R.; Dalla, E.; Brancolini, C. HDAC7-mediated control of tumour microenvironment maintains proliferative and stemness competence of human mammary epithelial cells. Mol. Oncol. 2019, 13, 1651–1668. [Google Scholar] [CrossRef] [Green Version]
- Shinke, G.; Yamada, D.; Eguchi, H.; Iwagami, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; Gotoh, K.; Kobayashi, S.; et al. Role of histone deacetylase 1 in distant metastasis of pancreatic ductal cancer. Cancer Sci. 2018, 109, 2520–2531. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.S.; Yang, X.Z.; Wen, Y.F.; Mail, S.J.; Wang, M.H.; Zhang, M.Y.; Zheng, X.F.; Wang, H.Y. Overexpressed HDAC4 is associated with poor survival and promotes tumor progression in esophageal carcinoma. Aging 2016, 8, 1236–1249. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Cao, F.; Yu, X.; Zhou, P.; Di, Q.; Lei, J.; Tai, Y.; Wu, H.; Li, X.; Wang, X.; et al. The expression of HDAC7 in cancerous gastric tissues is positively associated with distant metastasis and poor patient prognosis. Clin. Transl. Oncol. 2017, 19, 1045–1054. [Google Scholar] [CrossRef]
- Lagger, G.; O’Carroll, D.; Rembold, M.; Khier, H.; Tischler, J.; Weitzer, G.; Schuettengruber, B.; Hauser, C.; Brunmeir, R.; Jenuwein, T.; et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002, 21, 2672–2681. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, C.M.; Luo, Y.; Yin, Z.; Zhang, M.; Zhu, W.; Wang, T.; Floss, T.; Goettlicher, M.; Noppinger, P.R.; Wurst, W.; et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat. Med. 2007, 13, 324–331. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.L.; Chua, K.; et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Telles, E.; Karl, M.; Cheng, F.; Luetteke, N.; Sotomayor, E.M.; Miller, R.H.; Seto, E. Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Sci. Alliance 2018, 1, e201800039. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [Green Version]
- Schwer, B.; Verdin, E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Bordone, L.; Cohen, D.; Robinson, A.; Motta, M.C.; van Veen, E.; Czopik, A.; Steele, A.D.; Crowe, H.; Marmor, S.; Luo, J.; et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 2007, 6, 759–767. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, N.; Liu, B.; Ling, J.; Yang, W.; Pang, X.; Li, T. Pracinostat (SB939), a histone deacetylase inhibitor, suppresses breast cancer metastasis and growth by inactivating the IL-6/STAT3 signalling pathways. Life Sci. 2020, 248, 117469. [Google Scholar] [CrossRef] [PubMed]
- Kusaczuk, M.; Bartoszewicz, M.; Cechowska-Pasko, M. Phenylbutyric Acid: Simple structure—Multiple effects. Curr. Pharm. Des. 2015, 21, 2147–2166. [Google Scholar] [CrossRef] [PubMed]
- Kaletsch, A.; Pinkerneil, M.; Hoffmann, M.J.; Jaguva Vasudevan, A.A.; Wang, C.; Hansen, F.K.; Wiek, C.; Hanenberg, H.; Gertzen, C.; Gohlke, H.; et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenet. 2018, 10, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, V.; Bret, C.; Altucci, L.; Mai, A.; Duraffourd, C.; Loubersac, J.; Harmand, P.O.; Bonnet, S.; Valente, S.; Maudelonde, T.; et al. Specific activity of class II histone deacetylases in human breast cancer cells. Mol. Cancer Res. 2008, 6, 1908–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawas, A.; Radeski, D.; O’Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: A perspective review. Ther. Adv. Hematol. 2015, 6, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Barbarotta, L.; Hurley, K. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar]
- Baertsch, M.A.; Hillengass, J.; Blocka, J.; Schonland, S.; Hegenbart, U.; Goldschmidt, H.; Raab, M.S. Efficacy and tolerability of the histone deacetylase inhibitor panobinostat in clinical practice. Hematol. Oncol. 2018, 36, 210–216. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic acid pathway: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2013, 23, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Munster, P.; Marchion, D.; Bicaku, E.; Lacevic, M.; Kim, J.; Centeno, B.; Daud, A.; Neuger, A.; Minton, S.; Sullivan, D. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: Phase I/II trial of valproic acid and epirubicin/FEC. Clin. Cancer Res. 2009, 15, 2488–2496. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Murphy, B.; Miller, R.; Menon, V.; Banik, N.L.; Giglio, P.; Lindhorst, S.M.; Varma, A.K.; Vandergrift, W.A., 3rd; Patel, S.J.; et al. Mechanisms and clinical significance of histone deacetylase inhibitors: Epigenetic glioblastoma therapy. Anticancer Res. 2015, 35, 615–625. [Google Scholar]
