Crosstalk of the Caspase Family and Mammalian Target of Rapamycin Signaling

Abstract
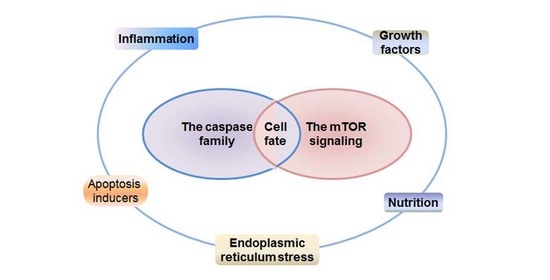
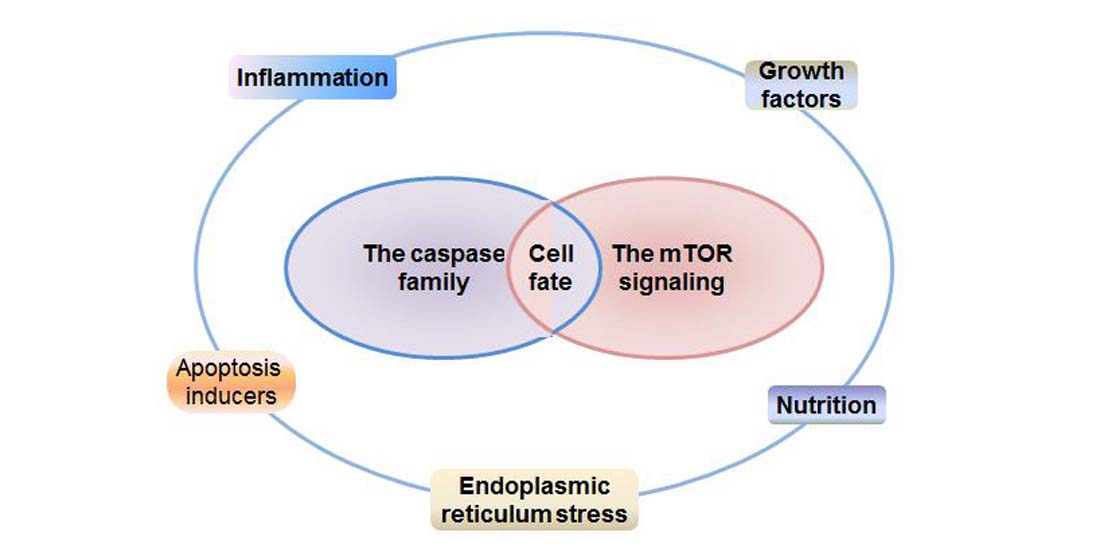
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Caspase Family
2.1. Apoptosis Response
2.2. Inflammation and Endoplasmic Reticulum (ER) Stress Response
3. The Multiple Functions of mTOR Signaling in Cells
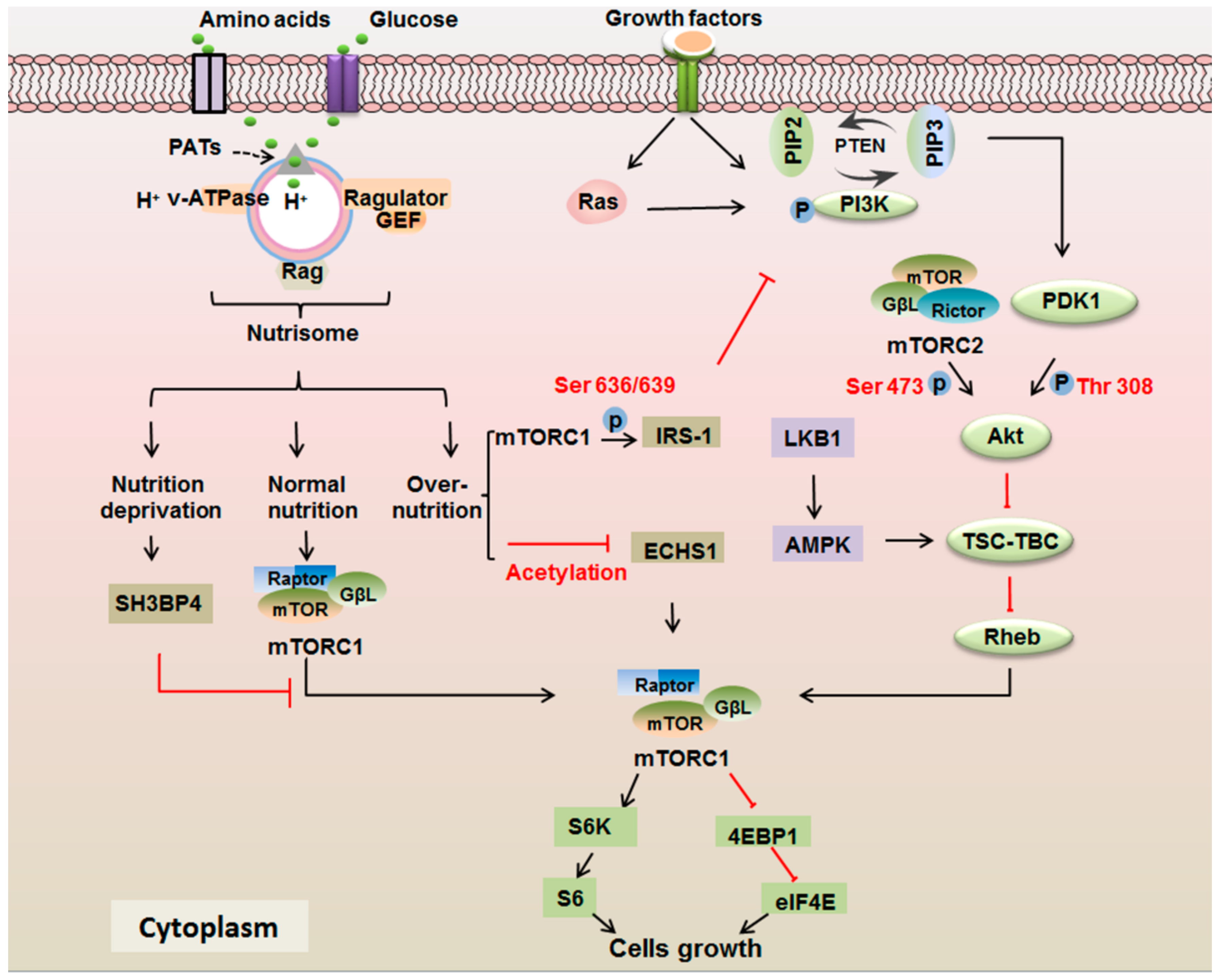
3.1. mTOR Signaling Responds to Growth Factors
3.2. mTOR Signaling Responds to the Nutrients
4. The Interplay between mTOR Signaling and the Caspase Family
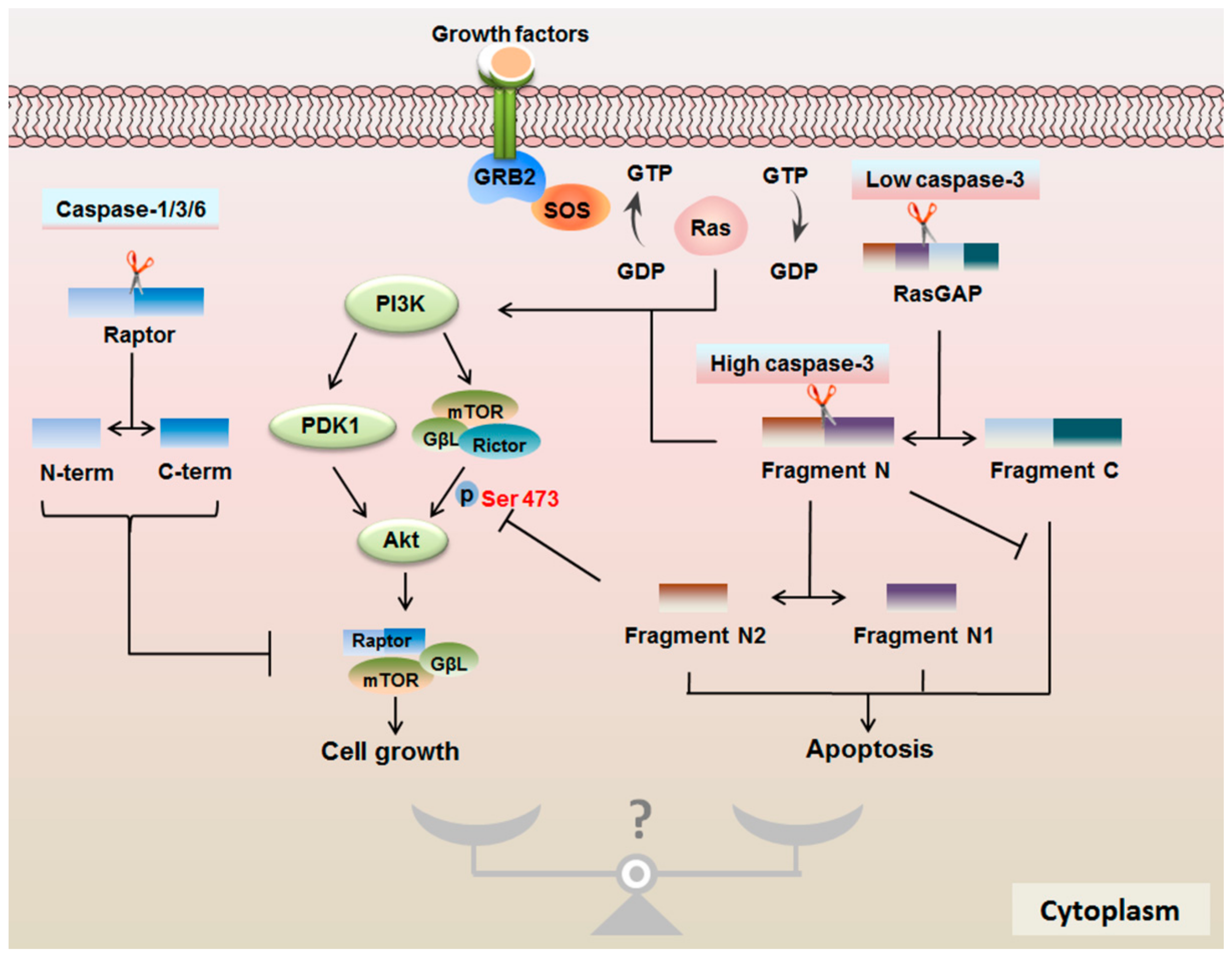
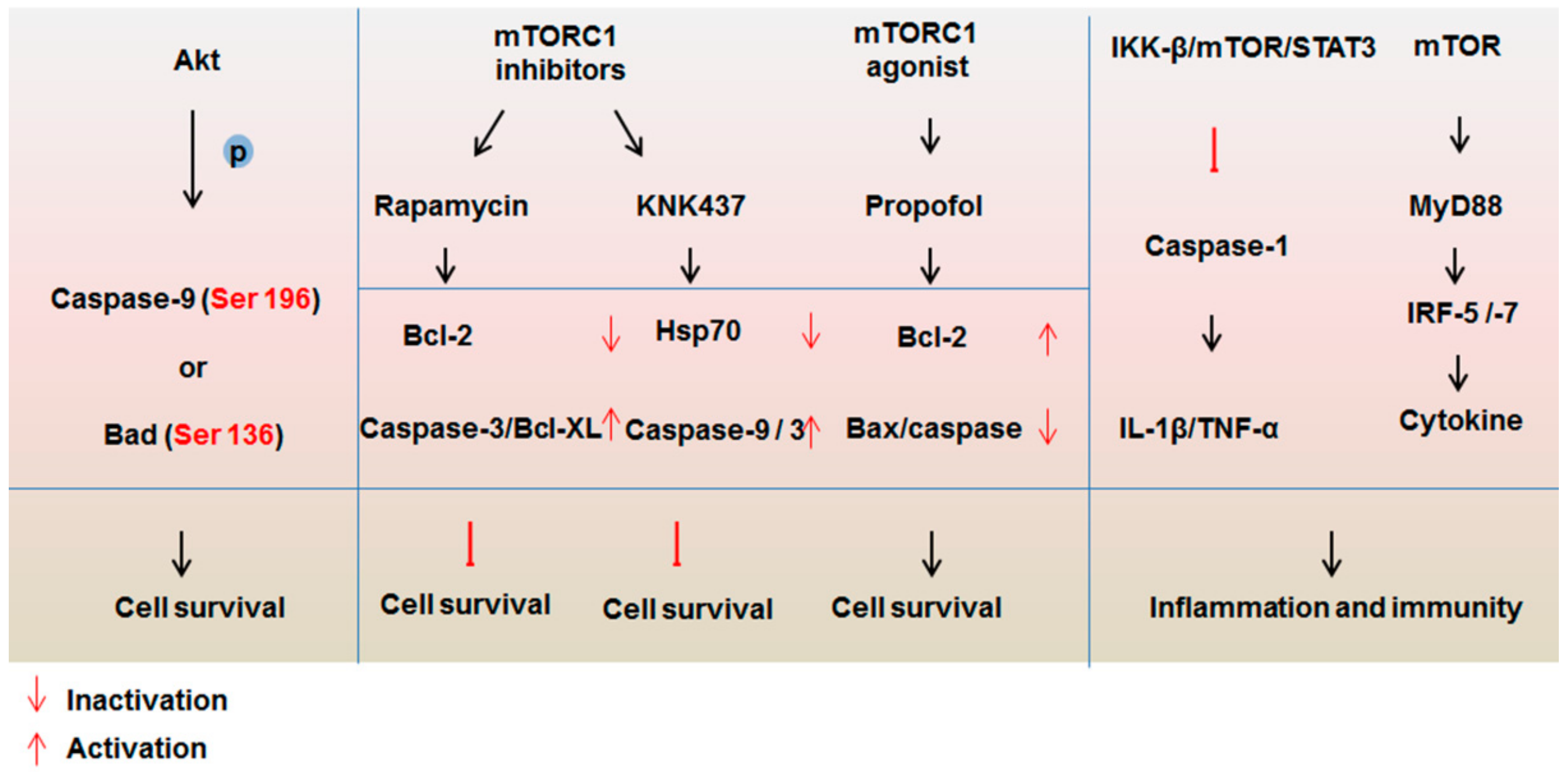
4.1. Impact of mTOR Signaling on the Caspase Family
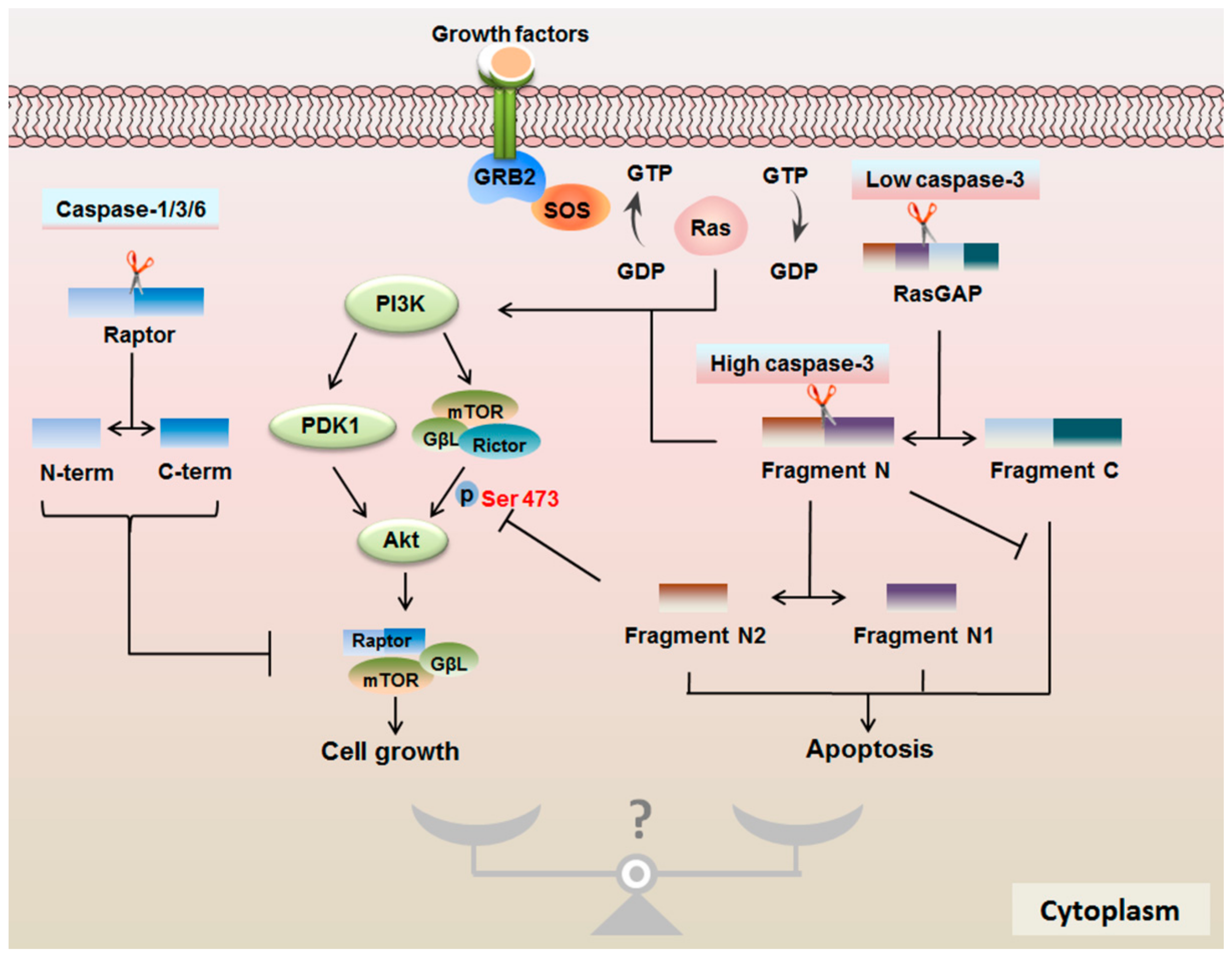
4.2. Protease Properties of Caspases are Important for mTOR Signaling
4.3. RasGAP Controls the Cell Fate in a Caspase-3 Activity-Dependent Manner
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blokhina, O.; Virolainen, E.; Fagerstedt, K.V. Antioxidants, oxidative damage and oxygen deprivation stress: A review. Ann. Bot. 2003, 91, 179–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seematter, G.; Binnert, C.; Martin, J.L.; Tappy, L. Relationship between stress, inflammation and metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Viganò, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, J. Programmed cell death: The paths to suicide. Trends Neurosci. 1992, 15, 278–280. [Google Scholar] [CrossRef]
- Alayev, A.; Holz, M.K. mTOR signaling for biological control and cancer. J. Cell. Physiol. 2013, 228, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, F.; Heit, A.; Dreher, S.; Eisenacher, K.; Mages, J.; Haas, T.; Krug, A.; Janssen, K.P.; Kirschning, C.J.; Wagner, H. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur. J. Immunol. 2008, 38, 2981–2992. [Google Scholar] [CrossRef]
- Yang, J.Y.; Widmann, C. Antiapoptotic signaling generated by caspase-induced cleavage of RasGAP. Mol. Cell. Biol. 2001, 21, 5346–5358. [Google Scholar] [CrossRef] [Green Version]
- Poreba, M.; Strozyk, A.; Salvesen, G.S.; Drag, M. Caspase substrates and inhibitors. Cold Spring Harb. Perspect. Biol. 2013, 5, a008680. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999, 6, 1028–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhivotovsky, B.; Orrenius, S. Caspase-2 function in response to DNA damage. Biochem. Biophys. Res. Commun. 2005, 331, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.H. Structural Features of Caspase-Activating Complexes. Int. J. Mol. Sci. 2012, 13, 4807–4818. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [Green Version]
- Fiandalo, M.V.; Kyprianou, N. Caspase control: Protagonists of cancer cell apoptosis. Exp. Oncol. 2012, 34, 165–175. [Google Scholar]
- Cory, S.; Huang, D.C.; Adams, J.M. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 8590–8607. [Google Scholar] [CrossRef] [Green Version]
