
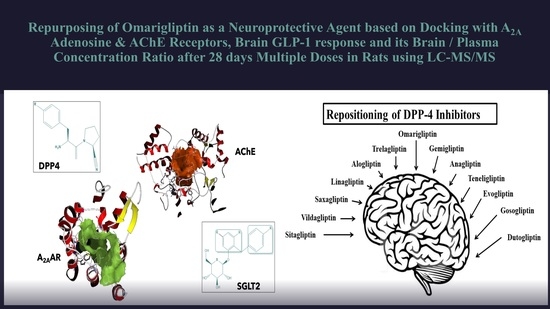
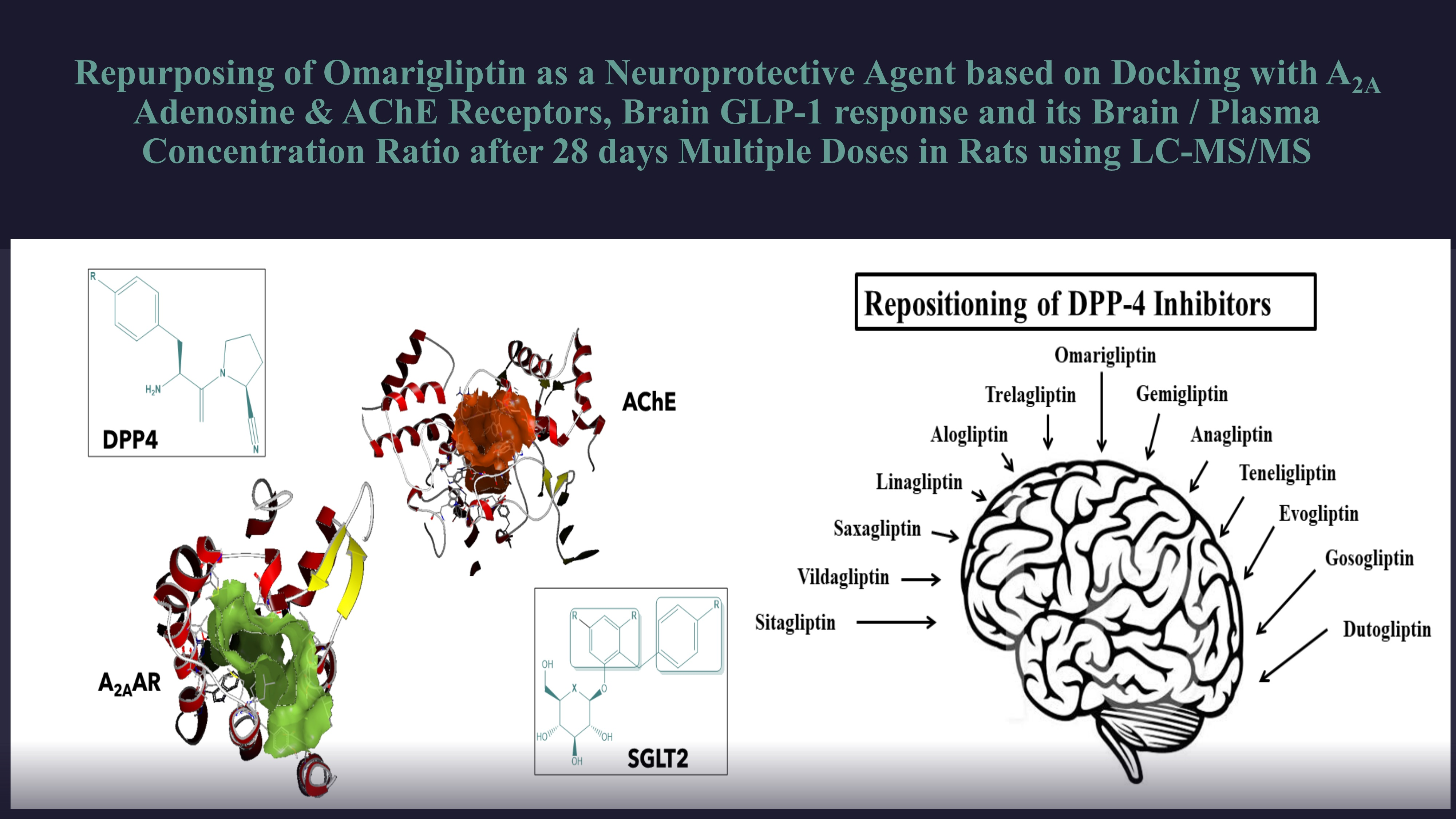
Repurposing of Omarigliptin as a Neuroprotective Agent Based on Docking with A2A Adenosine and AChE Receptors, Brain GLP-1 Response and Its Brain/Plasma Concentration Ratio after 28 Days Multiple Doses in Rats Using LC-MS/MS

 , ,
, ,
Abstract
:
1. Introduction
2. Methods
2.1. Docking Study of OMR, Other 12 DPP-4 Inhibitors and 11 SGLT-2 Inhibitors with A2A Adenosine Receptor (A2AAR) and Acetylcholinesterase (AChE) Receptor
2.2. Chemicals and Reagents
2.3. Biological Samples after Ethical Approval, Determination of Brain GLP-1 Concentration, and Multiple Dose In Vivo BBB Crossing Test
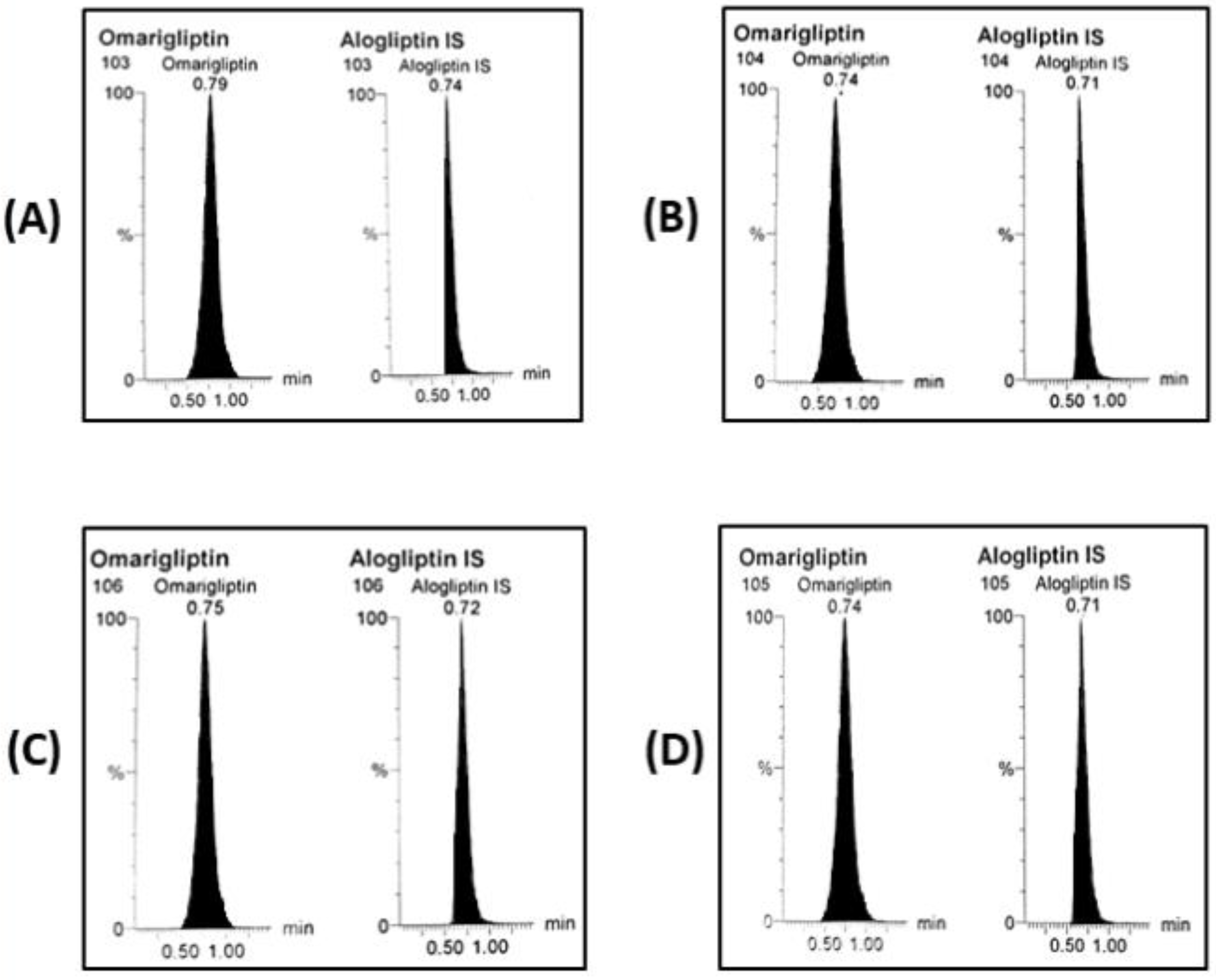
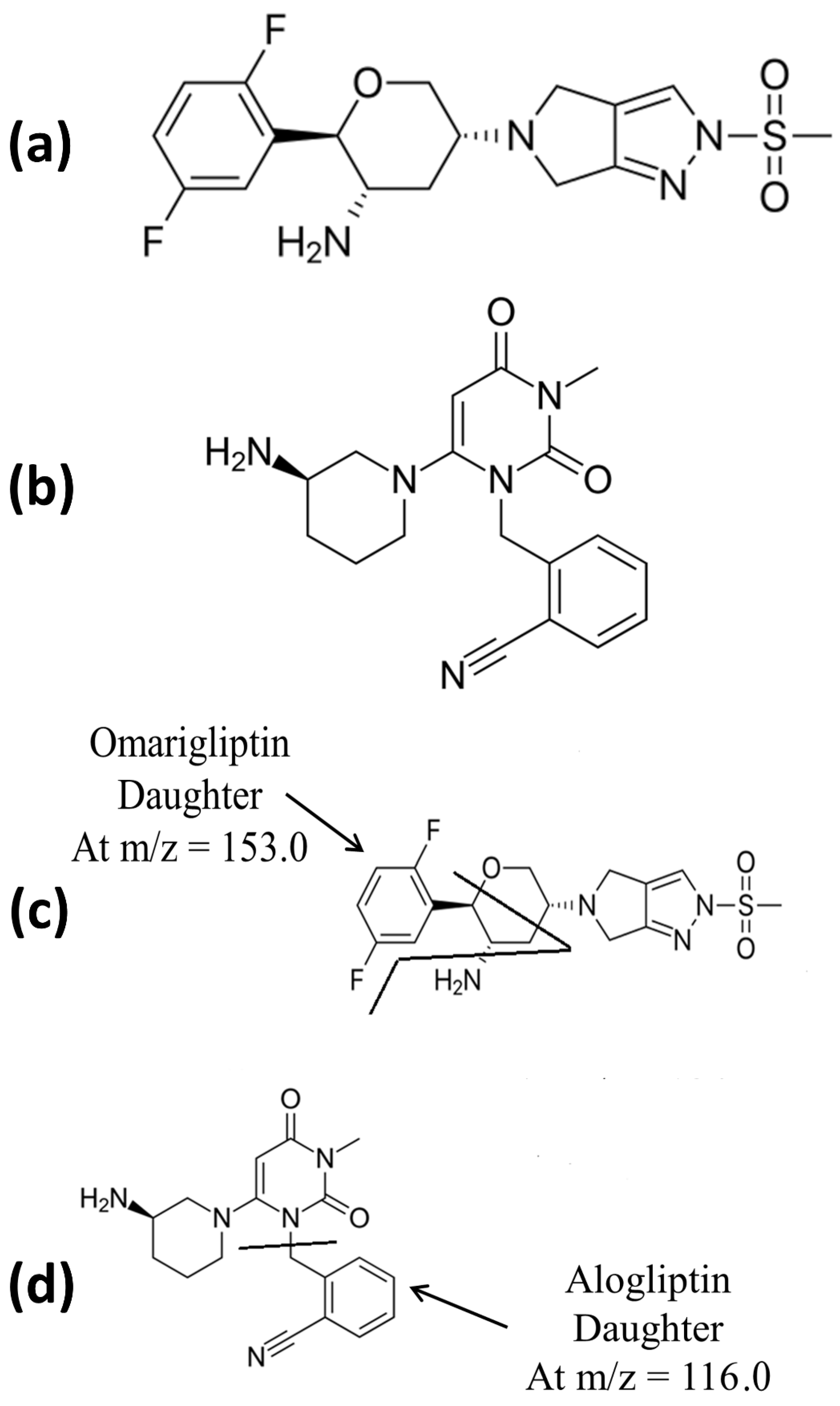
2.4. LC-MS/MS Conditions
2.5. LC-MS/MS Calibrators, QC Samples, and Sample Preparation
2.6. LC-MS/MS Bioanalytical Validation
3. Results and Discussion
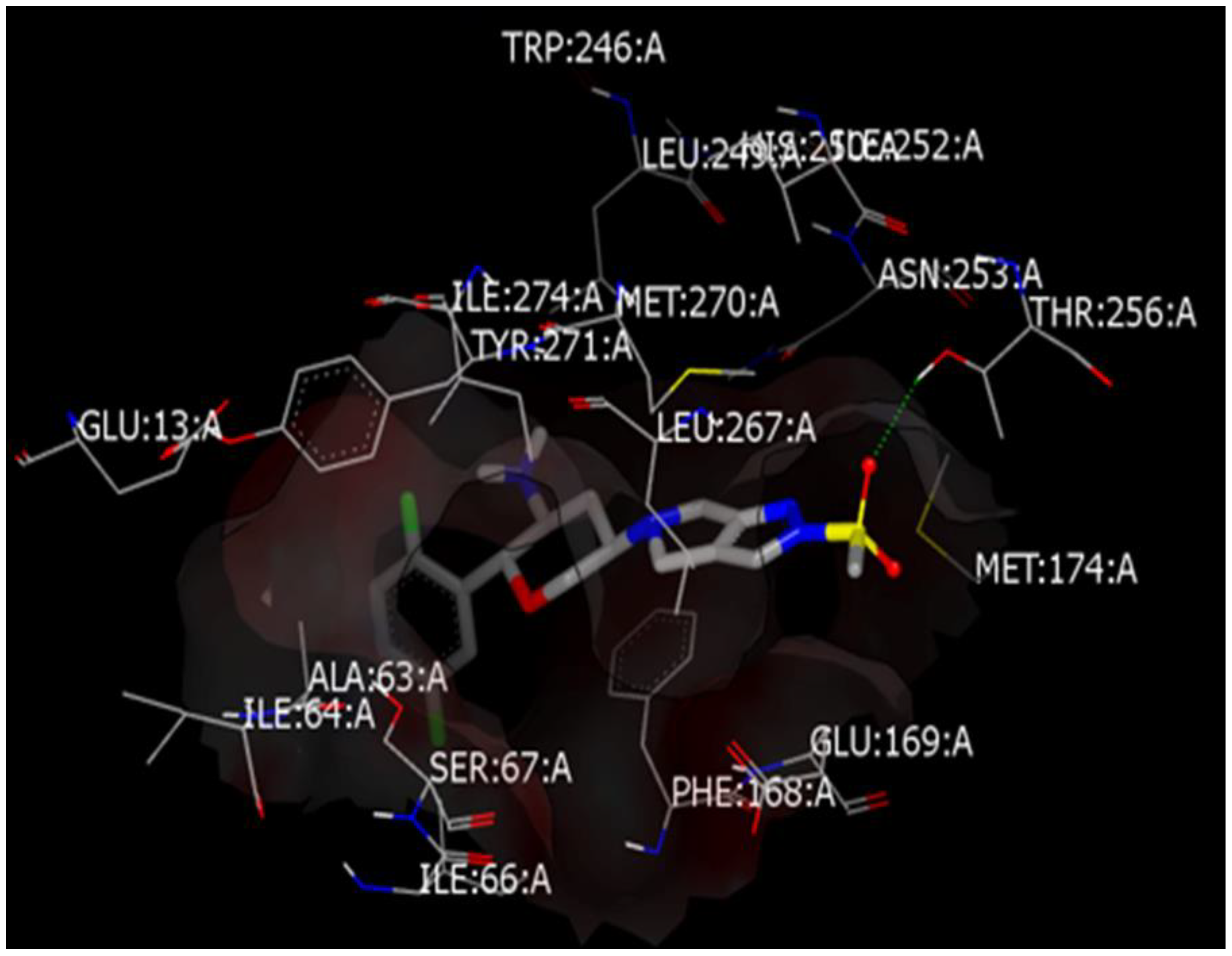
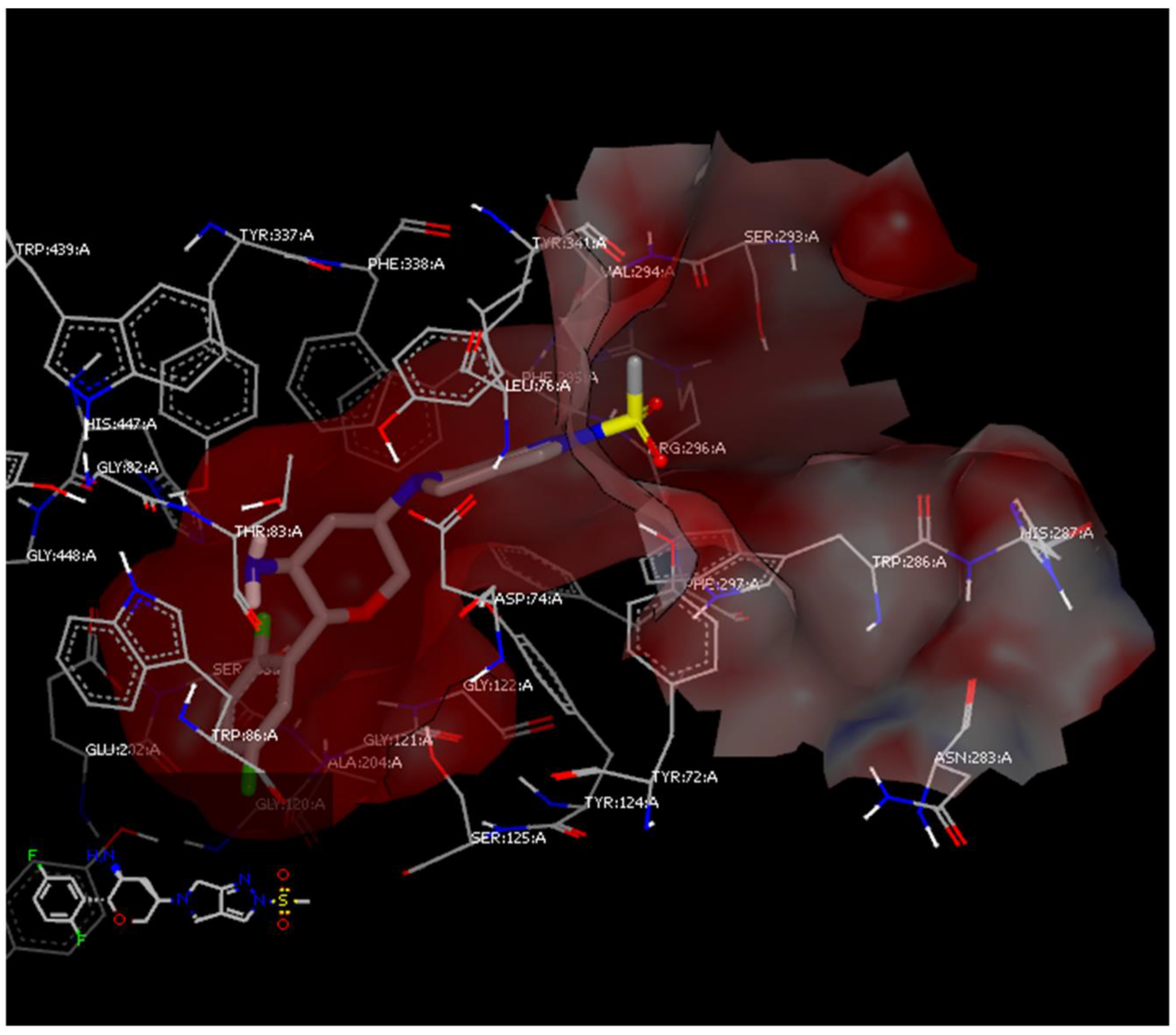
3.1. Comparative Docking Study (OMR, DPP-4 Inhibitors, and SGLT-2 Inhibitors) with A2AAR and AChE Receptors
3.1.1. Comparative Docking Study of DPP-4 Inhibitors with A2AAR that Support Repurposing of OMR for Parkinson’s’ Disease
3.1.2. Off-Label Neuroprotective Effect of DPP-4 Inhibitors and SGLT-2 Inhibitors and How the Brain Is Insulin Dependent
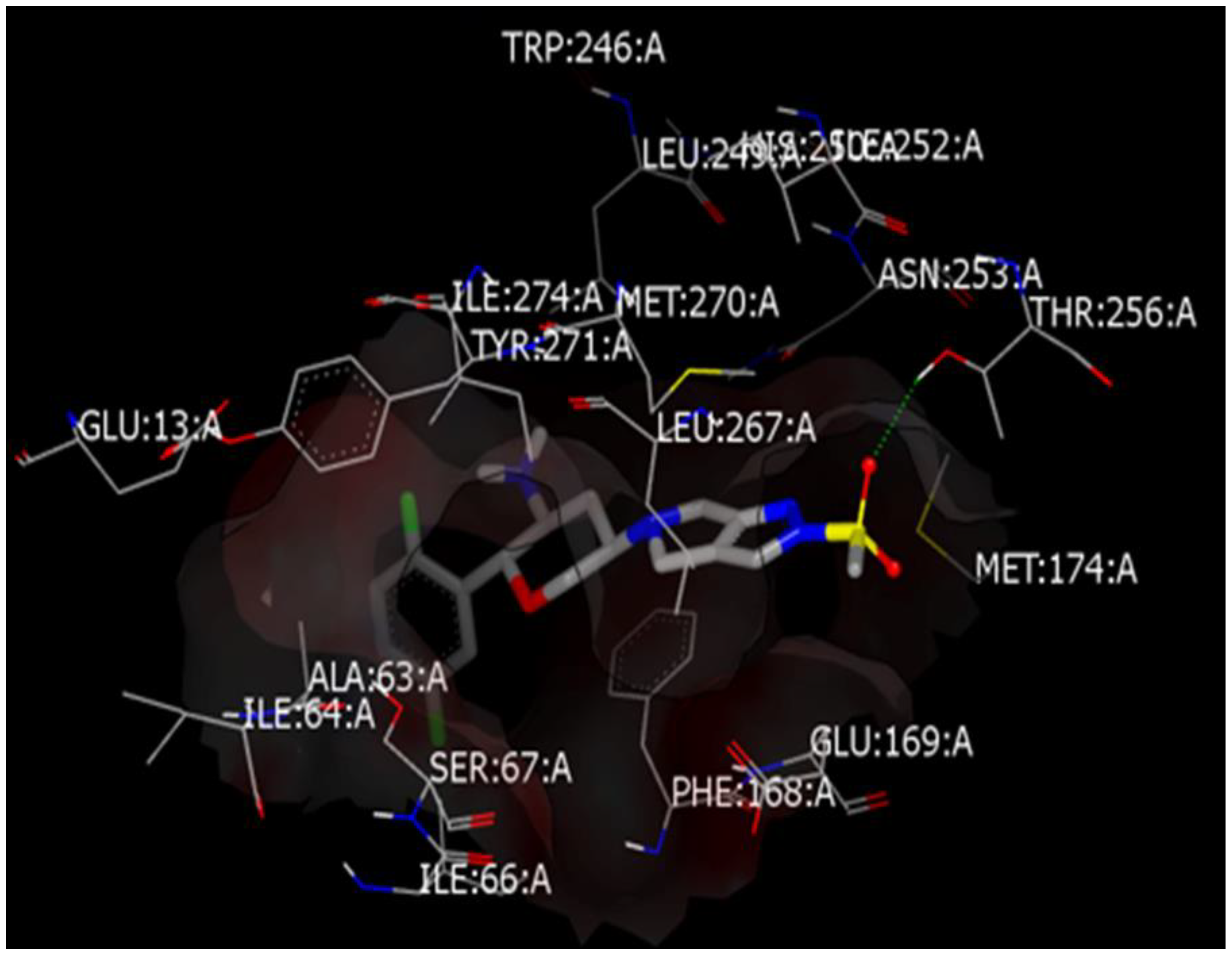
3.1.3. Docking Study of OMR (and 12 Other DPP-4 Inhibitors) with AChE Receptor
3.1.4. Comparative Docking Study of Gliflozins with A2AAR and AChE Receptors
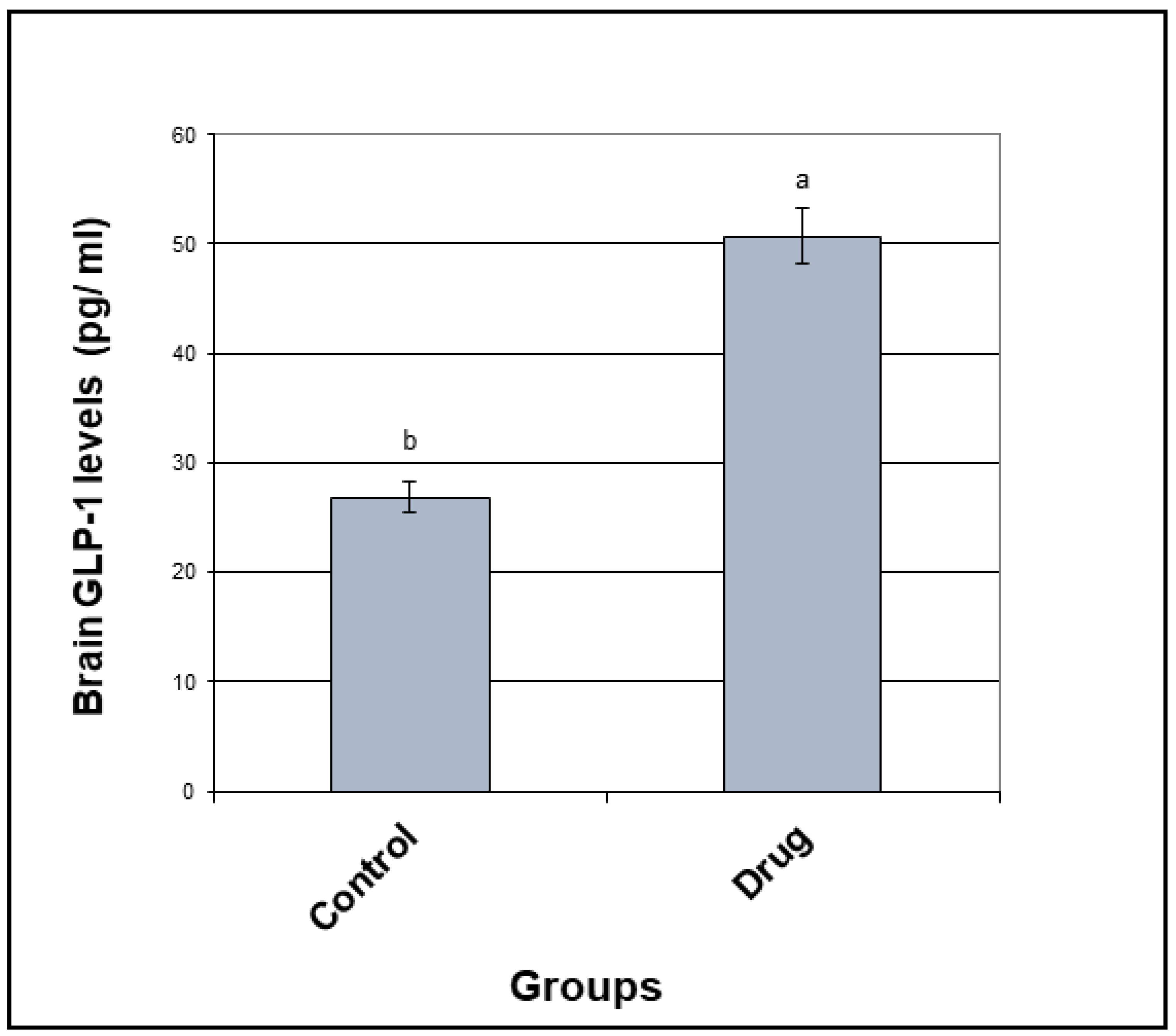
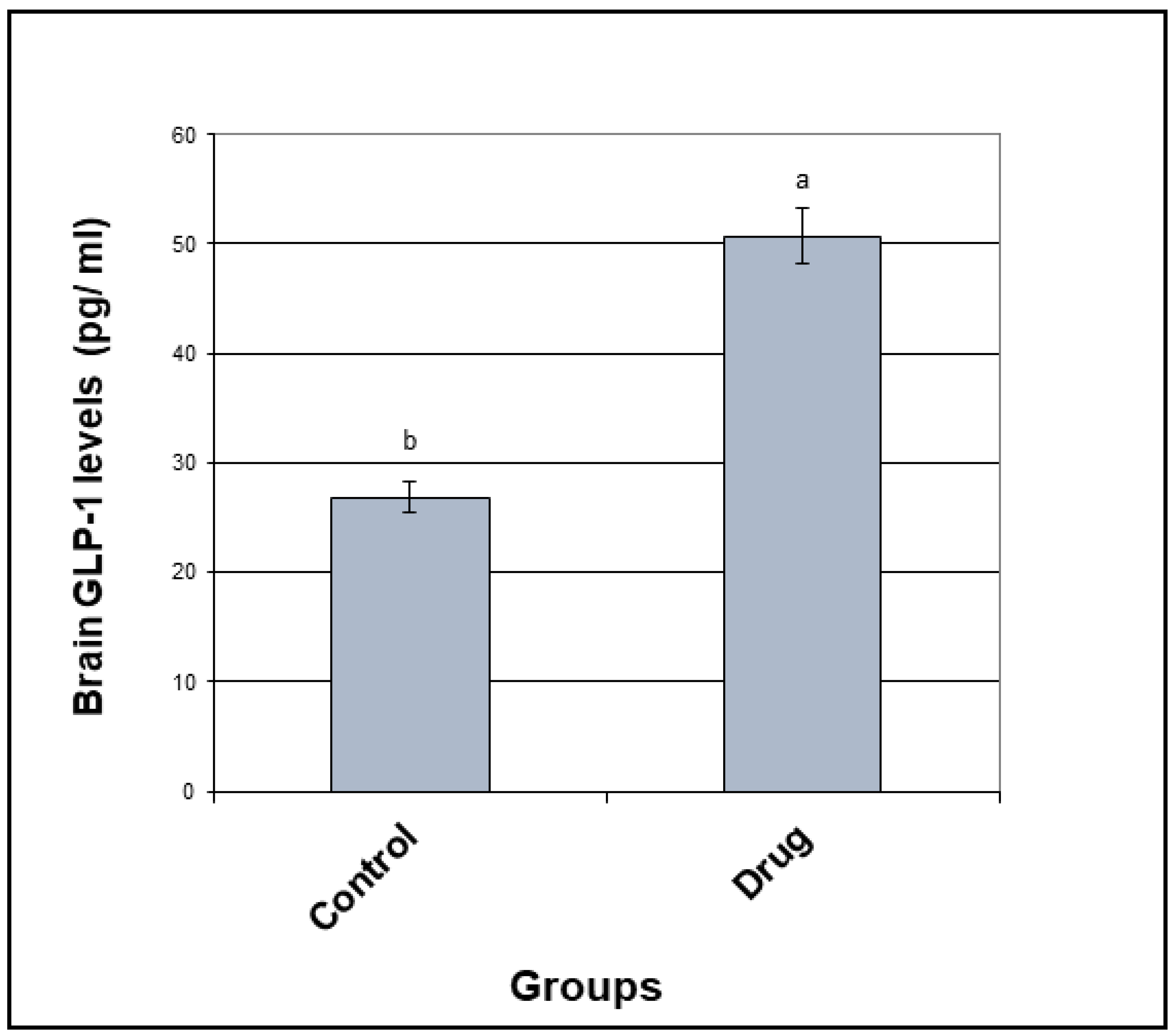
3.2. GLP-1 Concentration
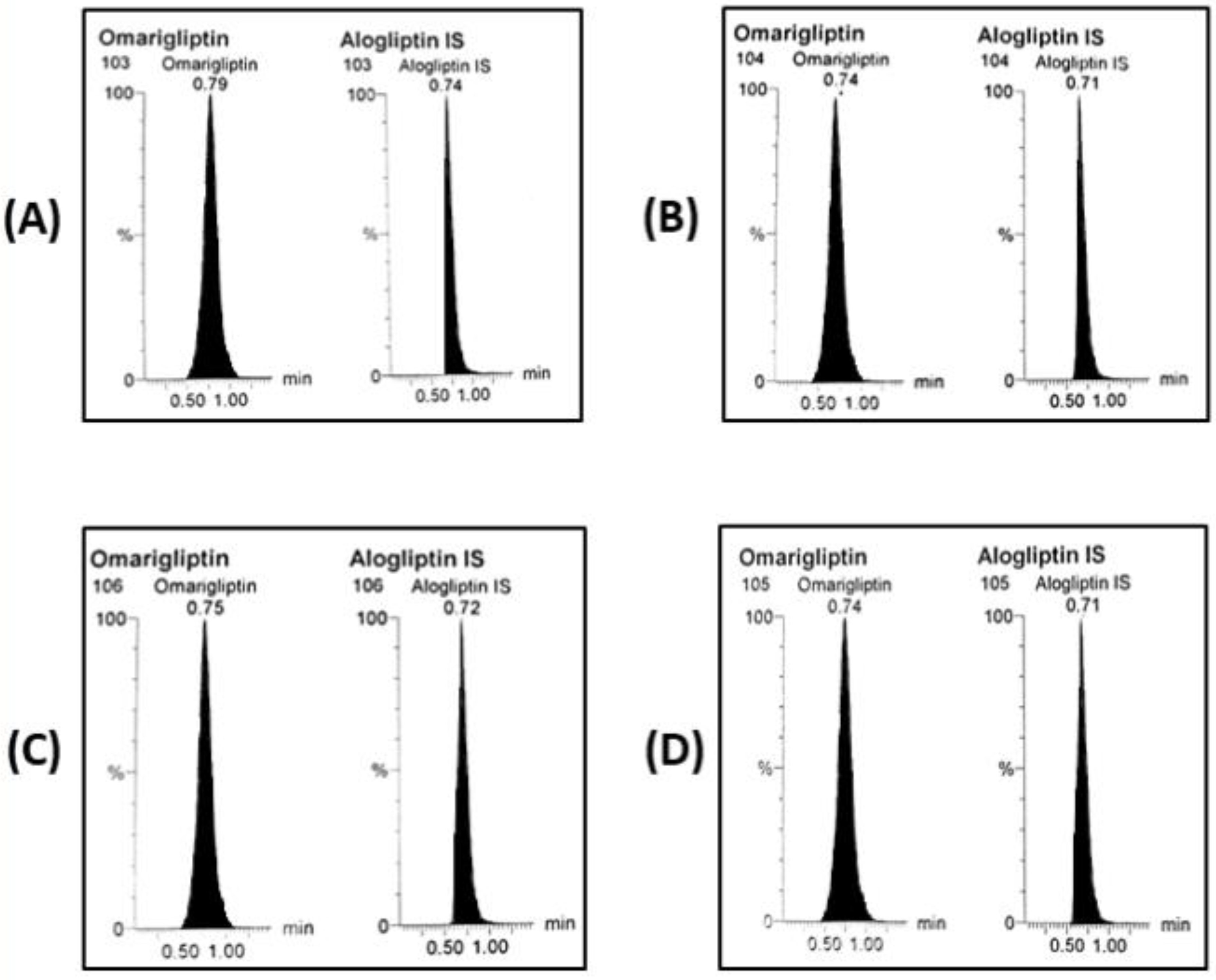
3.3. BBB Crossing After 28 Days of Multiple Doses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ye, H.; Wei, J.; Tang, K.; Feuers, R.; Hong, H. Drug repositioning through network pharmacology. Curr. Top. Med. Chem. 2016, 16, 3646–3656. [Google Scholar] [CrossRef]