- Kim, Y.S.; Cha, H.; Kim, H.J.; Cho, J.M.; Kim, H.R. The Anti-Fibrotic Effects of CG-745, an HDAC Inhibitor, in Bleomycin and PHMG-Induced Mouse Models. Molecules 2019, 24, 2792. [Google Scholar] [CrossRef] [Green Version]
- Wells, C.E.; Bhaskara, S.; Stengel, K.R.; Zhao, Y.; Sirbu, B.; Chagot, B.; Cortez, D.; Khabele, D.; Chazin, W.J.; Cooper, A.; et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS ONE 2013, 8, e68915. [Google Scholar] [CrossRef]
- Saito, S.; Zhuang, Y.; Shan, B.; Danchuk, S.; Luo, F.; Korfei, M.; Guenther, A.; Lasky, J.A. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFbeta-PI3K-Akt pathway. PLoS ONE 2017, 12, e0186615. [Google Scholar] [CrossRef] [Green Version]
- Deskin, B.; Yin, Q.; Zhuang, Y.; Saito, S.; Shan, B.; Lasky, J.A. Inhibition of HDAC6 Attenuates Tumor Growth of Non-Small Cell Lung Cancer. Transl. Oncol. 2020, 13, 135–145. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Pulya, S.; Amin, S.A.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol. Res. 2021, 163, 105274. [Google Scholar] [CrossRef]
- Hu, J.; Jing, H.; Lin, H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 2014, 6, 945–966. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Ma, T.; Zhu, Q.; Xu, Y.; Zha, X. Recent advances in inhibitors of sirtuin1/2: An update and perspective. Future Med. Chem. 2018, 10, 907–934. [Google Scholar] [CrossRef]
- Kee, H.J.; Kook, H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J. Biomed. Biotechnol. 2011, 2011, 928326. [Google Scholar] [CrossRef] [PubMed]
- Chelladurai, P.; Boucherat, O.; Stenmark, K.; Kracht, M.; Seeger, W.; Bauer, U.M.; Bonnet, S.; Pullamsetti, S.S. Targeting histone acetylation in pulmonary hypertension and right ventricular hypertrophy. Br. J. Pharmacol. 2021, 178, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.M.; Golden-Mason, L.; Ferguson, B.S.; Schuetze, K.B.; Cavasin, M.A.; Demos-Davies, K.; Yeager, M.E.; Stenmark, K.R.; McKinsey, T.A. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J. Mol. Cell. Cardiol. 2014, 67, 112–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, Z.; Fitzsimmons, R.L.; Reid, R.C.; Ramnath, D.; Clouston, A.; Gupta, P.K.; Irvine, K.M.; Powell, E.E.; Schroder, K.; Stow, J.L.; et al. Inhibitors of class I histone deacetylases attenuate thioacetamide-induced liver fibrosis in mice by suppressing hepatic type 2 inflammation. Br. J. Pharmacol. 2019, 176, 3775–3790. [Google Scholar] [CrossRef]
- Xiong, C.; Guan, Y.; Zhou, X.; Liu, L.; Zhuang, M.A.; Zhang, W.; Zhang, Y.; Masucci, M.V.; Bayliss, G.; Zhao, T.C.; et al. Selective inhibition of class IIa histone deacetylases alleviates renal fibrosis. FASEB J. 2019, 33, 8249–8262. [Google Scholar] [CrossRef]
- Jones, D.L.; Haak, A.J.; Caporarello, N.; Choi, K.M.; Ye, Z.; Yan, H.; Varelas, X.; Ordog, T.; Ligresti, G.; Tschumperlin, D.J. TGFbeta-induced fibroblast activation requires persistent and targeted HDAC-mediated gene repression. J. Cell Sci. 2019, 132, jcs233486. [Google Scholar] [CrossRef] [Green Version]
- Coward, W.R.; Watts, K.; Feghali-Bostwick, C.A.; Jenkins, G.; Pang, L. Repression of IP-10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol. Cell. Biol. 2010, 30, 2874–2886. [Google Scholar] [CrossRef] [Green Version]
- Sanders, Y.Y.; Tollefsbol, T.O.; Varisco, B.M.; Hagood, J.S. Epigenetic regulation of thy-1 by histone deacetylase inhibitor in rat lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2011, 45, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Korfei, M.; Stelmaszek, D.; MacKenzie, B.; Skwarna, S.; Chillappagari, S.; Bach, A.C.; Ruppert, C.; Saito, S.; Mahavadi, P.; Klepetko, W.; et al. Comparison of the antifibrotic effects of the pan-histone deacetylase-inhibitor panobinostat versus the IPF-drug pirfenidone in fibroblasts from patients with idiopathic pulmonary fibrosis. PLoS ONE 2018, 13, e0207915. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Shan, B.; Klingsberg, R.C.; Qin, X.; Lasky, J.A. Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L864–L870. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Li, Y.; Jiang, H.; Xiong, J.; Xu, J.; Qin, H.; Liu, B. Prevention of Pulmonary Fibrosis via Trichostatin A (TSA) in Bleomycin Induced Rats. Sarcoidosis Vasc. Diffus. Lung Dis. 2014, 31, 219–226. [Google Scholar]
