

Integrated Machine Learning Algorithms-Enhanced Predication for Cervical Cancer from Mass Spectrometry-Based Proteomics Data
 , ,
, ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Serum Proteomics Workflow Summary
2.1.1. MALDI-TOF-Based Proteomic Profiling and Mass Spectrometry Data Preprocessing for Serum Samples
Patients and Samples
Serum Sample Processing and MALDI-TOF MS Analysis
Raw Data Preprocessing
Peak Feature Extraction, Normalization, and Integrative Data Matrix Construction
2.1.2. Machine Learning-Driven Proteomic Data Analysis: From Dimensionality Reduction to Biomarker Candidate Prioritization
Dimension Reduction Analysis
Machine Learning
Feature Selection
2.2. Annotated Biomarker-Driven Machine Learning and Clinical Validation for Cervical Cancer Diagnostics
2.2.1. Integrated Proteomic Data Processing: High-Resolution Peptide Identification via Orbitrap MS and Unsupervised Clustering for Pattern Discovery
Peptide Identification
Unsupervised Clustering
2.2.2. Comprehensive Biomarker Evaluation: ROC-Driven Threshold Optimization and Decision Curve Analysis for Clinical Utility Validation
Roc Evaluation of Unique Indicator
Decision Curve Analysis
2.3. Bioinformatics Analysis and Clinical Validation of Public Data
2.3.1. Bioinformatics Analysis
2.3.2. Clinical Validation of Public Data
3. Results
3.1. General Information
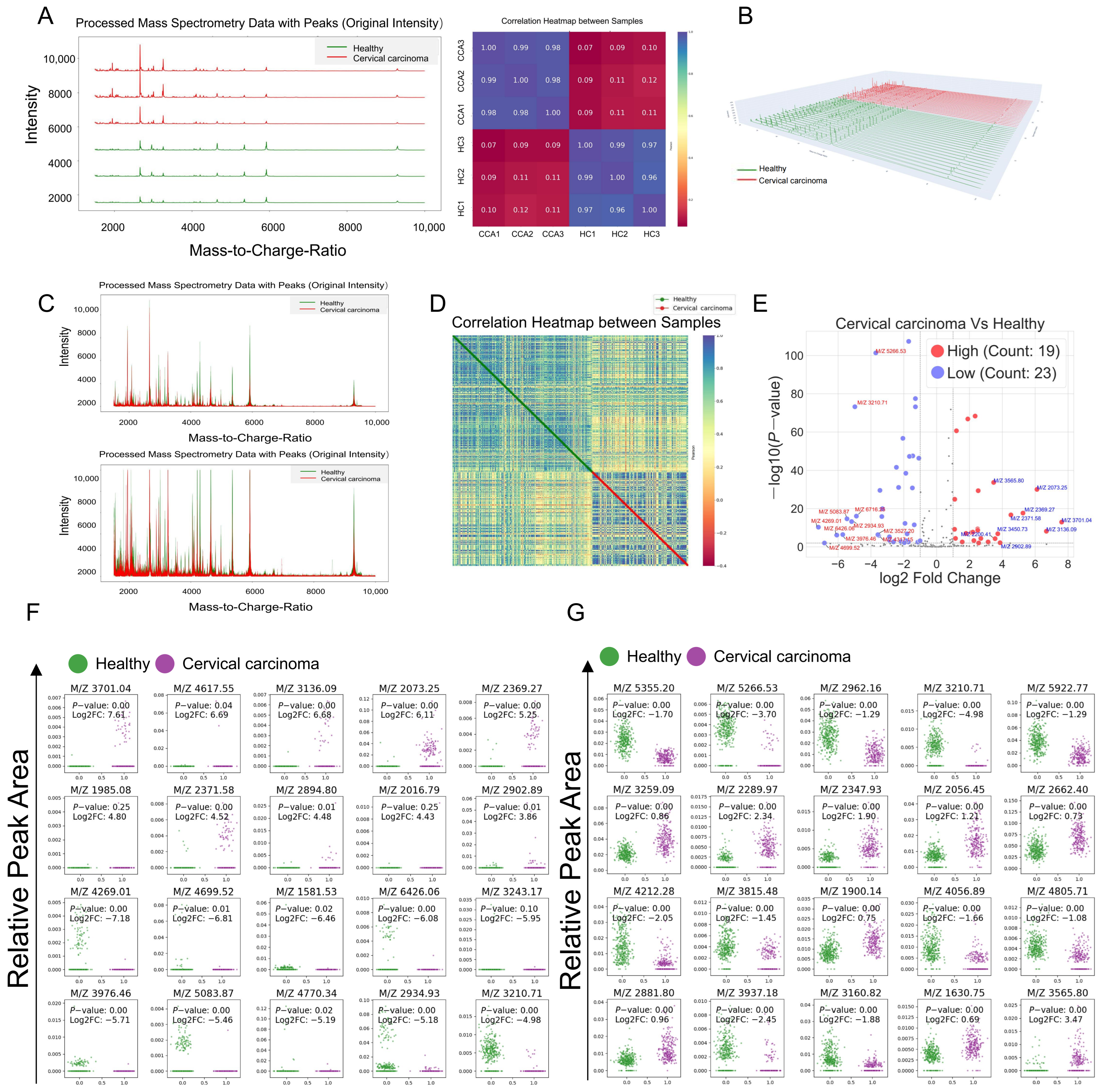
3.2. Mass Spectrometry Data Processing and Differential Polypeptide Screening
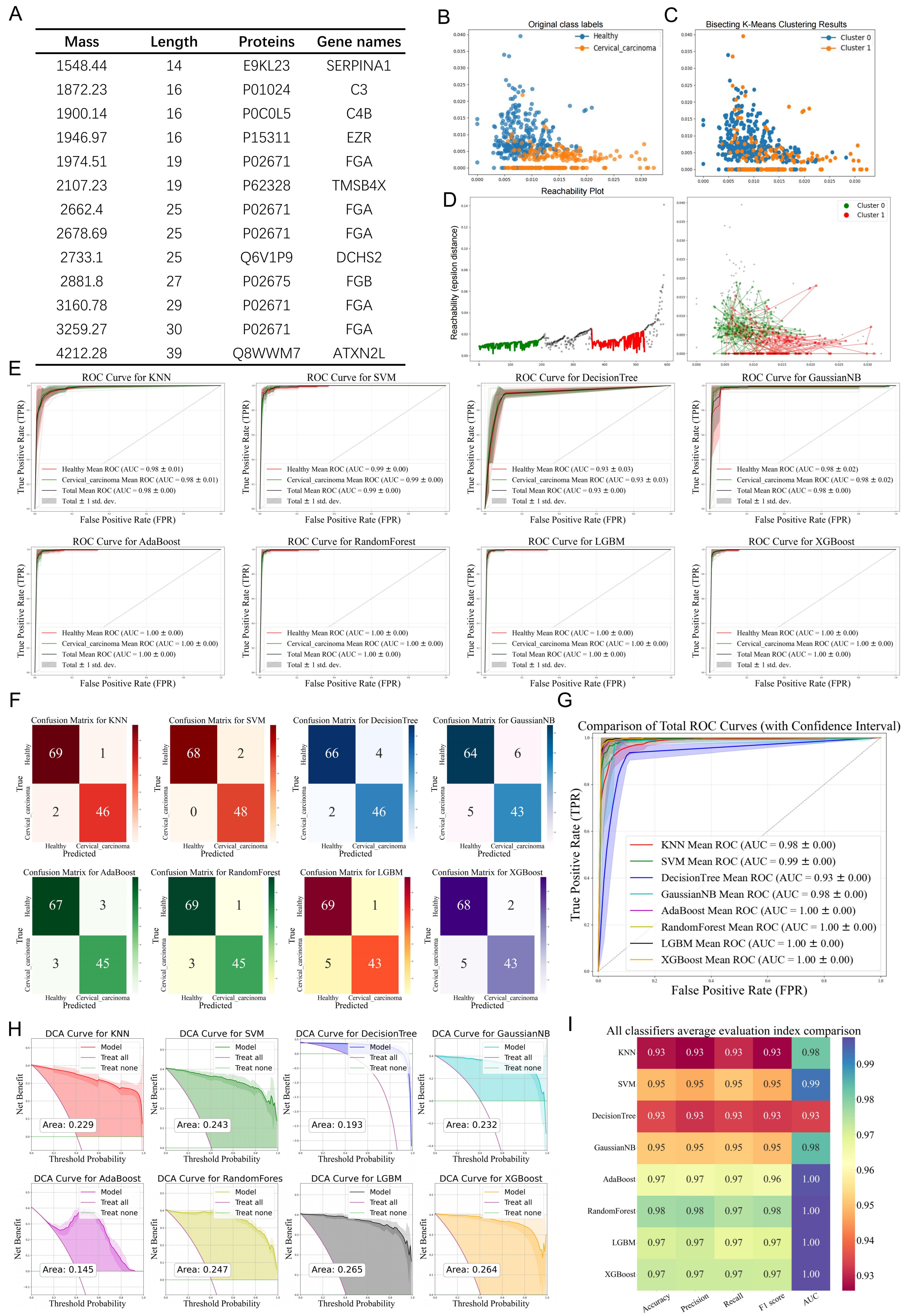
3.3. Evaluation of Diagnostic Efficiency Based on Different Machine Learning Algorithms
3.4. Feature Selection of Different Machine Learning Algorithms
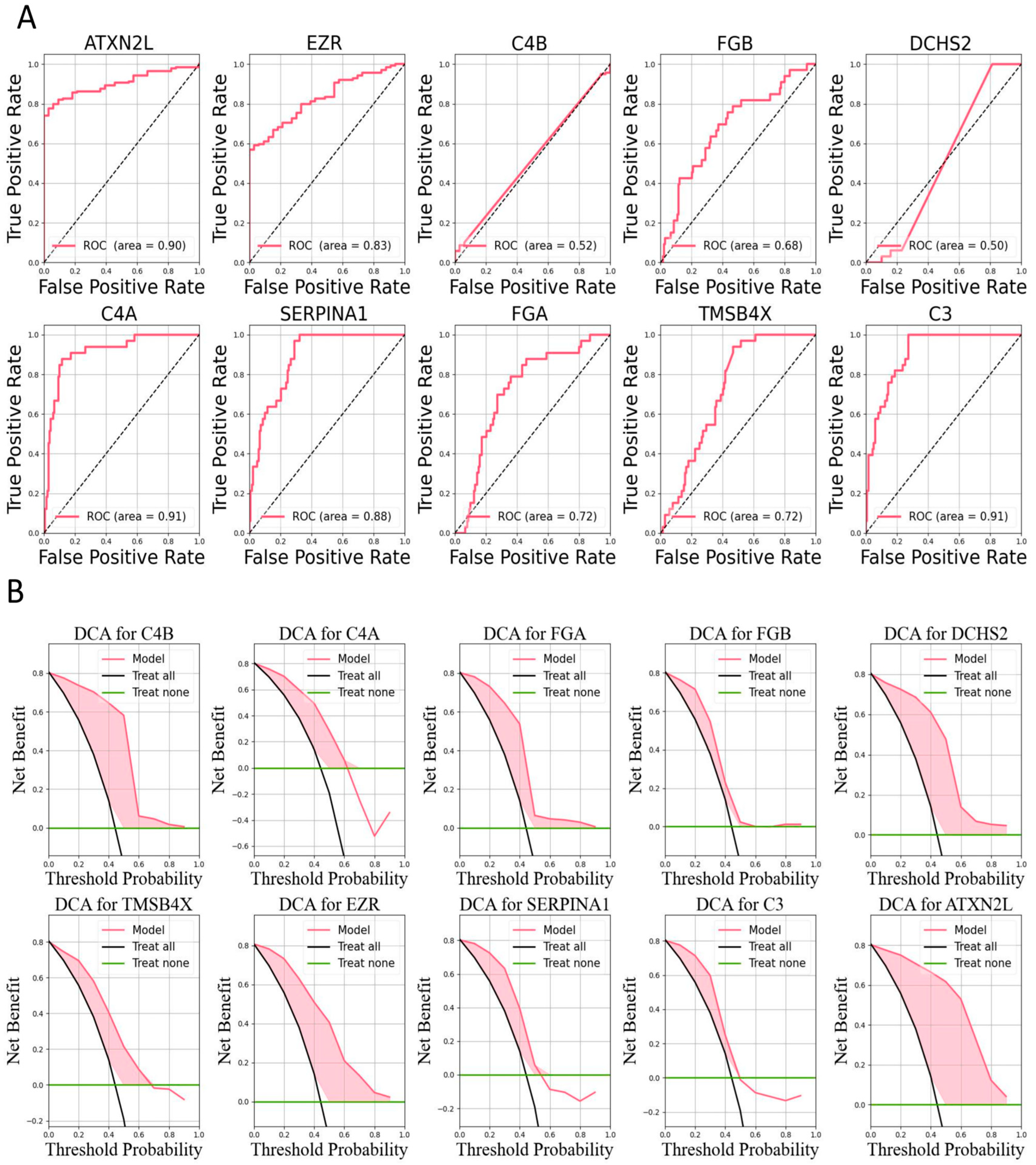
3.5. Analysis of Clinical Indicators of Single Differential Polypeptide
3.6. Identification of Differential Polypeptides and Evaluation of Their Diagnostic Efficacy
3.7. Bioinformatics Analysis of Differentially Expressed Peptides