- Siu, W.P.; Pun, P.B.; Latchoumycandane, C.; Boelsterli, U.A. Bax-mediated mitochondrial outer membrane permeabilization (MOMP), distinct from the mitochondrial permeability transition, is a key mechanism in diclofenac-induced hepatocyte injury: Multiple protective roles of cyclosporin A. Toxicol. Appl. Pharm. 2008, 227, 451–461. [Google Scholar] [CrossRef]
- Scott, F.L.; Denault, J.B.; Riedl, S.J.; Shin, H.; Renatus, M.; Salvesen, G.S. XIAP inhibits caspase-3 and -7 using two binding sites: Evolutionarily conserved mechanism of IAPs. EMBO J. 2005, 24, 645–655. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Hegde, R.; Saleh, A.; Datta, P.; Shiozaki, E.; Chai, J.; Lee, R.A.; Robbins, P.D.; Fernandes-Alnemri, T.; Shi, Y.; et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001, 410, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Kruidering, M.; Evan, G.I. Caspase-8 in apoptosis: The beginning of “the end”? Iubmb Life 2000, 50, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Death receptor-induced cell killing. Cell Signal 2004, 16, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chun, H.J.; Wong, W.; Spencer, D.M.; Lenardo, M.J. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 13884–13888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Lee, Y.; Kim, W.; Ko, H.; Choi, H.; Kim, K. Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 2005, 24, 3532–3542. [Google Scholar] [CrossRef] [Green Version]
- Paroni, G.; Henderson, C.; Schneider, C.; Brancolini, C. Caspase-2-induced apoptosis is dependent on caspase-9, but its processing during UV- or tumor necrosis factor-dependent cell death requires caspase-3. J. Biol. Chem. 2001, 276, 21907–21915. [Google Scholar] [CrossRef] [Green Version]
- Lassus, P.; Opitz-Araya, X.; Lazebnik, Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 2002, 297, 1352–1354. [Google Scholar] [CrossRef]
- Oliver, T.G.; Meylan, E.; Chang, G.P.; Xue, W.; Burke, J.R.; Humpton, T.J.; Hubbard, D.; Bhutkar, A.; Jacks, T. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol. Cell 2011, 43, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Tinel, A.; Janssens, S.; Lippens, S.; Cuenin, S.; Logette, E.; Jaccard, B.; Quadroni, M.; Tschopp, J. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. EMBO J. 2007, 26, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Paroni, G.; Henderson, C.; Schneider, C.; Brancolini, C. Caspase-2 can trigger cytochrome C release and apoptosis from the nucleus. J. Biol. Chem. 2002, 277, 15147–15161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Zafar, I.; Kim, J.; Schrier, R.W.; Edelstein, C.L. Caspase-3 gene deletion prolongs survival in polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Ruchaud, S.; Korfali, N.; Villa, P.; Kottke, T.J.; Dingwall, C.; Kaufmann, S.H.; Earnshaw, W.C. Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 2002, 21, 1967–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.; Sung, B.J.; Cho, Y.S.; Kim, H.J.; Ha, N.C.; Hwang, J.I.; Chung, C.W.; Jung, Y.K.; Oh, B.H. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry 2001, 40, 1117–1123. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Wanderer, A.A. Inflammasome and IL-1beta-mediated disorders. Curr. Allergy Asthma Rep. 2010, 10, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Poyet, J.L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, V.; LeBlanc, A. Inflammasome-mediated activation of caspase-1 and caspase-6 in primary human neurons. Alzheimer’s Dement. 2012, 8, P303. [Google Scholar] [CrossRef]