- Vora, P.K.; Somani, R.R.; Jain, M.H. Drug repositioning: An approach for drug discovery. Mini-Rev. Org. Chem. 2016, 13, 363–376. [Google Scholar] [CrossRef]
- Mehndiratta, M.M.; Wadhai, S.A.; Tyagi, B.K.; Gulati, N.S.; Sinha, M. Drug repositioning. Int. J. Epilepsy 2016, 3, 91–94. [Google Scholar] [CrossRef]
- Kim, T.W. Drug Repositioning Approaches for the Discovery of New Therapeutics for Alzheimer’s Disease. Neurotherapeutics 2015, 12, 132–142. [Google Scholar] [CrossRef]
- Corbett, A.; Williams, G.; Ballard, C. Drug repositioning: An opportunity to develop novel treatments for Alzheimer’s disease. Pharmaceuticals 2013, 6, 1304–1321. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.M.S.; Bain, S.C. Omarigliptin for the treatment of type 2 diabetes mellitus. Expert Opin. Pharmacother. 2016, 17, 1947–1952. [Google Scholar] [CrossRef]
- Jain, L.; Chain, A.S.Y.; Tatosian, D.A.; Hing, J.; Passarell, J.A.; Kauh, E.A.; Lai, E. Pharmacokinetic–pharmacodynamic (dipeptidyl peptidase-4 inhibition) model to support dose rationale in diabetes patients, including those with renal impairment, for once-weekly administered omarigliptin. Br. J. Clin. Pharmacol. 2019, 85, 2759–2771. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Friedman, E.; Addy, C.; Wakana, A.; Tatosian, D.; Matsumoto, Y.; Suzuki, H.; Kauh, E. Single and multiple dose pharmacokinetics and pharmacodynamics of omarigliptin, a novel, once-weekly dipeptidyl peptidase-4 inhibitor, in healthy Japanese men. J. Diabetes Investig. 