- Rao, S.S.; Zhang, X.Y.; Shi, M.J.; Xiao, Y.; Zhang, Y.Y.; Wang, Y.Y.; Zhang, C.Z.; Shao, S.J.; Liu, X.M.; Guo, B. Suberoylanilide hydroxamic acid attenuates paraquat-induced pulmonary fibrosis by preventing Smad7 from deacetylation in rats. J. Thorac. Dis. 2016, 8, 2485–2494. [Google Scholar] [CrossRef] [Green Version]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim. Biophys. Acta. 2007, 1773, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Kabel, A.M.; Omar, M.S.; Elmaaboud, M.A.A. Amelioration of bleomycin-induced lung fibrosis in rats by valproic acid and butyrate: Role of nuclear factor kappa-B, proinflammatory cytokines and oxidative stress. Int. Immunopharmacol. 2016, 39, 335–342. [Google Scholar] [CrossRef]
- Jiang, X.; Fang, G.; Dong, L.; Jin, P.; Ding, L.; Zhang, H.; Fan, J.; Mao, S.; Fan, X.; Gong, Y.; et al. Chemical chaperone 4-phenylbutyric acid alleviates the aggregation of human familial pulmonary fibrosis-related mutant SP-A2 protein in part through effects on GRP78. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3546–3557. [Google Scholar] [CrossRef]
- Zhao, H.; Qin, H.Y.; Cao, L.F.; Chen, Y.H.; Tan, Z.X.; Zhang, C.; Xu, D.X. Phenylbutyric acid inhibits epithelial-mesenchymal transition during bleomycin-induced lung fibrosis. Toxicol. Lett. 2015, 232, 213–220. [Google Scholar] [CrossRef]
- Sanders, Y.Y.; Liu, H.; Scruggs, A.M.; Duncan, S.R.; Huang, S.K.; Thannickal, V.J. Epigenetic Regulation of Caveolin-1 Gene Expression in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2017, 56, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Gao, Q.; Zhang, L.; Ding, Y.; Wang, H.; Cao, W. Inhibiting HDAC3 (Histone Deacetylase 3) Aberration and the Resultant Nrf2 (Nuclear Factor Erythroid-Derived 2-Related Factor-2) Repression Mitigates Pulmonary Fibrosis. Hypertension 2021, 78, e15–e25. [Google Scholar] [CrossRef]
- Wilborn, J.; Crofford, L.J.; Burdick, M.D.; Kunkel, S.L.; Strieter, R.M.; Peters-Golden, M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J. Clin. Investig. 1995, 95, 1861–1868. [Google Scholar] [CrossRef] [Green Version]
- Honda, S.; Lewis, Z.A.; Shimada, K.; Fischle, W.; Sack, R.; Selker, E.U. Heterochromatin protein 1 forms distinct complexes to direct histone deacetylation and DNA methylation. Nat. Struct. Mol. Biol. 2012, 19, 471–477. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Kanno, Y.; Chen, X.; Levy, D.E. Cell signaling. Stat acetylation—A key facet of cytokine signaling? Science 2005, 307, 217–218. [Google Scholar] [CrossRef]
- Gupta, M.; Han, J.J.; Stenson, M.; Wellik, L.; Witzig, T.E. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012, 26, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Cotto, M.; Cabanillas, F.; Tirado, M.; Garcia, M.V.; Pacheco, E. Epigenetic therapy of lymphoma using histone deacetylase inhibitors. Clin. Transl. Oncol. 2010, 12, 401–409. [Google Scholar] [CrossRef]
- Tang, S.; Cheng, B.; Zhe, N.; Ma, D.; Xu, J.; Li, X.; Guo, Y.; Wu, W.; Wang, J. Histone deacetylase inhibitor BG45-mediated HO-1 expression induces apoptosis of multiple myeloma cells by the JAK2/STAT3 pathway. Anticancer Drugs 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Barter, M.J.; Pybus, L.; Litherland, G.J.; Rowan, A.D.; Clark, I.M.; Edwards, D.R.; Cawston, T.E.; Young, D.A. HDAC-mediated control of ERK- and PI3K-dependent TGF-beta-induced extracellular matrix-regulating genes. Matrix Biol. 2010, 29, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, B.; Schupp, J.C.; Plappert, L.; Terwolbeck, O.; Kayser, G.; Engelhard, P.; Adams, T.S.; Zweigerdt, R.; Kempf, H.; Lienenklaus, S.; et al. Airway Basal Cells show a dedifferentiated KRT17highPhenotype and promote Fibrosis in Idiopathic Pulmonary Fibrosis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Paroni, G.; Mizzau, M.; Henderson, C.; Del Sal, G.; Schneider, C.; Brancolini, C. Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol. Biol. Cell 2004, 15, 2804–2818. [Google Scholar] [CrossRef] [Green Version]