- Sagulenko, V.; Vitak, N.; Vajjhala, P.R.; Vince, J.E.; Stacey, K.J. Caspase-1 Is an Apical Caspase Leading to Caspase-3 Cleavage in the AIM2 Inflammasome Response, Independent of Caspase-8. J. Mol. Biol. 2018, 430, 238–247. [Google Scholar] [CrossRef]
- Bian, Z.M.; Elner, S.G.; Khanna, H.; Murga-Zamalloa, C.A.; Patil, S.; Elner, V.M. Expression and functional roles of caspase-5 in inflammatory responses of human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8646–8656. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid-Burgk, J.L.; Gaidt, M.M.; Schmidt, T.; Ebert, T.S.; Bartok, E.; Hornung, V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol. 2015, 45, 2911–2917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, K.; Li, Z.; Guo, B. ER Stress-induced Inflammasome Activation Contributes to Hepatic Inflammation and Steatosis. J. Clin. Cell Immunol. 2016, 7, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [Green Version]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef] [Green Version]
- Cheung, H.H.; Lynn Kelly, N.; Liston, P.; Korneluk, R.G. Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: A role for the IAPs. Exp. Cell Res. 2006, 312, 2347–2357. [Google Scholar] [CrossRef]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef]
- Upton, J.P.; Austgen, K.; Nishino, M.; Coakley, K.M.; Hagen, A.; Han, D.; Papa, F.R.; Oakes, S.A. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol. Cell. Biol. 2008, 28, 3943–3951. [Google Scholar] [CrossRef] [Green Version]
- Sandow, J.J.; Dorstyn, L.; O’Reilly, L.A.; Tailler, M.; Kumar, S.; Strasser, A.; Ekert, P.G. ER stress does not cause upregulation and activation of caspase-2 to initiate apoptosis. Cell Death Differ. 2014, 21, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.M.; Elner, S.G.; Elner, V.M. Dual involvement of caspase-4 in inflammatory and ER stress-induced apoptotic responses in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2009, 50, 6006–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamuro, A.; Kishino, T.; Ohshima, Y.; Yoshioka, Y.; Kimura, T.; Kasai, A.; Maeda, S. Caspase-4 Directly Activates Caspase-9 in Endoplasmic Reticulum Stress-Induced Apoptosis in SH-SY5Y Cells. J. Pharm. Sci. 2011, 115, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wei, J.; Li, Y.; He, X.; Zhou, Q.; Yan, J.; Zhang, J.; Liu, Y.; Liu, Y.; Shu, H.B. Transmembrane Protein 214 (TMEM214) mediates endoplasmic reticulum stress-induced caspase 4 enzyme activation and apoptosis. J. Biol. Chem. 2013, 288, 17908–17917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Glab, J.A.; Doerflinger, M.; Nedeva, C.; Jose, I.; Mbogo, G.W.; Paton, J.C.; Paton, A.W.; Kueh, A.J.; Herold, M.J.; Huang, D.C.S.; et al. DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 2017, 24, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Pinedo, C.; López-Rivas, A. A role for caspase-8 and TRAIL-R2/DR5 in ER-stress-induced apoptosis. Cell Death Differ. 2017, 25, 226. [Google Scholar] [CrossRef]