2016, 8, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Tatosian, D.A.; Marricco, N.C.; Glasgow, X.S.; DeGroot, B.; Dunnington, K.; George, L.; Gendrano, I.N.; Johnson-Levonas, A.O.; Swearingen, D.; Kauh, E.A. Thorough QTc study confirms early pharmacokinetics/QTc Modeling: A supratherapeutic dose of omarigliptin, a once-weekly DPP-4 Inhibitor, does not prolong the QTc interval. Clin. Pharmacol. Drug Dev. 2016, 5, 383–392. [Google Scholar] [CrossRef]
- Addy, C.; Tatosian, D.A.; Glasgow, X.S.; Iii, I.N.G.; Sisk, C.M.; Kauh, E.; Stoch, S.A.; Wagner, J.A. Effects of age, sex, and obesity on the single-dose pharmacokinetics of omarigliptin in healthy Subjects. Clin. Pharmacol. Drug Dev. 2016, 5, 374–382. [Google Scholar] [CrossRef]
- Addy, C.; Tatosian, D.; Glasgow, X.S.; Gendrano, I.N.; Kauh, E.; Martucci, A.; Johnson-Levonas, A.O.; Selverian, D.; Matthews, C.Z.; Gutierrez, M.; et al. Pharmacokinetic and pharmacodynamic effects of multiple-dose administration of omarigliptin, a once-weekly Dipeptidyl Peptidase-4 Inhibitor, in obese Participants with and without type 2 diabetes mellitus. Clin. Ther. 2016, 38, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Addy, C.; Tatosian, D.; Glasgow, X.S.; Iii, I.N.G.; Robberechts, M.; Haazen, W.; De Hoon, J.; Depré, M.; Martucci, A.; et al. Pharmacokinetics and pharmacodynamics of omarigliptin, a once-weekly dipeptidyl peptidase-4 (DPP-4) inhibitor, after single and multiple doses in Healthy Subjects. J. Clin. Pharmacol. 2016, 56, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Tatosian, D.; Mcintosh, I.; Caceres, M.; Matthews, C.; Samuel, K.; Selverian, D.; Kumar, S.; Kauh, E. Absorption, metabolism and excretion of [14C] omarigliptin, a once-weekly DPP-4 inhibitor, in humans. Xenobiotica 2018, 48, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Mowaka, S.; Ashoush, N.; Tadros, M.M.; El Zahar, N.M.; Ayoub, B.M. Enhanced extraction technique of omarigliptin from human plasma—Applied to biological samples from healthy human volunteers. Molecules 2020, 25, 4232. [Google Scholar] [CrossRef]