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.Y.A.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Han, B.F.; Zheng, R.S.; Zeng, H.M.; Wang, S.M.; Sun, K.X.; Chen, R.; Li, L.; Wei, W.Q.; He, J. Cancer incidence and mortality in China, 2022. J. Natl. Cancer Ctr. 2024, 4, 47–53. [Google Scholar] [CrossRef]
- Cubie, H.A.; Campbell, C. Cervical cancer screening-The challenges of complete pathways of care in low-income countries: Focus on Malawi. Womens Health 2020, 16, 1745506520914804. [Google Scholar] [CrossRef]
- Xia, C.F.; Basu, P.; Kramer, B.S.; Li, H.; Qu, C.F.; Yu, X.Q.; Canfell, K.; Qiao, Y.L.; Armstrong, B.K.; Chen, W.Q. Cancer screening in China: A steep road from evidence to implementation. Lancet Public Health 2023, 8, E996–E1005. [Google Scholar] [CrossRef]
- Perkins, R.B.; Wentzensen, N.; Guido, R.S.; Schiffman, M. Cervical Cancer Screening A Review. Jama J. Am. Med. Assoc. 2023, 330, 547–558. [Google Scholar] [CrossRef]
- Choi, S.; Ismail, A.; Pappas-Gogos, G.; Boussios, S. HPV and Cervical Cancer: A Review of Epidemiology and Screening Uptake in the UK. Pathogens 2023, 12, 298. [Google Scholar] [CrossRef]
- Khan, M.J.; Werner, C.L.; Darragh, T.M.; Guido, R.S.; Mathews, C.; Moscicki, A.B.; Mitchell, M.M.; Schiffman, M.; Wentzensen, N.; Massad, L.S.; et al. ASCCP Colposcopy Standards: Role of Colposcopy, Benefits, Potential Harms, and Terminology for Colposcopic Practice. J. Low. Genit. Tract. Di 2017, 21, 223–229. [Google Scholar] [CrossRef]
- Saslow, D.; Solomon, D.; Lawson, H.W.; Killackey, M.; Kulasingam, S.L.; Cain, J.; Garcia, F.A.R.; Moriarty, A.T.; Waxman, A.G.; Wilbur, D.C.; et al. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer J. Clin. 2012, 62, 147–172. [Google Scholar] [CrossRef]
- Bourgioti, C.; Chatoupis, K.; Rodolakis, A.; Antoniou, A.; Tzavara, C.; Koutoulidis, V.; Moulopoulos, L.A. Incremental prognostic value of MRI in the staging of early cervical cancer: A prospective study and review of the literature. Clin. Imag. 2016, 40, 72–78. [Google Scholar] [CrossRef]