- Scott, F.L.; Fuchs, G.J.; Boyd, S.E.; Denault, J.B.; Hawkins, C.J.; Dequiedt, F.; Salvesen, G.S. Caspase-8 cleaves histone deacetylase 7 and abolishes its transcription repressor function. J. Biol. Chem. 2008, 283, 19499–19510. [Google Scholar] [CrossRef] [Green Version]
- Meja, K.K.; Rajendrasozhan, S.; Adenuga, D.; Biswas, S.K.; Sundar, I.K.; Spooner, G.; Marwick, J.A.; Chakravarty, P.; Fletcher, D.; Whittaker, P.; et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am. J. Respir. Cell Mol. Biol. 2008, 39, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Osoata, G.O.; Yamamura, S.; Ito, M.; Vuppusetty, C.; Adcock, I.M.; Barnes, P.J.; Ito, K. Nitration of distinct tyrosine residues causes inactivation of histone deacetylase 2. Biochem. Biophys. Res. Commun. 2009, 384, 366–371. [Google Scholar] [CrossRef]
- Min, T.; Bodas, M.; Mazur, S.; Vij, N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. 2011, 89, 577–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zheng, Y.; Yuan, H.; Liu, Y.; Wen, X. Effects of dynamic changes in histone acetylation and deacetylase activity on pulmonary fibrosis. Int. Immunopharmacol. 2017, 52, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Jiao, L.; Yu, N.; Duan, H.; Yu, Y.; Bai, Y. Histone Deacetylase 3-Mediated Inhibition of microRNA-19a-3p Facilitates the Development of Rheumatoid Arthritis-Associated Interstitial Lung Disease. Front. Physiol. 2020, 11, 549656. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, M.; Heldin, C.H.; Ericsson, J.; Gronroos, E. The balance between acetylation and deacetylation controls Smad7 stability. J. Biol. Chem. 2005, 280, 21797–21803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, E.R.; Haitchi, H.M.; Thatcher, T.H.; Sime, P.J.; Kottmann, R.M.; Ganesan, A.; Packham, G.; O’Reilly, K.M.; Davies, D.E. Spiruchostatin A inhibits proliferation and differentiation of fibroblasts from patients with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 687–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, O.H.; Zhu, P.; Ostendorff, H.P.; Golebiewski, M.; Tiefenbach, J.; Peters, M.A.; Brill, B.; Groner, B.; Bach, I.; Heinzel, T.; et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003, 22, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Wang, L.; Yao, C.; Ni, Z.; Liu, F.; Gong, C.; Zhu, X.; Yan, X.; Watowich, S.S.; Lee, D.A.; et al. The histone deacetylase inhibitor valproic acid inhibits NKG2D expression in natural killer cells through suppression of STAT3 and HDAC3. Sci. Rep. 2017, 7, 45266. [Google Scholar] [CrossRef]
- Noguchi, S.; Eitoku, M.; Moriya, S.; Kondo, S.; Kiyosawa, H.; Watanabe, T.; Suganuma, N. Regulation of Gene Expression by Sodium Valproate in Epithelial-to-Mesenchymal Transition. Lung 2015, 193, 691–700. [Google Scholar] [CrossRef]
- Kamio, K.; Azuma, A.; Usuki, J.; Matsuda, K.; Inomata, M.; Nishijima, N.; Itakura, S.; Hayashi, H.; Kashiwada, T.; Kokuho, N.; et al. XPLN is modulated by HDAC inhibitors and negatively regulates SPARC expression by targeting mTORC2 in human lung fibroblasts. Pulm. Pharmacol. Ther. 2017, 44, 61–69. [Google Scholar] [CrossRef]
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Gunther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 2229. [Google Scholar] [CrossRef] [Green Version]
- Petkova, D.K.; Clelland, C.A.; Ronan, J.E.; Lewis, S.; Knox, A.J. Reduced expression of cyclooxygenase (COX) in idiopathic pulmonary fibrosis and sarcoidosis. Histopathology 2003, 43, 381–386. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [Green Version]
- Waltregny, D.; De Leval, L.; Glenisson, W.; Ly Tran, S.; North, B.J.; Bellahcene, A.; Weidle, U.; Verdin, E.; Castronovo, V. Expression of histone deacetylase 8, a class I histone deacetylase, is restricted to cells showing smooth muscle differentiation in normal human tissues. Am. J. Pathol. 2004, 165, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Waltregny, D.; Glenisson, W.; Tran, S.L.; North, B.J.; Verdin, E.; Colige, A.; Castronovo, V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005, 19, 966–968. [Google Scholar] [CrossRef]
- Borok, Z.; Horie, M.; Flodby, P.; Wang, H.; Liu, Y.; Ganesh, S.; Firth, A.L.; Minoo, P.; Li, C.; Beers, M.F.; et al. Grp78 Loss in Epithelial Progenitors Reveals an Age-linked Role for Endoplasmic Reticulum Stress in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 198–211. [Google Scholar] [CrossRef]