- Attallah, M.; Mowaka, S.; Elkady, E.F.; Fouad, M.A.; Ayoub, B.M. Analysis and bio-analysis of omarigliptin, trelagliptin and alogliptin: Applied to biological samples and degradation kinetic study. Microchem. J. 2019, 148, 253–261. [Google Scholar] [CrossRef]
- Li, M.-F.; Hu, X.-X.; Ma, A.-Q. Ultra-high pressure liquid chromatography–tandem mass spectrometry method for the determination of omarigliptin in rat plasma and its application to a pharmacokinetic study in rats. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef]
- Ayoub, B.M.; Mowaka, S.; Safar, M.M.; Ashoush, N.; Arafa, M.G.; Michel, H.E.; Tadros, M.M.; Elmazar, M.M.; Mousa, S.A. Repositioning of omarigliptin as a once-weekly intranasal anti-parkinsonian Agent. Sci. Rep. 2018, 8, 8959. [Google Scholar] [CrossRef]
- Tan, X. Omarigliptin for the treatment of type 2 diabetes. Endocrine 2016, 54, 24–31. [Google Scholar] [CrossRef]
- Breen, K.C.; Drutyte, G. Non-motor symptoms of Parkinson’s disease: The patient’s perspective. J. Neural Transm. 2013, 120, 531–535. [Google Scholar] [CrossRef] [Green Version]
- Mima, A. Incretin-based therapy for prevention of diabetic vascular complications. J. Diabetes Res. 2015, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ashraghi, M.R.; Pagano, G.; Polychronis, S.; Niccolini, F.; Politis, M. Parkinson’s disease, diabetes and cognitive impairment. Recent Pat. Endocr. Metab. Immune Drug Discov. 2016, 10, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Dellavalle, B.; Brix, G.S.; Brock, B.; Gejl, M.; Rungby, J.; Larsen, A. Oral administration of sitagliptin activates creb and is neuroprotective in murine model of brain trauma. Front. Pharmacol. 2016, 7, 450. [Google Scholar] [CrossRef] [PubMed]