- Lam, M.; Lawrence, D.A.; Ashkenazi, A.; Walter, P. Confirming a critical role for death receptor 5 and caspase-8 in apoptosis induction by endoplasmic reticulum stress. Cell Death Differ. 2018, 25, 1530–1531. [Google Scholar] [CrossRef] [Green Version]
- Martín-Pérez, R.; Palacios, C.; Yerbes, R.; Cano-González, A.; Iglesias-Serret, D.; Gil, J.; Reginato, M.J.; López-Rivas, A. Activated ERBB2/HER2 Licenses Sensitivity to Apoptosis upon Endoplasmic Reticulum Stress through a PERK-Dependent Pathway. Cancer Res. 2014, 74, 1766–1777. [Google Scholar] [CrossRef] [Green Version]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Glidden, E.J.; Gray, L.G.; Vemuru, S.; Li, D.; Harris, T.E.; Mayo, M.W. Multiple site acetylation of Rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J. Biol. Chem. 2012, 287, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; White, E.P.; Mehnert, J.M. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS ONE 2013, 8, e55096. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Golub, T.R.; Sabatini, D.M. The immunosuppressant rapamycin mimics a starvation-like signal distinct from amino acid and glucose deprivation. Mol. Cell. Biol. 2002, 22, 5575–5584. [Google Scholar] [CrossRef] [Green Version]
- Ebner, M.; Sinkovics, B.; Szczygiel, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell. Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, R.C.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Sun, X.J.; Wang, L.M.; Zhang, Y.; Yenush, L.; Myers, M.G., Jr.; Glasheen, E.; Lane, W.S.; Pierce, J.H.; White, M.F. Role of IRS-2 in insulin and cytokine signalling. Nature 1995, 377, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Geer, P.; Henkemeyer, M.; Jacks, T.; Pawson, T. Aberrant Ras regulation and reduced p190 tyrosine phosphorylation in cells lacking p120-Gap. Mol. Cell. Biol. 1997, 17, 1840–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Vogt, P.K. Hot-spot mutations in p110alpha of phosphatidylinositol 3-kinase (pI3K): Differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle 2010, 9, 596–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Inoki, K.; Opland, D.; Munzberg, H.; Villanueva, E.C.; Faouzi, M.; Ikenoue, T.; Kwiatkowski, D.J.; Macdougald, O.A.; Myers, M.G., Jr.; et al. Critical roles for the TSC-mTOR pathway in beta-cell function. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1013–E1022. [Google Scholar] [CrossRef] [Green Version]