- Chernov, D.Y.; Tikhonovskaya, O.A.; Logvinov, S.; Petrov, I.A.; Yuriev, S.Y.; Zhdankina, A.A.; Gerasimov, A.; Zingalyuk, I.; Mikheenko, G.A. Challenges in the diagnosis of cervical pathologies. Byulleten Sib. Med. 2023, 22, 201–209. [Google Scholar] [CrossRef]
- Ogilvie, G.; Nakisige, C.; Huh, W.K.; Mehrotra, R.; Franco, E.L.; Jeronimo, J. Optimizing secondary prevention of cervical cancer: Recent advances and future challenges. Int. J. Gynecol. Obstet. 2017, 138, 15–19. [Google Scholar] [CrossRef]
- Chen, E.I.; Yates, J.R. Cancer proteomics by quantitative shotgun proteomics. Mol. Oncol. 2007, 1, 144–159. [Google Scholar] [CrossRef]
- Jafari, A.; Babajani, A.; Rezaei-Tavirani, M. Multiple Sclerosis Biomarker Discoveries by Proteomics and Metabolomics Approaches. Biomark. Insights 2021, 16, 1–14. [Google Scholar] [CrossRef]
- Das, V.; Kalita, J.; Pal, M. Predictive and prognostic biomarkers in colorectal cancer: A systematic review of recent advances and challenges. Biomed. Pharmacother. 2017, 87, 8–19. [Google Scholar] [CrossRef]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. CA125 and Ovarian Cancer: A Comprehensive Review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef]
- Van Poppel, H.; Albreht, T.; Basu, P.; Hogenhout, R.; Collen, S.; Roobol, M. Serum PSA-based early detection of prostate cancer in Europe and globally: Past, present and future. Nat. Rev. Urol. 2022, 19, 562–572. [Google Scholar] [CrossRef]
- Goossens, N.; Nakagawa, S.; Sun, X.C.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256–269. [Google Scholar] [CrossRef]
- Wu, L.; Qu, X.G. Cancer biomarker detection: Recent achievements and challenges. Chem. Soc. Rev. 2015, 44, 2963–2997. [Google Scholar] [CrossRef]
- Vargas, A.J.; Harris, C.C. Biomarker development in the precision medicine era: Lung cancer as a case study. Nat. Rev. Cancer 2016, 16, 525–537. [Google Scholar] [CrossRef]