- Badawi, G.A.; El Fattah, M.A.A.; Zaki, H.F.; El Sayed, M.I. Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology 2017, 25, 369–382. [Google Scholar] [CrossRef]
- Nader, M.A.; Ateyya, H.; El-Shafey, M.; El-Sherbeeny, N.A. Sitagliptin enhances the neuroprotective effect of pregabalin against pentylenetetrazole-induced acute epileptogenesis in mice: Implication of oxidative, inflammatory, apoptotic and autophagy pathways. Neurochem. Int. 2018, 115, 11–23. [Google Scholar] [CrossRef]
- Abdelsalam, R.M.; Safar, M.M. Neuroprotective effects of vildagliptin in rat rotenone Parkinson’s disease model: Role of RAGE-NFκB and Nrf2-antioxidant signaling pathways. J. Neurochem. 2015, 133, 700–707. [Google Scholar] [CrossRef]
- Nassar, N.N.; Al-Shorbagy, M.Y.; Arab, H.H.; Abdallah, D.M. Saxagliptin: A novel antiparkinsonian approach. Neuropharmacology 2015, 89, 308–317. [Google Scholar] [CrossRef]
- Lin, C.L.; Huang, C.N. The neuroprotective effects of the anti-diabetic drug linagliptin against Aβ-induced neurotoxicity. Neural Regen. Res. 2016, 11, 236–237. [Google Scholar] [PubMed]
- Duarte, A.I.; Candeias, E.; Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Plácido, A.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Crosstalk between diabetes and brain: Glucagon-like peptide-1 mimetics as a promising therapy against neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Shantikumar, S.; Satheeshkumar, N.; Srinivas, R. Pharmacokinetic and protein binding profile of peptidomimetic DPP-4 inhibitor—Teneligliptin in rats using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1002, 194–200. [Google Scholar] [CrossRef] [PubMed]
- FDA. Bioanalytical Method Validation, Guidance for Industry, U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM). 2018. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 1 August 2020).