- Santiago Lima, A.J.; Hoogeveen-Westerveld, M.; Nakashima, A.; Maat-Kievit, A.; van den Ouweland, A.; Halley, D.; Kikkawa, U.; Nellist, M. Identification of regions critical for the integrity of the TSC1-TSC2-TBC1D7 complex. PLoS ONE 2014, 9, e93940. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS Motif-Mediated Raptor Binding Regulates 4E-BP1 Multisite Phosphorylation and Function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Thoreen, C.C. Many roads from mTOR to eIF4F. Biochem. Soc. Trans. 2013, 41, 913–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, M.; De Rossi, M.; Barberi, L.; Musaro, A. IL-6 impairs myogenic differentiation by downmodulation of p90RSK/eEF2 and mTOR/p70S6K axes, without affecting AKT activity. BioMed Res. Int. 2014, 2014, 206026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Efeyan, A.; Zoncu, R.; Chang, S.; Gumper, I.; Snitkin, H.; Wolfson, R.L.; Kirak, O.; Sabatini, D.D.; Sabatini, D.M. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 2013, 493, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Guan, K.L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci 2013, 38, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Efeyan, A.; Sabatini, D.M. Nutrients and growth factors in mTORC1 activation. Biochem. Soc. Trans. 2013, 41, 902–905. [Google Scholar] [CrossRef]
- Fan, S.J.; Goberdhan, D.C.I. PATs and SNATs: Amino Acid Sensors in Disguise. Front. Pharm. 2018, 9, 640. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Saqcena, M.; Menon, D.; Patel, D.; Mukhopadhyay, S.; Chow, V.; Foster, D.A. Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS ONE 2013, 8, e74157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.M.; Stone, M.; Hwang, T.H.; Kim, Y.G.; Dunlevy, J.R.; Griffin, T.J.; Kim, D.H. SH3BP4 is a negative regulator of amino acid-Rag GTPase-mTORC1 signaling. Mol. Cell 2012, 46, 833–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Bukhari, S.A.; Ali, M.; Lee, H.W. Upstream signalling of mTORC1 and its hyperactivation in type 2 diabetes (T2D). BMB Rep. 2017, 50, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.K.; Qu, Y.Y.; Lin, Y.; Wu, X.H.; Chen, H.Z.; Wang, X.; Zhou, K.Q.; Wei, Y.; Guo, F.; Yao, C.F.; et al. Enoyl-CoA hydratase-1 regulates mTOR signaling and apoptosis by sensing nutrients. Nat. Commun. 2017, 8, 464. [Google Scholar] [CrossRef] [PubMed]
- Tzatsos, A.; Kandror, K.V. Nutrients Suppress Phosphatidylinositol 3-Kinase/Akt Signaling via Raptor-Dependent mTOR-Mediated Insulin Receptor Substrate 1 Phosphorylation. Mol. Cell. Biol. 2006, 26, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.P.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.B.; Ellingson, W.J.; Lamb, J.D.; Chesser, D.G.; Winder, W.W. Long-chain acyl-CoA esters inhibit phosphorylation of AMP-activated protein kinase at threonine-172 by LKB1/STRAD/MO25. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1055–E1061. [Google Scholar] [CrossRef] [Green Version]
- Lacher, M.D.; Pincheira, R.; Zhu, Z.; Camoretti-Mercado, B.; Matli, M.; Warren, R.S.; Castro, A.F. Rheb activates AMPK and reduces p27Kip1 levels in Tsc2-null cells via mTORC1-independent mechanisms: Implications for cell proliferation and tumorigenesis. Oncogene 2010, 29, 6543–6556. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef] [Green Version]