- Borrebaeck, C.A.K. Precision diagnostics: Moving towards protein biomarker signatures of clinical utility in cancer. Nat. Rev. Cancer 2017, 17, 199–204. [Google Scholar] [CrossRef]
- Jafari, A.; Farahani, M.; Abdollahpour-Alitappeh, M.; Manzari-Tavakoli, A.; Yazdani, M.; Rezaei-Tavirani, M. Unveiling diagnostic and therapeutic strategies for cervical cancer: Biomarker discovery through proteomics approaches and exploring the role of cervical cancer stem cells. Front. Oncol. 2024, 13, 1277772. [Google Scholar] [CrossRef]
- Acs, B.; Rantalainen, M.; Hartman, J. Artificial intelligence as the next step towards precision pathology. J. Intern. Med. 2020, 288, 62–81. [Google Scholar] [CrossRef]
- Bera, K.; Schalper, K.A.; Rimm, D.L.; Velcheti, V.; Madabhushi, A. Artificial intelligence in digital pathology-new tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 2019, 16, 703–715. [Google Scholar] [CrossRef]
- Echle, A.; Rindtorff, N.T.; Brinker, T.J.; Luedde, T.; Pearson, A.T.; Kather, J.N. Deep learning in cancer pathology: A new generation of clinical biomarkers. Br. J. Cancer 2021, 124, 686–696. [Google Scholar] [CrossRef]
- Yang, J.L.; Ju, J.; Guo, L.; Ji, B.B.; Shi, S.F.; Yang, Z.X.; Gao, S.L.; Yuan, X.; Tian, G.; Liang, Y.B.; et al. Prediction of HER2-positive breast cancer recurrence and metastasis risk from histopathological images and clinical information via multimodal deep learning. Comput. Struct. Biotec 2022, 20, 333–342. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Hu, D.Y.; Qu, H.; Tian, Y.; Cui, X.Y. Application of Deep Learning in Histopathology Images of Breast Cancer: A Review. Micromachines 2022, 13, 2197. [Google Scholar] [CrossRef]
- Bhattacharya, I.; Khandwala, Y.S.; Vesal, S.; Shao, W.; Yang, Q.Y.; Soerensen, S.J.C.; Fan, R.E.; Ghanouni, P.; Kunder, C.A.; Brooks, J.D.; et al. A review of artificial. intelligence in prostate cancer detection on imaging. Ther. Adv. Urol. 2022, 14, 17562872221128791. [Google Scholar] [CrossRef]