- Jing, X.; Sun, W.; Yang, X.; Huang, H.; Wang, P.; Luo, Q.; Xia, S.; Fang, C.; Zhang, Q.; Guo, J.; et al. CCAAT/enhancer-binding protein (C/EBP) homologous protein promotes alveolar epithelial cell senescence via the nuclear factor-kappa B pathway in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2021, 143, 106142. [Google Scholar] [CrossRef]
- Yao, C.; Carraro, G.; Konda, B.; Guan, X.; Mizuno, T.; Chiba, N.; Kostelny, M.; Kurkciyan, A.; David, G.; McQualter, J.L.; et al. Sin3a regulates epithelial progenitor cell fate during lung development. Development 2017, 144, 2618–2628. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tian, Y.; Morley, M.P.; Lu, M.M.; Demayo, F.J.; Olson, E.N.; Morrisey, E.E. Development and regeneration of Sox2+ endoderm progenitors are regulated by a Hdac1/2-Bmp4/Rb1 regulatory pathway. Dev. Cell 2013, 24, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Frank, D.B.; Morley, M.P.; Zhou, S.; Wang, X.; Lu, M.M.; Lazar, M.A.; Morrisey, E.E. HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17-92 and TGF-beta Signaling Regulation. Dev. Cell 2016, 36, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Saito, S.; Sanchez, C.G.; Zhuang, Y.; Gongora Rosero, R.E.; Shan, B.; Luo, F.; Lasky, J.A. TGF-beta1 stimulates HDAC4 nucleus-to-cytoplasm translocation and NADPH oxidase 4-derived reactive oxygen species in normal human lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L936–L944. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Davis, F.J.; Gupta, M.; Camoretti-Mercado, B.; Schwartz, R.J.; Gupta, M.P. Calcium/calmodulin-dependent protein kinase activates serum response factor transcription activity by its dissociation from histone deacetylase, HDAC4. Implications in cardiac muscle gene regulation during hypertrophy. J. Biol. Chem. 2003, 278, 20047–20058. [Google Scholar] [CrossRef] [Green Version]
- Backs, J.; Song, K.; Bezprozvannaya, S.; Chang, S.; Olson, E.N. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Investig. 2006, 116, 1853–1864. [Google Scholar] [CrossRef]
- Kang, D.H.; Yin, G.N.; Choi, M.J.; Song, K.M.; Ghatak, K.; Minh, N.N.; Kwon, M.H.; Seong, D.H.; Ryu, J.K.; Suh, J.K. Silencing Histone Deacetylase 7 Alleviates Transforming Growth Factor-beta1-Induced Profibrotic Responses in Fibroblasts Derived from Peyronie’s Plaque. World J. Mens Health 2018, 36, 139–146. [Google Scholar] [CrossRef]
- Hemmatazad, H.; Rodrigues, H.M.; Maurer, B.; Brentano, F.; Pileckyte, M.; Distler, J.H.; Gay, R.E.; Michel, B.A.; Gay, S.; Huber, L.C.; et al. Histone deacetylase 7, a potential target for the antifibrotic treatment of systemic sclerosis. Arthritis Rheum. 2009, 60, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.S.; Wen, H.C.; Weng, C.M.; Lee, H.S.; Chen, B.C.; Lin, C.H. Histone deacetylase 7 mediates endothelin-1-induced connective tissue growth factor expression in human lung fibroblasts through p300 and activator protein-1 activation. J. Biomed. Sci. 2021, 28, 38. [Google Scholar] [CrossRef]
- Sarvari, P.; Chelladurai, P.; Korfei, M.; Olson, E.; Gunther, A.; Seeger, W.; Pullamsetti, S.S. The role of histone deacetylase 9 in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, A2396. [Google Scholar]
- Mannaerts, I.; Eysackers, N.; Onyema, O.O.; Van Beneden, K.; Valente, S.; Mai, A.; Odenthal, M.; van Grunsven, L.A. Class II HDAC inhibition hampers hepatic stellate cell activation by induction of microRNA-29. PLoS ONE 2013, 8, e55786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Liu, Y.; Zhu, B.; Ding, K.; Yao, T.P.; Chen, F.; Zhan, L.; Xu, P.; Ehrlich, M.; Liang, T.; et al. Loss of alpha-Tubulin Acetylation Is Associated with TGF-beta-induced Epithelial-Mesenchymal Transition. J. Biol. Chem. 2016, 291, 5396–5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5(+) basal cells. Nat. Cell Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef]
- Korfei, M.; Schmitt, S.; Ruppert, C.; Henneke, I.; Markart, P.; Loeh, B.; Mahavadi, P.; Wygrecka, M.; Klepetko, W.; Fink, L.; et al. Comparative proteomic analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF) and lung transplant donor lungs. J. Proteome Res. 2011, 10, 2185–2205. [Google Scholar] [CrossRef]