- Krogsgaard, M.; Li, Q.J.; Sumen, C.; Huppa, J.B.; Huse, M.; Davis, M.M. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature 2005, 434, 238–243. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Chen, Z.; Yang, L.; Xing, X.; Ma, X.; Yang, Z. Both PI3K- and mTOR-signaling pathways take part in CVB3-induced apoptosis of Hela cells. DNA Cell Biol. 2013, 32, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; Liu, J.J.; Lu, M.Q.; Cai, C.J.; Yang, Y.; Li, H.; Xu, C.; Chen, G.H. Rapamycin inhibits cell growth by induction of apoptosis on hepatocellular carcinoma cells in vitro. Transpl. Immunol. 2007, 17, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Calastretti, A.; Rancati, F.; Ceriani, M.C.; Asnaghi, L.; Canti, G.; Nicolin, A. Rapamycin increases the cellular concentration of the BCL-2 protein and exerts an anti-apoptotic effect. Eur. J. Cancer 2001, 37, 2121–2128. [Google Scholar] [CrossRef]
- Inoue, H.; Uyama, T.; Hayashi, J.; Watanabe, A.; Kobayashi, K.; Tadokoro, T.; Yamamoto, Y. N-Formyl-3,4-methylenedioxy-benzylidene-gamma-butyrolaetam, KNK437 induces caspase-3 activation through inhibition of mTORC1 activity in Cos-1 cells. Biochem. Biophys. Res. Commun. 2010, 395, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xia, Y.; Xu, Z.; Deng, X. Propofol Suppressed Hypoxia/Reoxygenation-Induced Apoptosis in HBVSMC by Regulation of the Expression of Bcl-2, Bax, Caspase3, Kir6.1, and p-JNK. Oxid. Med. Cell Longev. 2016, 2016, 1518738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Tintignac, L.A.; Baudry, A.; Hemmings, B.A. Protein kinase B/Akt at a glance. J. Cell Sci. 2005, 118, 5675–5678. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Juin, P.; Hueber, A.O.; Littlewood, T.; Evan, G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999, 13, 1367–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rano, T.A.; Timkey, T.; Peterson, E.P.; Rotonda, J.; Nicholson, D.W.; Becker, J.W.; Chapman, K.T.; Thornberry, N.A. A combinatorial approach for determining protease specificities: Application to interleukin-1beta converting enzyme (ICE). Chem. Biol. 1997, 4, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, H.; Kodandapani, L.; Mittl, P.R.; Marco, S.D.; Krebs, J.F.; Wu, J.C.; Tomaselli, K.J.; Grütter, M.G. The three-dimensional structure of caspase-8: An initiator enzyme in apoptosis. Structure 1999, 7, 1125–1133. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, R.; Jelakovic, S.; Campbell, A.J.; Li, Z.Z.; Asgian, J.L.; Powers, J.C.; Grütter, M.G. Exploring the S4 and S1 prime subsite specificities in caspase-3 with aza-peptide epoxide inhibitors. Biochemistry 2006, 45, 9059–9067. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Mittl, P.R.; Jelakovic, S.; Grutter, M.G. Extended substrate recognition in caspase-3 revealed by high resolution X-ray structure analysis. J. Mol. Biol. 2006, 359, 1378–1388. [Google Scholar] [CrossRef]
- Seaman, J.E.; Julien, O.; Lee, P.S.; Rettenmaier, T.J.; Thomsen, N.D.; Wells, J.A. Cacidases: Caspases can cleave after aspartate, glutamate and phosphoserine residues. Cell Death Differ. 2016, 23, 1717–1726. [Google Scholar] [CrossRef] [Green Version]