- Xue, Y.-J.; Pursley, J.; Arnold, M.E. A simple 96-well liquid–liquid extraction with a mixture of acetonitrile and methyl t-butyl ether for the determination of a drug in human plasma by high-performance liquid chromatography with tandem mass spectrometry. J. Pharm. Biomed. Anal. 2004, 34, 369–378. [Google Scholar] [CrossRef]
- Ramalingam, P.; Ko, Y.T. A validated LC-MS/MS method for quantitative analysis of curcumin in mouse plasma and brain tissue and its application in pharmacokinetic and brain distribution studies. J. Chromatogr. B 2014, 969, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Minocha, M.; Khurana, V.; Mitra, A.K. Determination of pazopanib (GW-786034) in mouse plasma and brain tissue by liquid chromatography-tandem mass spectrometry (LC/MS-MS). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 901, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Terry, A.V., Jr.; Bartlett, M.G. Determination of the lipophilic antipsychotic drug ziprasidone in rat plasma and brain tissue using liquid chromatography-tandem mass spectrometry. Biomed. Chromatogr. 2008, 22, 770–778. [Google Scholar] [CrossRef] [PubMed]
- US-FDA. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Adult Healthy Volunteer; US Food and Drug Administration: Rockville, MD, USA, 2005. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf (accessed on 1 August 2020).
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 2426. [Google Scholar] [CrossRef]
- Meissner, W.G.; Frasier, M.; Gasser, T.; Goetz, C.G.; Lozano, A.M.; Piccini, P.; Obeso, J.A.; Rascol, O.; Schapira, A.H.V.; Voon, V.; et al. Priorities in Parkinson’s disease research. Nat. Rev. Drug Discov. 2011, 10, 377–393. [Google Scholar] [CrossRef]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiteh, M.; Zeifman, A.; Saarinen, M.; Svenningsson, P.; Brea, J.M.; Loza, M.I.; Carlsson, J. Docking screens for dual inhibitors of disparate drug targets for Parkinson’s disease. J. Med. Chem. 2018, 61, 5269–5278. [Google Scholar] [CrossRef]
- Carlsson, J.; Yoo, L.; Gao, Z.-G.; Irwin, J.J.; Shoichet, B.K.; Jacobson, K.A. Structure-based discovery of A2A adenosine receptor ligands. J. Med. Chem. 2010, 53, 3748–3755. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.P.; Brown, S.P.; Warren, G.L.; Muchmore, S.W. POSIT: Flexible shape-guided docking for pose prediction. J. Chem. Inf. Model. 2015, 55, 1771–1780. [Google Scholar] [CrossRef]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: A balanced overview. Diabetologia 1992, 35, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Juillerat-Jeanneret, L. Dipeptidyl peptidase IV and its inhibitors: Therapeutics for type 2 diabetes and what else? J. Med. Chem. 2014, 57, 2197–2212. [Google Scholar] [CrossRef]
- Lasserson, D.; Mant, J. The role of dipeptidyl peptidase-4 inhibitors. BMJ 2012, 344, e1213. [Google Scholar] [CrossRef] [PubMed]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef]
- Lee, S.; Zabolotny, J.M.; Huang, H.; Lee, H.; Kim, Y.-B. Insulin in the nervous system and the mind: Functions in metabolism, memory, and mood. Mol. Metab. 2016, 5, 589–601. [Google Scholar] [CrossRef]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef] [Green Version]
- Plum, L.; Schubert, M.; Brüning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef]
- DeFronzo, R.A. From the triumvirate to the “ominous octet”: A new paradigm for the treatment of type 2 diabetes mellitus. Clin. Diabetol. 2009, 10, 101–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintana, H.; Apaijai, N.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Effects of metformin on learning and memory behaviors and brain mitochondrial functions in high fat diet induced insulin resistant rats. Life Sci. 2012, 91, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Sripetchwandee, J.; Pipatpiboon, N.; Chattipakorn, N.; Chattipakorn, S. Combined therapy of iron chelator and antioxidant completely restores brain dysfunction induced by iron toxicity. PLoS ONE 2014, 9, e85115. [Google Scholar] [CrossRef] [Green Version]
- Pratchayasakul, W.; Kerdphoo, S.; Petsophonsakul, P.; Pongchaidecha, A.; Chattipakorn, N.; Chattipakorn, S.C. Effects of high-fat diet on insulin receptor function in rat hippocampus and the level of neuronal corticosterone. Life Sci. 2011, 88, 619–627. [Google Scholar] [CrossRef]
- Yu, A.S.; Hirayama, B.A.; Timbol, G.; Liu, J.; Diez-Sampedro, A.; Kepe, V.; Satyamurthy, N.; Huang, S.-C.; Wright, E.M.; Barrio, J.R. Regional distribution of SGLT activity in rat brain in vivo. Am. J. Physiol. Physiol. 2013, 304, C240–C247. [Google Scholar] [CrossRef]
- Hierro-Bujalance, C.; Infante-Garcia, C.; Del Marco, A.; Herrera, M.; Carranza-Naval, M.J.; Suarez, J.; Alves-Martinez, P.; Lubian-Lopez, S.; Garcia-Alloza, M. Empagliflozin reduces vascular damage and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Alzheimer’s Res. Ther. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Ridderstråle, M.; Svaerd, R.; Zeller, C.; Kim, G.; Woerle, H.-J.; Broedl, U.C. Rationale, design and baseline characteristics of a 4-year (208-week) phase III trial of empagliflozin, an SGLT2 inhibitor, versus glimepiride as add-on to metformin in patients with type 2 diabetes mellitus with insufficient glycemic control. Cardiovasc. Diabetol. 2013, 12, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Millar, P.; Pathak, N.; Parthsarathy, V.; Bjourson, A.J.; O’Kane, M.; Pathak, V.; Moffett, R.C.; Flatt, P.R.; Gault, V.A. Metabolic and neuroprotective effects of dapagliflozin and liraglutide in diabetic mice. J. Endocrinol. 2017, 234, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Gutniak, M.; Ørkov, C.; Holst, J.J.; Ahren, B.; Efendic, S. Antidiabetogenic effect of glucagon-like peptide-1 (7–36) amide in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 1992, 326, 1316–1322. [Google Scholar] [CrossRef]
- Elrick, H.; Stimmler, L.; Hlad, C.J., Jr.; Arai, Y. Plasma insulin response to oral and intravenous glucose administration. J. Clin. Endocrinol. Metab. 1964, 24, 1076–1082. [Google Scholar] [CrossRef]
- Creutzfeldt, W. The incretin concept today. Diabetologia 1979, 16, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, T.; Paschos, P.; Paletas, K.; Matthews, D.R.; Tsapas, A. Dipeptidyl peptidase-4 inhibitors for treatment of type 2 diabetes mellitus in the clinical setting: Systematic review and meta-analysis. BMJ 2012, 344, e1369. [Google Scholar] [CrossRef] [Green Version]
- Mishriky, B.M.; Cummings, D.M.; Tanenberg, R.J. The efficacy and safety of DPP4 inhibitors compared to sulfonylureas as add-on therapy to metformin in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2015, 109, 378–388. [Google Scholar] [CrossRef]