- Ganai, S.A. Panobinostat: The Small Molecule Metalloenzyme Inhibitor with Marvelous Anticancer Activity. Curr. Top. Med. Chem. 2016, 16, 427–434. [Google Scholar] [CrossRef]
- Frew, A.J.; Johnstone, R.W.; Bolden, J.E. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009, 280, 125–133. [Google Scholar] [CrossRef]
- Larsson, P.; Alwis, I.; Niego, B.; Sashindranath, M.; Fogelstrand, P.; Wu, M.C.; Glise, L.; Magnusson, M.; Daglas, M.; Bergh, N.; et al. Valproic acid selectively increases vascular endothelial tissue-type plasminogen activator production and reduces thrombus formation in the mouse. J. Thromb. Haemost. 2016, 14, 2496–2508. [Google Scholar] [CrossRef] [Green Version]
- Saluveer, O.; Larsson, P.; Ridderstrale, W.; Hrafnkelsdottir, T.J.; Jern, S.; Bergh, N. Profibrinolytic effect of the epigenetic modifier valproic acid in man. PLoS ONE 2014, 9, e107582. [Google Scholar] [CrossRef] [Green Version]
- Larsson, P.; Ulfhammer, E.; Magnusson, M.; Bergh, N.; Lunke, S.; El-Osta, A.; Medcalf, R.L.; Svensson, P.A.; Karlsson, L.; Jern, S. Role of histone acetylation in the stimulatory effect of valproic acid on vascular endothelial tissue-type plasminogen activator expression. PLoS ONE 2012, 7, e31573. [Google Scholar] [CrossRef] [Green Version]
- Svennerholm, K.; Haney, M.; Biber, B.; Ulfhammer, E.; Saluveer, O.; Larsson, P.; Omerovic, E.; Jern, S.; Bergh, N. Histone deacetylase inhibition enhances tissue plasminogen activator release capacity in atherosclerotic man. PLoS ONE 2015, 10, e0121196. [Google Scholar]
- Jiang, P.; Gil de Rubio, R.; Hrycaj, S.M.; Gurczynski, S.J.; Riemondy, K.A.; Moore, B.B.; Omary, M.B.; Ridge, K.M.; Zemans, R.L. Ineffectual Type 2-to-Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8(hi) Transitional State. Am. J. Respir. Crit. Care Med. 2020, 201, 1443–1447. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Pang, M.; Kothapally, J.; Mao, H.; Tolbert, E.; Ponnusamy, M.; Chin, Y.E.; Zhuang, S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2009, 297, F996–F1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Patel, P.; Jageshwar; Patel, V.K.; Jain, D.K.; Kamal, M.; Rajak, H. The Safety, Efficacy and Therapeutic Potential of Histone Deacetylase Inhibitors with Special Reference to Panobinostat in Gastrointestinal Tumors: A Review of Preclinical and Clinical Studies. Curr. Cancer Drug Targets 2018, 18, 720–736. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Borkiewicz, L.; Okon, E.; Kukula-Koch, W.; Afshan, S.; Halasa, M. Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers 2021, 13, 4700. [Google Scholar] [CrossRef]
- Claveria-Cabello, A.; Colyn, L.; Arechederra, M.; Urman, J.M.; Berasain, C.; Avila, M.A.; Fernandez-Barrena, M.G. Epigenetics in Liver Fibrosis: Could HDACs be a Therapeutic Target? Cells 2020, 9, 2321. [Google Scholar] [CrossRef] [PubMed]
- Bajbouj, K.; Al-Ali, A.; Ramakrishnan, R.K.; Saber-Ayad, M.; Hamid, Q. Histone Modification in NSCLC: Molecular Mechanisms and Therapeutic Targets. Int. J. Mol. Sci. 2021, 22, 11701. [Google Scholar] [CrossRef]
- Hervouet, E. The Promising Role of New Generation HDACis in Anti-Cancer Therapies. EBioMedicine 2018, 32, 6–7. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef] [Green Version]
- Khabele, D. The therapeutic potential of class I selective histone deacetylase inhibitors in ovarian cancer. Front. Oncol. 2014, 4, 111. [Google Scholar] [CrossRef] [Green Version]
- King, T.E., Jr.; Schwarz, M.I.; Brown, K.; Tooze, J.A.; Colby, T.V.; Waldron, J.A., Jr.; Flint, A.; Thurlbeck, W.; Cherniack, R.M. Idiopathic pulmonary fibrosis: Relationship between histopathologic features and mortality. Am. J. Respir. Crit. Care Med. 2001, 164, 1025–1032. [Google Scholar] [CrossRef]
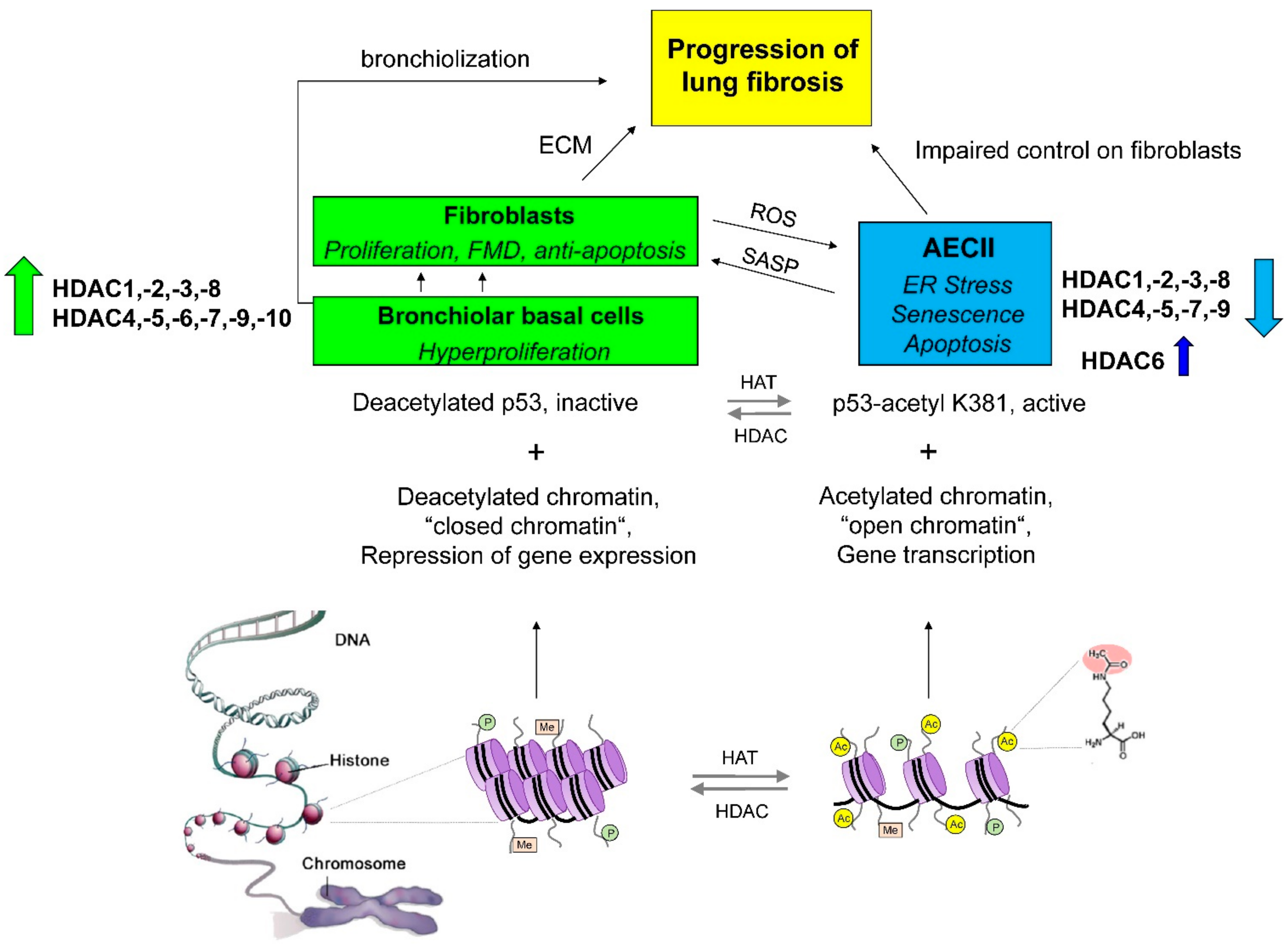
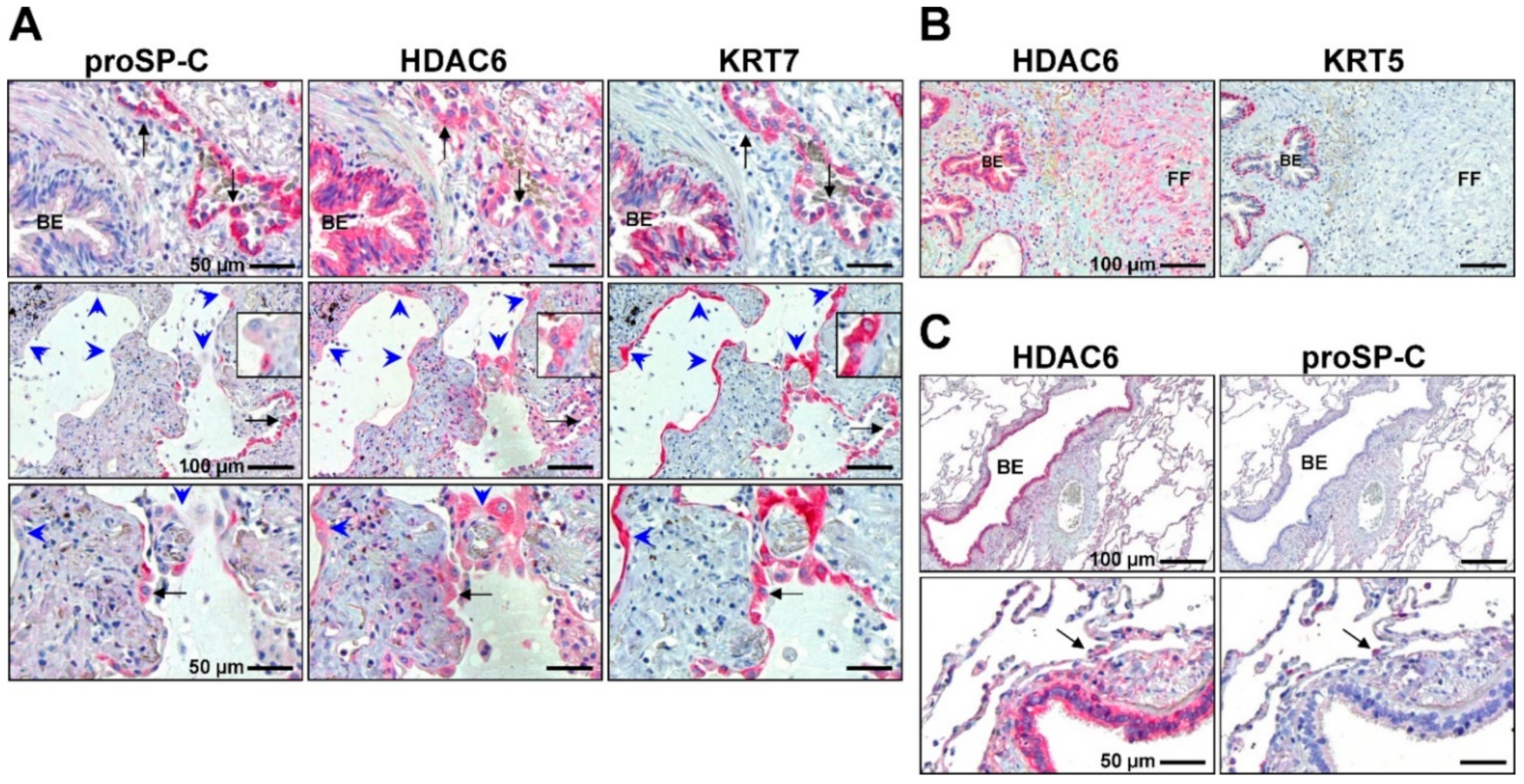
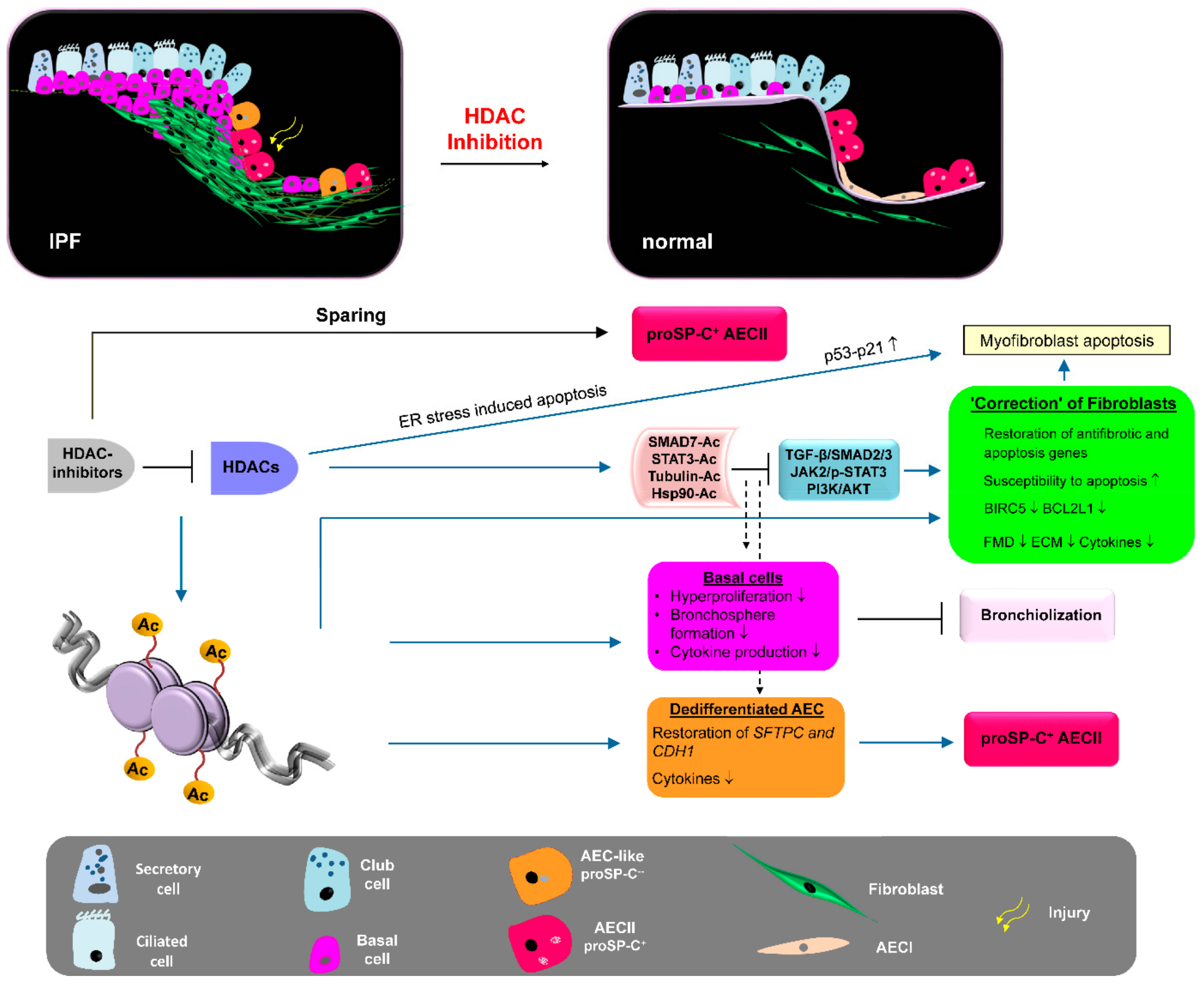

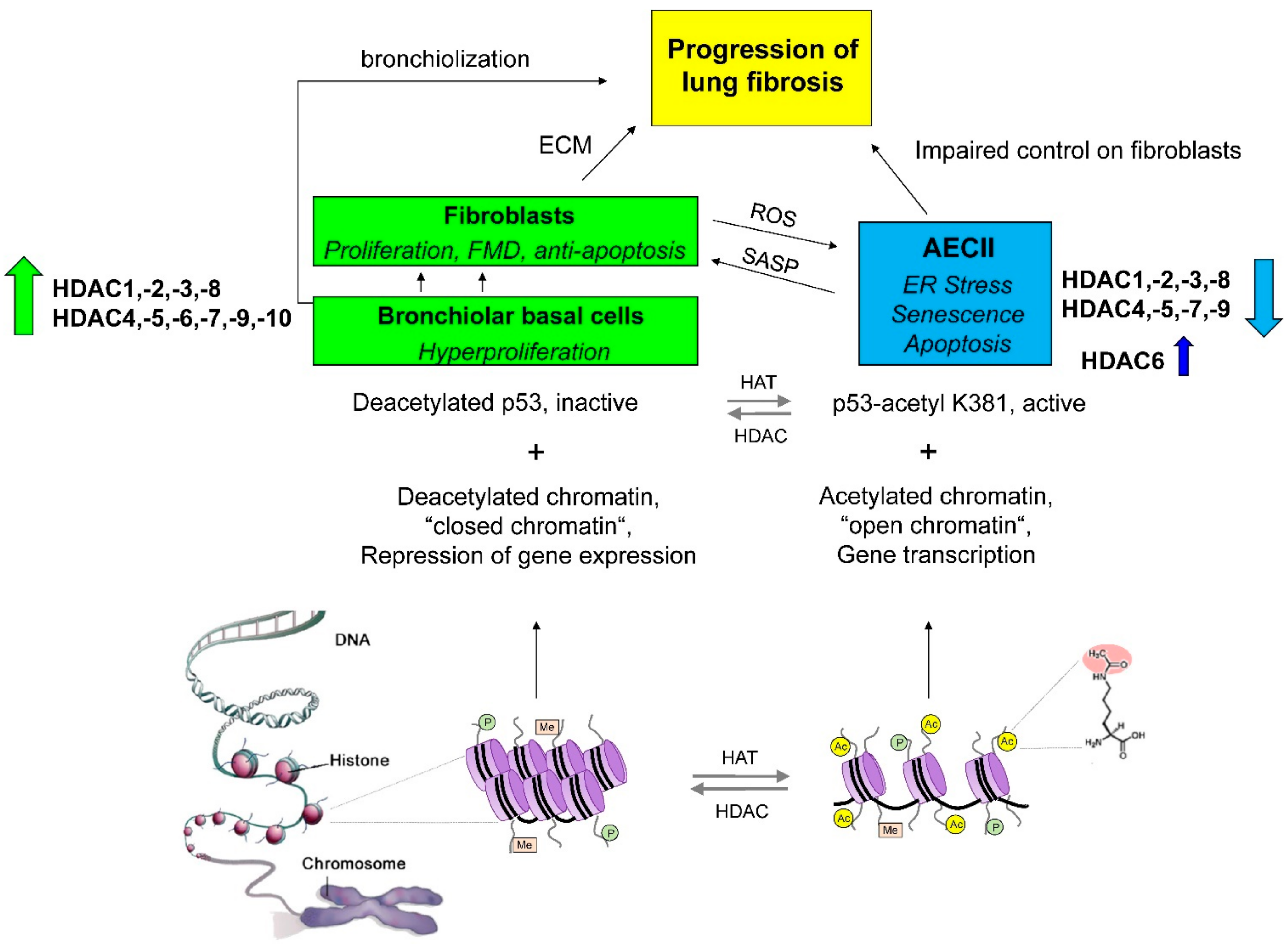{kind=link}
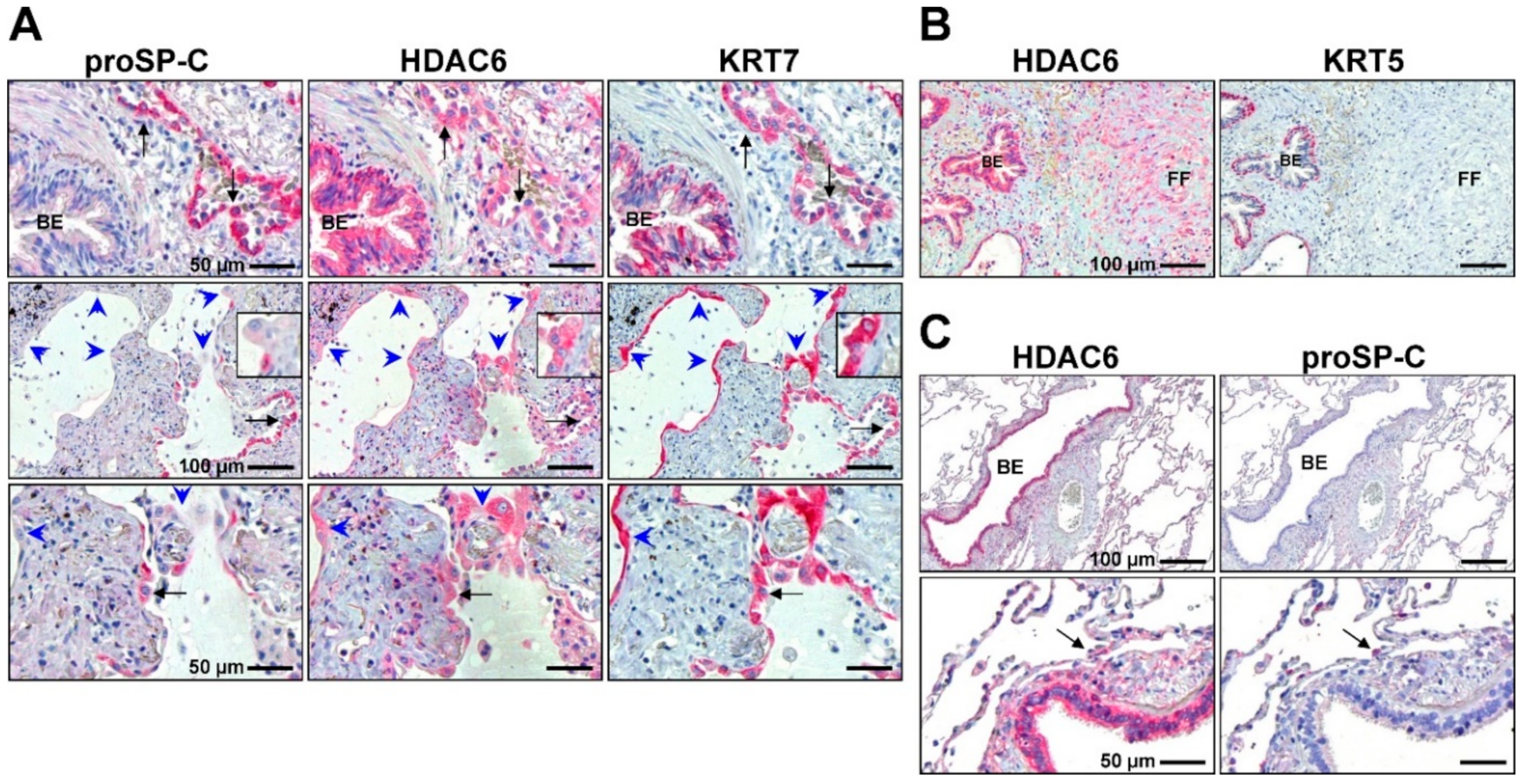{kind=link}
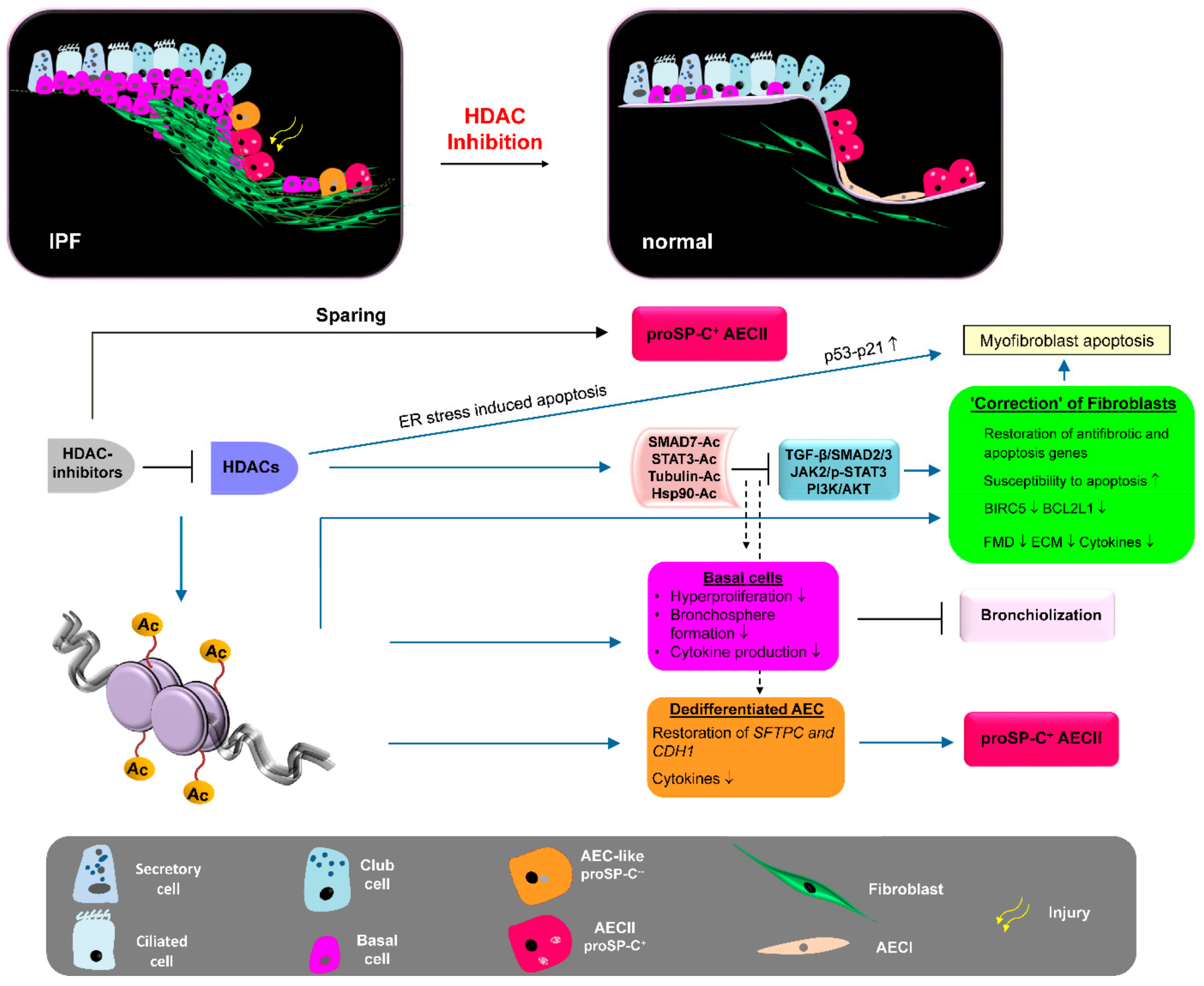{kind=link}
| Study | Lung Fibrosis Model | HDAC Inhibiton | Effect/Involved Molecules |
|---|---|---|---|
| Coward et al. (2009) [162] | TGF-β-treated IPF fibroblasts | Panobinostat (LBH589) pan-HDAC | H3 and H4 acetylation at COX2 promoter, derepression of COX2 expression |