- de Rooij, M.; van Poppel, H.; Barentsz, J.O. Risk Stratification and Artificial Intelligence in Early Magnetic Resonance Imaging-based Detection of Prostate Cancer. Eur. Urol. Focus. 2022, 8, 1187–1191. [Google Scholar] [CrossRef]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python (vol 33, pg 219, 2020). Nat. Methods 2020, 17, 352. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Mckinney, W. Data Structures for Statistical Computing in Python. SciPy 2010, 445, 51–56. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Ke, G.L.; Meng, Q.; Finley, T.; Wang, T.F.; Chen, W.; Ma, W.D.; Ye, Q.W.; Liu, T.Y. LightGBM: A Highly Efficient Gradient Boosting Decision Tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3149–3157. [Google Scholar]
- Chen, T.Q.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the Kdd’16: Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.I. A Unified Approach to Interpreting Model Predictions. Adv. Neural Inf. Process. Syst. 2017, 30, 4768–4777. [Google Scholar]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?” Explaining the Predictions of Any Classifier. In Proceedings of the Kdd’16: Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Yu, G.C. Thirteen years of clusterProfiler. Innov. Amst. 2024, 5, 100722. [Google Scholar] [CrossRef]
- Yu, J.; Gui, X.Q.; Zou, Y.H.; Liu, Q.; Yang, Z.C.; An, J.S.; Guo, X.; Wang, K.H.; Guo, J.M.; Huang, M.N.; et al. A proteogenomic analysis of cervical cancer reveals therapeutic and biological insights. Nat. Commun. 2024, 15, 10114. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, X.; Ip, F.C.; Chan, P.; Chen, Y.; Lai, N.C.H.; Cheung, K.; Lo, R.M.N.; Tong, E.P.S.; Wong, B.W.Y.; et al. Large-scale plasma proteomic profiling identifies a high-performance biomarker panel for Alzheimer’s disease screening and staging. Alzheimers Dement. 2022, 18, 88–102. [Google Scholar] [CrossRef]