- Desroches, A.; Denault, J.B. Caspase-7 uses RNA to enhance proteolysis of poly(ADP-ribose) polymerase 1 and other RNA-binding proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 21521–21528. [Google Scholar] [CrossRef] [Green Version]
- Ginalski, K.; Zhang, H.; Grishin, N.V. Raptor protein contains a caspase-like domain. Trends Biochem. Sci 2004, 29, 522–524. [Google Scholar] [CrossRef]
- Martin, R.; Desponds, C.; Eren, R.O.; Quadroni, M.; Thome, M.; Fasel, N. Caspase-mediated cleavage of raptor participates in the inactivation of mTORC1 during cell death. Cell Death Discov. 2016, 2, 16024. [Google Scholar] [CrossRef] [Green Version]
- Elia, A.; Henry-Grant, R.; Adiseshiah, C.; Marboeuf, C.; Buckley, R.J.; Clemens, M.J.; Mudan, S.; Pyronnet, S. Implication of 4E-BP1 protein dephosphorylation and accumulation in pancreatic cancer cell death induced by combined gemcitabine and TRAIL. Cell Death Dis. 2017, 8, 3204. [Google Scholar] [CrossRef] [PubMed]
- Widmann, C.; Gibson, S.; Johnson, G.L. Caspase-dependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti-apoptotic signals. J. Biol. Chem. 1998, 273, 7141–7147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adapala, N.S.; Barbe, M.F.; Tsygankov, A.Y.; Lorenzo, J.A.; Sanjay, A. Loss of Cbl-PI3K interaction enhances osteoclast survival due to p21-Ras mediated PI3K activation independent of Cbl-b. J. Cell Biochem. 2014, 115, 1277–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, M.F.; Koch, C.A.; Anderson, D.; Ellis, C.; England, L.; Martin, G.S.; Pawson, T. Src homology region 2 domains direct protein-protein interactions in signal transduction. Proc. Natl. Acad. Sci. USA 1990, 87, 8622–8626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulski, N.; Hall, M.N. TOR complex 2: A signaling pathway of its own. Trends Biochem. Sci. 2009, 34, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Datta, K.; Franke, T.F.; Chan, T.O.; Makris, A.; Yang, S.I.; Kaplan, D.R.; Morrison, D.K.; Golemis, E.A.; Tsichlis, P.N. AH/PH domain-mediated interaction between Akt molecules and its potential role in Akt regulation. Mol. Cell. Biol. 1995, 15, 2304–2310. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Walicki, J.; Abderrahmani, A.; Cornu, M.; Waeber, G.; Thorens, B.; Widmann, C. Expression of an uncleavable N-terminal RasGAP fragment in insulin-secreting cells increases their resistance toward apoptotic stimuli without affecting their glucose-induced insulin secretion. J. Biol. Chem. 2005, 280, 32835–32842. [Google Scholar] [CrossRef] [Green Version]
- Pamonsinlapatham, P.; Hadj-Slimane, R.; Lepelletier, Y.; Allain, B.; Toccafondi, M.; Garbay, C.; Raynaud, F. p120-Ras GTPase activating protein (RasGAP): A multi-interacting protein in downstream signaling. Biochimie 2009, 91, 320–328. [Google Scholar] [CrossRef]
- Yang, J.Y.; Michod, D.; Walicki, J.; Murphy, B.M.; Kasibhatla, S.; Martin, S.J.; Widmann, C. Partial cleavage of RasGAP by caspases is required for cell survival in mild stress conditions. Mol. Cell. Biol. 2004, 24, 10425–10436. [Google Scholar] [CrossRef] [Green Version]