| Huang et al. (2013) [163] | Lung fibroblasts of bleomycin mice, Primary IPF fibroblasts | Trichostatin A (TSA), vorinostat (SAHA) pan-HDAC | H3 acetylation at the Fas/FAS promoter, derepression of Fas/FAS expression |
| Sanders et al. (2014) [164] | Primary IPF fibroblasts, Bleomycin mouse model | Vorinostat (SAHA) pan-HDAC | In vitro: proliferation H3 and H4 acetylation, H3K9Ac ↑ BAK ↑ BID ↑ BCL2L1 ↓ In vivo: ameliorated lung fibrosis H3K9Ac ↑, Bak ↑ Bcl2l1 ↓ |
| Korfei et al. (2015) [165] | Primary IPF fibroblasts | Panobinostat (LBH589) pan-HDAC | Tubulin acetylation ↑, H3K27 acetylation, CIP1/p21 ↑, CHOP ↑, proliferation (CCND1) ↓, FMD (ACTA2, COL1A1, COL1A3, FN) ↓, surviving ↓ BCL-XL ↓ |
| Zhang et al. (2013) [167] | Primary IPF fibroblasts, Bleomycin mouse model | Vorinostat (SAHA) pan-HDAC | In vitro: H3 and H4 acetylation, COL3A1 (mRNA and protein) ↓ In vivo: ameliorated lung fibrosis, collagen-III ↓ |
| Ota et al. (2015) [168] | TGF-β-stimulated A549 cells, Bleomycin mouse model | Trichostatin A (TSA) | In vitro: EMT ↓ restoration of CDH1 expression. In vivo: Partial attenuation of fibrosis, restoration of AECII-Sftpc expression |
| Kim et al. (2019) [233] | Bleomycin mouse model, PHMG induced lung fibrosis | CG-745 Class I-HDAC + HDAC6 | Abrogation of bleomycin-fibrosis, H3 acetylation, Pai-1 ↓ α-Sma ↓ collagen-I ↓ BALF: Tnf-α ↓ Il-6 ↓ Attenuation of PHMG-fibrosis |
| Jones et al. (2019) [246] | TGF-β-treated primary IPF fibroblasts | Pracrinostat pan-HDAC, except HDAC6 | H3 acetylation at PGC1A promoter, derepression of PGC1A expression, HDAC7 signalling ↓, ACTA2 ↓ TNC ↓ IL6 ↓ PDGFA ↓ Inhibition of FMD |
| Coward et al. (2010) [247] | TGF-β-treated primary IPF fibroblasts | Panobinostat (LBH589) pan-HDAC | H3 and H4 acetylation at CXCL10 promoter, reduction of repressive H3K9Me3 at CXCL10 promoter, derepression of CXCL10 expression |
| Sanders et al. (2011) [248] | Fibrotic rat Thy1 (-) lung fibroblasts | Trichostatin A (TSA) | H3 and H4 acetylation, derepression of Thy1 (CD90) expression |
| Korfei et al. (2018) [249] | Primary IPF fibroblasts | Panobinostat (LBH589) pan-HDAC | Tubulin acetylation ↑, H3K27Ac ↑ STAT3-pTyr705 ↓, proliferation ↓, FMD ↓ ECM (pro-collagen-I) ↓ HDAC1 ↓ HDAC2 ↓ HDAC7 (mRNA and protein) ↓ |
| Guo et al. (2009) [250] | TGF-β-treated human normal lung fibroblasts | Trichostatin A (TSA) pan-HDAC | HDAC4 signalling ↓ ACTA2 ↓ COL1A1 ↓ CTGF ↑ (!) Inhibition of FMD, α-SMA ↓ AKT phosphorylation ↓ |
| Ye et al. (2014) [251] | Bleomycin rat model | Trichostatin A (TSA) pan-HDAC | Reduction of lung fibrosis, HDAC2 (mRNA and protein) ↓ |
| Rao et al. (2016) [252] | TGF-β-treated normal human lung fibroblasts (HFL1), paraquat-induced lung fibrosis in rats | Vorinostat (SAHA) pan-HDAC | In vitro and in vivo: SMAD7 acetylation and stabilization, SMAD3 dephosphorylation, FMD ↓ attenuation of lung fibrosis |
| Glenisson et al. (2007) [253] | TGF-β-treated primary normal skin fibroblasts (human) | Trichostatin A (TSA) pan-HDAC | HDAC4 signalling ↓ ACTA2/α-SMA ↓ Inhibition of FMD |
| Kabel et al. (2016) [254] | Bleomycin rat model | 4-phenyl-butyrate (4-PBA) Class I- and Class IIA-HDAC | attenuation of lung fibrosis, oxidative stress ↓ BALF: IL6 ↓ TGF-β ↓ TNF-α ↓ |
| Jiang et al. (2018) [255] | A549 cells overexpressing mutant SP-AG231V or SP-AF198S | (4-PBA) Class I- and Class IIA-HDAC | GRP78 ↑ suppressed protein aggregation, improved secretion |
| Zhao et al. (2015) [256] | Bleomycin mouse model | (4-PBA) Class I- and Class IIA-HDAC | ER stress ↓ EMT ↓ NK-κB (p65) ↓ cytokines ↓ α-SMA ↓ Col1a1 ↓ Col1a2 ↓ alleviation of lung fibrosis |
| Study | Lung Fibrosis Model | HDAC Inhibition | Effect/Involved Molecules |
|---|---|---|---|
| Korfei et al. (2015) [165] | Primary IPF fibroblasts | Valproic acid (VPA) HDAC1, -2 | H3K27 acetylation, CIP1 ↑, cell proliferation ↓ BIRC5/surviving ↓ collagen-I ↓ HDAC2 ↓ HDAC7 ↓ |
| Conforti et al. (2017) [169] | TGF-β-treated primary IPF fibroblasts, Bleomycin mouse model | Romidepsin (FK228) HDAC1, -2 | In vitro: H3 acetylation, CIP1 ↑ ACTA2 ↓ COL3A1 ↓ LOX ↓ HDAC4 ↓ inhibition of FMD. In vivo: inhibited lung fibrosis, ECM genes ↓ Lox protein ↓ |