- Del Prato, S.; Foley, J.E.; Kothny, W.; Kozlovski, P.; Stumvoll, M.; Paldánius, P.M.; Matthews, D.R. Study to determine the durability of glycaemic control with early treatment with a vildagliptin–metformin combination regimen vs. standard-of-care metformin monotherapy—the VERIFY trial: A randomized double-blind trial. Diabet. Med. 2014, 31, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.R.; Paldánius, P.M.; Proot, P.; Chiang, Y.; Stumvoll, M.; Del Prato, S. Glycaemic durability of an early combination therapy with vildagliptin and metformin versus sequential metformin monotherapy in newly diagnosed type 2 diabetes (VERIFY): A 5-year, multicentre, randomised, double-blind trial. Lancet 2019, 394, 1519–1529. [Google Scholar] [CrossRef]
- Saisho, Y. SGLT2 Inhibitors: The Star in the Treatment of Type 2 Diabetes? Diseases 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Chasis, H.; Jolliffe, N.; Smith, H.W. The action of phlorizin on the excretion of glucose, xylose, sucrose, creatinine and urea by man. J. Clin. Investig. 1933, 12, 1083–1090. [Google Scholar] [CrossRef]
- Nagahisa, T.; Saisho, Y. Cardiorenal protection: Potential of SGLT2 inhibitors and GLP-1 receptor agonists in the treatment of type 2 diabetes. Diabetes Ther. 2019, 10, 1733–1752. [Google Scholar] [CrossRef]
- Kosiborod, M.; Cavender, M.A.; Fu, A.Z.; Wilding, J.P.; Khunti, K.; Holl, R.W.; Norhammar, A.; Birkeland, K.I.; Jørgensen, M.E.; Thuresson, M.; et al. Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: The CVD-REAL study (comparative effectiveness of cardiovascular outcomes in new users of sodium-glucose cotranspo. Circulation 2017, 136, 249–259. [Google Scholar] [CrossRef]
- Buse, J.B.; Wexler, D.J.; Tsapas, A.; Rossing, P.; Mingrone, G.; Mathieu, C.; D’Alessio, D.A.; Davies, M.J. 2019 update to: Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2020, 63, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M.; Turk, E. The sodium/glucose cotransport family SLC5. Pflügers Arch. 2004, 447, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2010, 22, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, E.M.; Loo, D.D.F.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madaan, T.; Akhtar, M.; Najmi, A.K. Sodium glucose CoTransporter 2 (SGLT2) inhibitors: Current status and future perspective. Eur. J. Pharm. Sci. 2016, 93, 244–252. [Google Scholar] [CrossRef]
- Haider, K.; Pathak, A.; Rohilla, A.; Haider, R.; Ahmad, K.; Yar, M.S. Synthetic strategy and SAR studies of C-glucoside heteroaryls as SGLT2 inhibitor: A review. Eur. J. Med. Chem. 2019, 184, 111773. [Google Scholar] [CrossRef]
- Vallon, V.; Richter, K.; Blantz, R.C.; Thomson, S.; Osswald, H. Glomerular hyperfiltration in experimental diabetes mellitus: Potential role of tubular reabsorption. J. Am. Soc. Nephrol. 1999, 10, 2569–2576. [Google Scholar]
- Coady, M.J.; Wallendorff, B.; Gagnon, D.G.; Lapointe, J.-Y. Identification of a novel Na+/myo-inositol cotransporter. J. Biol. Chem. 2002, 277, 35219–35224. [Google Scholar] [CrossRef] [Green Version]
- Manoj, A.; Das, S.; Ramachandran, A.K.; Alex, A.T.; Joseph, A. SGLT2 inhibitors, an accomplished development in field of medicinal chemistry: An extensive review. Future Med. Chem. 2020, 12, 1961–1990. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Rathore, A.; Parwani, D.; Mallick, C.; Asati, V.; Agarwal, S.; Rajoriya, V.; Das, R.; Kashaw, S.K. An exhaustive perspective on structural insights of SGLT2 inhibitors: A novel class of antidiabetic agent. Eur. J. Med. Chem. 2020, 204, 112523. [Google Scholar] [CrossRef] [PubMed]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non–insulin-dependent diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Lowering plasma glucose concentration by inhibiting renal sodium–glucose cotransport. J. Intern. Med. 2014, 276, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Hopsu-Havu, V.K.; Glenner, G.G. A new dipeptide naphthylamidase hydrolyzing glycyl-prolyl-β-naphthylamide. Histochemie 1966, 7, 197–201. [Google Scholar] [CrossRef]
- Misumi, Y.; Hayashi, Y.; Arakawa, F.; Ikehara, Y. Molecular cloning and sequence analysis of human dipeptidyl peptidase IV, a serine proteinase on the cell surface. Biochim. Biophys. Acta BBA Gene Struct. Expr. 1992, 1131, 333–336. [Google Scholar] [CrossRef]
- Tanaka, T.; Camerini, D.; Seed, B.; Torimoto, Y.; Dang, N.H.; Kameoka, J.; Dahlberg, H.N.; Schlossman, S.F.; Morimoto, C. Cloning and functional expression of the T cell activation antigen CD26. J. Immunol. 1992, 149, 481–486. [Google Scholar]
- Abbott, C.A.; Baker, E.; Sutherland, G.R.; McCaughan, G.W. Genomic organization, exact localization, and tissue expression of the human CD26 (dipeptidyl peptidase IV) gene. Immunogenetics 1994, 40, 331–338. [Google Scholar] [CrossRef]
- Engel, M.; Hoffmann, T.; Wagner, L.; Wermann, M.; Heiser, U.; Kiefersauer, R.; Huber, R.; Bode, W.; DeMuth, H.-U.; Brandstetter, H. The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc. Natl. Acad. Sci. USA 2003, 100, 5063–5068. [Google Scholar] [CrossRef] [Green Version]
- Fukasawa, K.M.; Sahara, N.; Harada, M.; Kondo, Y.; Nagatsu, I. Immunohistochemical localization of dipeptidyl aminopeptidase IV in rat kidney, liver, and salivary glands. J. Histochem. Cytochem. 1981, 29, 337–343. [Google Scholar] [CrossRef]
- Deacon, C.F. What do we know about the secretion and degradation of incretin hormones? Regul. Pept. 2005, 128, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, H.; Kyono, K.; Shima, H.; Fukushima, C.; Sugiyama, S.; Inaka, K.; Yamamoto, A.; Shimizu, R. Crystallization and preliminary X-ray study of human dipeptidyl peptidase IV (DPPIV). Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 595–596. [Google Scholar] [CrossRef]
- Meissner, W.G.; Frasier, M.; Gasser, T.; Goetz, C.G.; Lozano, A.; Piccini, P.; Obeso, J.A.; Rascol, O.; Schapira, A.; Voon, V.; et al. Molecular characterization of dipeptidyl peptidase activity in serum: Soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur. J. Biochem. 2000, 267, 5608–5613. [Google Scholar]
- Cordero Óscar, J.; Salgado, F.J.; Nogueira, M. On the origin of serum CD26 and its altered concentration in cancer patients. Cancer Immunol. Immunother. 2009, 58, 1723–1747. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, I.; Nagatsu, T.; Yamamoto, T. Hydrolysis of amino acidβ-naphthylamides by aminopeptidases in human parotid saliva and human serum. Experientia 1968, 24, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Munakata, Y.; Iwata, S.; Ohnuma, K.; Kobayashi, S.; Dang, N.H.; Morimoto, C. Soluble CD26/dipeptidyl peptidase IV enhances transendothelial migration via its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cell Immunol. 2002, 215, 106–110. [Google Scholar] [CrossRef]