- Li, R.; Li, L.; Xu, Y.; Yang, J. Machine learning meets omics: Applications and perspectives. Brief. Bioinform. 2022, 23, bbab460. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.T.; You, J.; He, Y.; Zhang, Y.; Li, H.Y.; Wu, X.R.; Cheng, J.Y.; Guo, Y.; Long, Z.W.; Chen, Y.L.; et al. Atlas of the plasma proteome in health and disease in 53,026 adults. Cell 2025, 188, 253–271.e7. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.; Alli-Shaik, A.; Gunaratne, J. Machine Learning-Enhanced Extraction of Biomarkers for High-Grade Serous Ovarian Cancer from Proteomics Data. Sci. Data 2024, 11, 685. [Google Scholar] [CrossRef]
- He, Y.S.; Dai, X.; Chen, Y.Y.; Huang, S.Y. Comprehensive Analysis of Genomic and Expression Data Identified Potential Markers for Predicting Prognosis and Immune Response in CRC. Genet. Res. 2022, 2022, 1831211. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Zhao, L.; Guo, B.; Guo, A.; Ding, J.; Tong, D.; Wang, B.; Zhou, Z. Integrated Machine Learning Algorithms-Enhanced Predication for Cervical Cancer from Mass Spectrometry-Based Proteomics Data. Bioengineering 2025, 12, 269. https://doi.org/10.3390/bioengineering12030269
Zhang D, Zhao L, Guo B, Guo A, Ding J, Tong D, Wang B, Zhou Z. Integrated Machine Learning Algorithms-Enhanced Predication for Cervical Cancer from Mass Spectrometry-Based Proteomics Data. Bioengineering. 2025; 12(3):269. https://doi.org/10.3390/bioengineering12030269
Chicago/Turabian StyleZhang, Da, Lihong Zhao, Bo Guo, Aihong Guo, Jiangbo Ding, Dongdong Tong, Bingju Wang, and Zhangjian Zhou. 2025. "Integrated Machine Learning Algorithms-Enhanced Predication for Cervical Cancer from Mass Spectrometry-Based Proteomics Data" Bioengineering 12, no. 3: 269. https://doi.org/10.3390/bioengineering12030269
APA StyleZhang, D., Zhao, L., Guo, B., Guo, A., Ding, J., Tong, D., Wang, B., & Zhou, Z. (2025). Integrated Machine Learning Algorithms-Enhanced Predication for Cervical Cancer from Mass Spectrometry-Based Proteomics Data. Bioengineering, 12(3), 269. https://doi.org/10.3390/bioengineering12030269