| Chen et al. (2021) [170] | TGF-β-treated A549 cells, Bleomycin mouse model | Valproic acid HDAC1, -2 | In vitro and in vivo: SMAD2/3 deactivation, inhibition of EMT |
| Barter et al. (2010) [265] | TGF-β-treated embryonic mouse fibroblasts | Entinostat (MS-275) HDAC1, -3; HDAC3 siRNA | Inhibition of PI3K and ERK pathways Adam12 ↓ Timp1 ↓ |
| Davies et al. (2012) [275] | TGF-β-treated primary IPF fibroblasts | Spiruchostatin A HDAC1, -2 | H3 acetylation, CIP1 ↑ cell proliferation ↓ inhibition of FMD |
| Noguchi et al. (2015) [278] | TGF-β-treated A549 cells | Valproic acid HDAC1, -2 | H3K27 acetylation, partial inhibition of EMT |
| Kabel et al. (2016) [254] | Bleomycin rat model | Valproic acid HDAC1, -2 | attenuation of lung fibrosis, oxidative stress ↓ BALF: IL6 ↓ TGF-β ↓ TNF-α ↓ |
| Kamio et al. (2017) [279] | TGF-β-treated normal human lung fibroblasts | Entinostat (MS-275): HDAC1, -3 | Restoration of ARHGEF3/XPLN (negative regulator of SPARC) ↑ SPARC ↓ |
| Saito et al. (2019) [171] | TGF-β-treated normal human lung fibroblasts, Bleomycin mouse model | NCC170 HDAC8 | In vitro: H3K27 acetylation, restoration of PPARG expression, ECM protein production ↓ In vivo: inhibited lung fibrosis |
| Chen et al. (2021) [258] | Bleomycin mouse model | RGFP966 HDAC3 | H3 acetylation at the Nrf2 promoter, restoration of Nrf2 expression, E-cadherin expression ↑ ECM proteins ↓ |
| Yuan et al. (2020) [273] | RA-ILD mouse model | HDAC3 siRNA | miR-19a-3p ↑ IL17RA ↓ IL17/IL17RA signalling ↓ Inflammation ↓ attenuation of lung fibrosis |
| Study | Model | HDAC Inhibiton | Effect/Involved Molecules |
|---|---|---|---|
| Jones et al. (2019) [246] | TGF-β-treated primary IPF fibroblasts | HDAC7 siRNA | ACTA2 ↓ CTGF ↓ NOX4 ↓ HDAC2 ↓ HDAC6 ↓ HDAC8 ↓ HDAC10 ↓ HDAC9 ↑ Derepression of PGC1A expression |
| Guo et al. (2009) [250] | TGF-β-treated human normal lung fibroblasts | HDAC4 siRNA | ACTA2 ↓ Inhibition of FMD, AKT phosphorylation ↓ |
| Glenisson et al. (2007) [253] | TGF-β-treated primary normal skin fibroblasts (human) | HDAC4 siRNA | ACTA2/α-SMA ↓ FMD ↓ Upregulation of TGIF1 and TGIF2 (= Inhibitors of TGF-β signalling) |
| Kang et al. (2018) [294] | TGF-β-treated fibroblasts isolated from PD plaque | HDAC7 siRNA | Inhibition of SMAD2/3 activation, FMD ↓ ECM protein production ↓ |
| Hua et al. (2021) [296] | Endothelin-treated human normal lung fibroblasts (WI-38) | HDAC7 siRNA | Deacetylation of AP-1, AP-1 activity ↓ α-SMA ↓ CTGF ↓ |
| Study | Lung Fibrosis Model | HDAC Inhibition | Effect/Involved Molecules |
|---|---|---|---|
| Campiani et al. (2021) [174] | Organoid cultures derived from IPF basal cells, Ex vivo human lung model of fibrosis | “Compound 6h” HDAC6 | Basal cell proliferation ↓ Bronchosphere formation ↓ Tubulin acetylation ↑ TGF-β dependent ECM synthesis ↓ |
| Shan et al. (2008) [190] | TGF-β-stimulated A549 cells | Tubacin HDAC6 HDAC6 siRNA | Tubulin-hyperacetylation, restoration of E-cadherin, SMAD3 phosphorylation ↓ PAI1 ↓ COL1A1 ↓ inhibition of EMT |
| Deskin et al. (2016) [191] | TGF-β-stimulated A549 cells | Tubacin HDAC6 HDAC6 siRNA | Abrogation of TGF-β induced Notch1 signalling (HEY1, HES1 ↓) Acetylation of HSP90 (Ac-K294) p38 pathway ↓ |
| Saito et al. (2017) [235] | TGF-β-stimulated human normal lung fibroblasts, Bleomycin mouse model | Tubastatin A HDAC6 | In vitro and in vivo: Tubulin hyperacetylation, inhibition of PI3K-AKT pathway, FMD ↓ ECM ↓ amelioration of lung fibrosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korfei, M.; Mahavadi, P.; Guenther, A. Targeting Histone Deacetylases in Idiopathic Pulmonary Fibrosis: A Future Therapeutic Option. Cells 2022, 11, 1626. https://doi.org/10.3390/cells11101626
Korfei M, Mahavadi P, Guenther A. Targeting Histone Deacetylases in Idiopathic Pulmonary Fibrosis: A Future Therapeutic Option. Cells. 2022; 11(10):1626. https://doi.org/10.3390/cells11101626
Chicago/Turabian StyleKorfei, Martina, Poornima Mahavadi, and Andreas Guenther. 2022. "Targeting Histone Deacetylases in Idiopathic Pulmonary Fibrosis: A Future Therapeutic Option" Cells 11, no. 10: 1626. https://doi.org/10.3390/cells11101626
APA StyleKorfei, M., Mahavadi, P., & Guenther, A. (2022). Targeting Histone Deacetylases in Idiopathic Pulmonary Fibrosis: A Future Therapeutic Option. Cells, 11(10), 1626. https://doi.org/10.3390/cells11101626