- Zhong, J.; Rao, X.; Rajagopalan, S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: Potential implications in cardiovascular disease. Atherosclerosis 2013, 226, 305–314. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Porte, D. Diabetes, obesity, and the brain. Science 2005, 307, 375–379. [Google Scholar] [CrossRef]
- Cunnane, S.; Courchesne-Loyer, A.; Vandenberghe, C.; St-Pierre, V.; Fortier, M.; Hennebelle, M.; Croteau, E.; Bocti, C.; Fulop, T.; Castellano, C.-A. Can ketones help rescue brain fuel supply in later life? Implications for cognitive health during aging and the treatment of Alzheimer’s disease. Front. Mol. Neurosci. 2016, 9, 53. [Google Scholar] [CrossRef]
- Zilberter, Y.; Zilberter, M. The vicious circle of hypometabolism in neurodegenerative diseases: Ways and mechanisms of metabolic correction. J. Neurosci. Res. 2017, 95, 2217–2235. [Google Scholar] [CrossRef] [Green Version]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 mobilization supports energetic demands of active synapses. Neuron 2017, 93, 606–615. [Google Scholar] [CrossRef]
- Frere, S.; Slutsky, I. Alzheimer’s disease: From firing instability to homeostasis network collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef] [Green Version]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What happens with the circuit in Alzheimer’s disease in mice and humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef]
- Yu, L.; Shen, Z.; Wang, C.; Yu, Y. Efficient coding and energy efficiency are promoted by balanced excitatory and inhibitory synaptic currents in neuronal network. Front. Cell. Neurosci. 2018, 12, 123. [Google Scholar] [CrossRef] [Green Version]
- Oyarzabal, A.; Marin-Valencia, I. Synaptic energy metabolism and neuronal excitability, in sickness and health. J. Inherit. Metab. Dis. 2019, 42, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.-T.; Dikmen, H.O.; Schilling, S.; Chausse, B.; Lewen, A.; Hollnagel, J.-O.; Kann, O. Priming of microglia with IFN-γ slows neuronal gamma oscillations in situ. Proc. Natl. Acad. Sci. USA 2019, 116, 4637–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
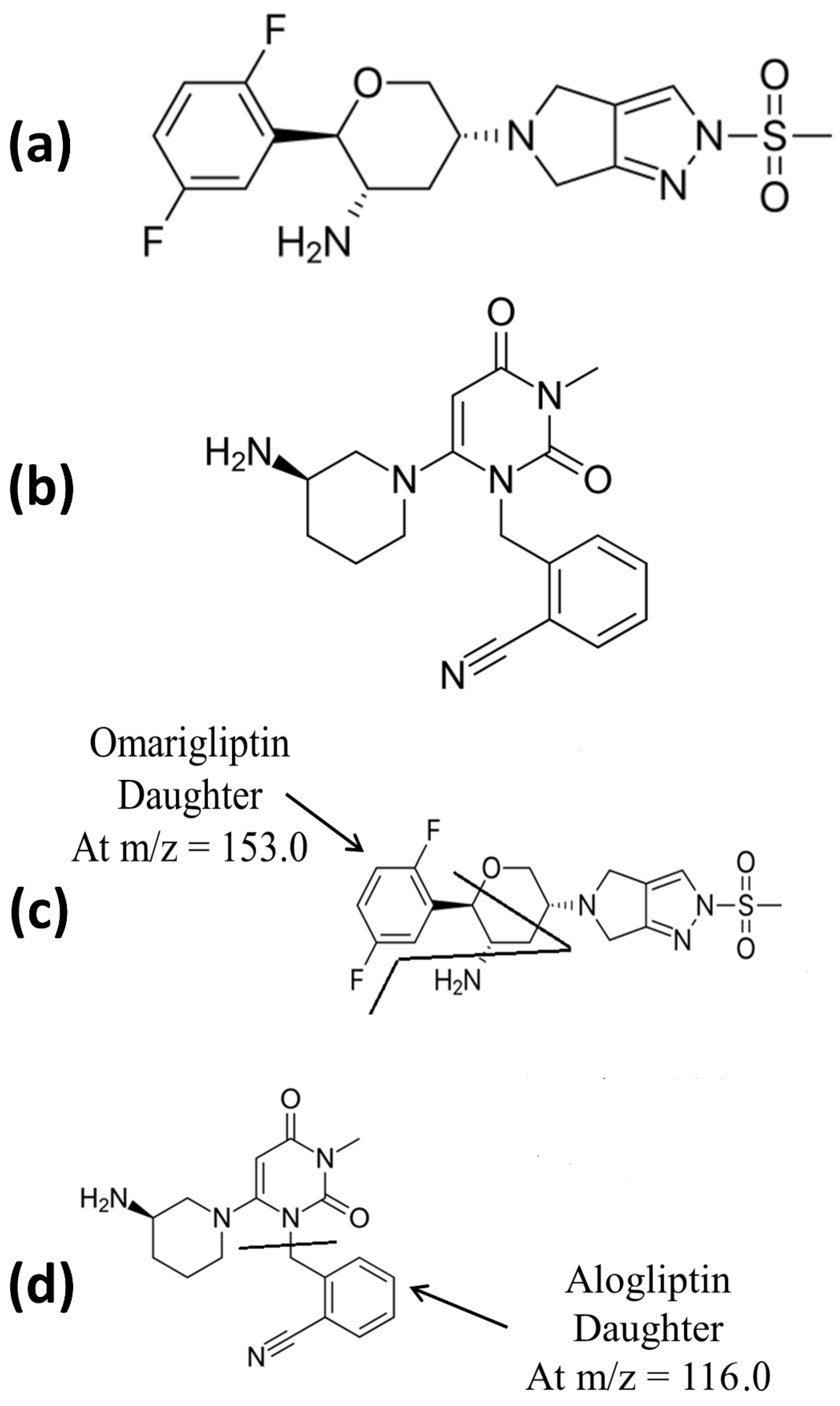


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SGLT2 Inhibitors | Score | DPP4 Inhibitors | Score | |
|---|---|---|---|---|
| AChE Receptors | Ipragliflozin | −18.0826 | Linagliptin | −16.7927 |
| Luseogliflozin | −17.4729 | Sitagliptin | −14.6245 | |
| Canagliflozin | −16.6481 | Gemigliptin | −14.3007 | |
| Dapagliflozin | −16.4979 | Anagliptin | −14.2372 | |
| Empagliflozin | −15.7074 | Gosogliptin | −13.9488 | |
| Sotagliflozin | −15.5315 | Teneligliptin | −13.7557 | |
| Tofogliflozin | −15.1791 | Saxagliptin | −13.2396 | |
| Ertugliflozin | −14.4763 | Omarigliptin | −13.1468 | |
| Sergliflozin | −13.4309 | Alogliptin | −13.1226 | |
| Remogliflozin | −12.2717 | Evogliptin | −13.0186 | |
| Ligand | −9.8658 | Vildagliptin | −12.727 | |
| Dutogliptin | −12.1961 | |||
| Ligand | −11.0175 | |||
| Trelagliptin | N/A | |||
| A2A AR Receptors | Canagliflozin | −13.5012 | Linagliptin | −11.6503 |
| Empagliflozin | −13.0962 | Alogliptine | −11.14 | |
| Ertugliflozin | −12.9675 | Ligand | −10.7851 | |
| Luseogliflozin | −12.2521 | Trelagliptin | −10.778 | |
| Ipragliflozin | −12.1433 | Sitagliptin | −10.3922 | |
| Dapagliflozin | −12.0988 | Tenegliptin | −10.2282 | |
| Sotagliflozin | −11.9531 | Gosogliptin | −9.8775 | |
| Tofogliflozin | −11.2769 | Evogliptin | −9.8116 | |
| Sergliflozin | −11.2085 | Geminigliptin | −9.7432 | |
| Ligand | −10.7851 | Anagliptin | −9.2179 | |
| Remogliflozin | −8.7495 | Vildagliptin | −9.0521 | |
| Dutogliptin | −8.5373 | |||
| Omarigliptin | −8.4469 | |||
| Saxagliptin | −8.3109 | |||
| Accuracy and Precision | Parameters | LLOQ | LQC | MQC | HQC |
|---|---|---|---|---|---|
| Rats’ plasma intra-day | Mean of R% | 118.72 | 99.47 | 91.43 | 103.42 |
| Bias | 18.72 | −0.53 | −8.57 | 3.42 | |
| S.D. | 13.03 | 10.95 | 7.21 | 8.76 | |
| C.V. (%RSD) | 10.97 | 11.01 | 7.89 | 8.47 | |
| Rats’ plasma inter-day | Mean of R% | 99.81 | 101.75 | 91.09 | 101.35 |
| Bias | −0.19 | 1.75 | −8.91 | 1.35 | |
| S.D. | 17.78 | 9.60 | 6.68 | 8.24 | |
| C.V. (%RSD) | 17.81 | 9.43 | 7.33 | 8.13 | |
| Rats’ brain intra-day | Mean of R% | 86.99 | 87.96 | 110.25 | 109.62 |
| Bias | −13.01 | −12.04 | 10.25 | 9.62 | |
| S.D. | 13.29 | 10.25 | 5.67 | 5.79 | |
| C.V. (%RSD) | 15.28 | 9.62 | 5.14 | 5.29 | |
| Rats’ brain inter-day | Mean of R% | 83.23 | 90.61 | 99.80 | 107.24 |
| Bias | −16.77 | −9.39 | −0.20 | 7.24 | |
| S.D. | 14.04 | 8.18 | 9.26 | 5.59 | |
| C.V. (%RSD) | 16.88 | 9.03 | 9.28 | 5.21 |
| Stability Studies | Parameters | Recoveries of LQC | Recoveries of HQC |
|---|---|---|---|
| Rats’ plasma | Auto-sampler stability | 100.58 ± 0.41 | 100.58 ± 0.41 |
| Short-term stability | 97.66 ± 1.66 | 109.83 ± 6.95 | |
| Long-term stability | 91.43 ± 6.06 | 105.17 ± 3.66 | |
| Freeze–thaw stability | 107.06 ± 4.99 | 111.14 ± 7.88 | |
| Rats’ brain | Auto-sampler stability | 101.57 ± 1.11 | 104.97 ± 3.51 |
| Short-term stability | 96.60 ± 2.40 | 106.40 ± 4.53 | |
| Long-term stability | 97.98 ± 1.42 | 112.86 ± 9.09 | |
| Freeze–thaw stability | 119.73 ± 13.95 | 119.57 ± 13.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayoub, B.M.; Michel, H.E.; Mowaka, S.; Hendy, M.S.; Tadros, M.M. Repurposing of Omarigliptin as a Neuroprotective Agent Based on Docking with A2A Adenosine and AChE Receptors, Brain GLP-1 Response and Its Brain/Plasma Concentration Ratio after 28 Days Multiple Doses in Rats Using LC-MS/MS. Molecules 2021, 26, 889. https://doi.org/10.3390/molecules26040889
Ayoub BM, Michel HE, Mowaka S, Hendy MS, Tadros MM. Repurposing of Omarigliptin as a Neuroprotective Agent Based on Docking with A2A Adenosine and AChE Receptors, Brain GLP-1 Response and Its Brain/Plasma Concentration Ratio after 28 Days Multiple Doses in Rats Using LC-MS/MS. Molecules. 2021; 26(4):889. https://doi.org/10.3390/molecules26040889
Chicago/Turabian StyleAyoub, Bassam M., Haidy E. Michel, Shereen Mowaka, Moataz S. Hendy, and Mariam M. Tadros. 2021. "Repurposing of Omarigliptin as a Neuroprotective Agent Based on Docking with A2A Adenosine and AChE Receptors, Brain GLP-1 Response and Its Brain/Plasma Concentration Ratio after 28 Days Multiple Doses in Rats Using LC-MS/MS" Molecules 26, no. 4: 889. https://doi.org/10.3390/molecules26040889