- Cailliau, K.; Lescuyer, A.; Burnol, A.F.; Cuesta-Marban, A.; Widmann, C.; Browaeys-Poly, E. RasGAP Shields Akt from Deactivating Phosphatases in Fibroblast Growth Factor Signaling but Loses This Ability Once Cleaved by Caspase-3. J. Biol. Chem. 2015, 290, 19653–19665. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Walicki, J.; Michod, D.; Dubuis, G.; Widmann, C. Impaired Akt activity down-modulation, caspase-3 activation, and apoptosis in cells expressing a caspase-resistant mutant of RasGAP at position 157. Mol. Biol. Cell 2005, 16, 3511–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, H.; Kuroda, S.; Tanaka, M.; Matsuzaki, H.; Ono, Y.; Kameyama, K.; Haga, T.; Kikkawa, U. Molecular cloning and characterization of a new member of the RAC protein kinase family: Association of the pleckstrin homology domain of three types of RAC protein kinase with protein kinase C subspecies and beta gamma subunits of G proteins. Biochem. Biophys. Res. Commun. 1995, 216, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Vanli, G.; Peltzer, N.; Dubuis, G.; Widmann, C. The activity of the anti-apoptotic fragment generated by the caspase-3/p120 RasGAP stress-sensing module displays strict Akt isoform specificity. Cell Signal 2014, 26, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Cron, P.; Thompson, V.; Good, V.M.; Hess, D.; Hemmings, B.A.; Barford, D. Molecular Mechanism for the Regulation of Protein Kinase B/Akt by Hydrophobic Motif Phosphorylation. Mol. Cell 2002, 9, 1227–1240. [Google Scholar] [CrossRef]
- Zhuravleva, E.; Tschopp, O.; Hemmings, B.A. Role of PKB/Akt in Liver Diseases. In Signaling Pathways in Liver Diseases; Springer: Berlin/Heidelberg, Germany, 2010; pp. 243–259. [Google Scholar] [CrossRef]
- Peltzer, N.; Vanli, G.; Yang, J.Y.; Widmann, C. Role of mTOR, Bad, and Survivin in RasGAP Fragment N-Mediated Cell Protection. PLoS ONE 2013, 8, e68123. [Google Scholar] [CrossRef]
- Bulat, N.; Jaccard, E.; Peltzer, N.; Khalil, H.; Yang, J.Y.; Dubuis, G.; Widmann, C. RasGAP-derived fragment N increases the resistance of beta cells towards apoptosis in NOD mice and delays the progression from mild to overt diabetes. PLoS ONE 2011, 6, e22609. [Google Scholar] [CrossRef]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lamb, R.; Hulit, J.; Howell, A.; Gandara, R.; Sartini, M.; Rubin, E.; Lisanti, M.P.; Sotgia, F. Mitochondrial dysfunction in breast cancer cells prevents tumor growth: Understanding chemoprevention with metformin. Cell Cycle 2013, 12, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Peltzer, N.; Walicki, J.; Yang, J.Y.; Dubuis, G.; Gardiol, N.; Held, W.; Bigliardi, P.; Marsland, B.; Liaudet, L.; et al. Caspase-3 protects stressed organs against cell death. Mol. Cell. Biol. 2012, 32, 4523–4533. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.; Xie, Y.; Si, J.; Gan, L.; Li, H.; Sun, C.; Di, C.; Zhang, J.; Huang, G.; Zhang, X.; et al. Crosstalk of the Caspase Family and Mammalian Target of Rapamycin Signaling. Int. J. Mol. Sci. 2021, 22, 817. https://doi.org/10.3390/ijms22020817
Yan J, Xie Y, Si J, Gan L, Li H, Sun C, Di C, Zhang J, Huang G, Zhang X, et al. Crosstalk of the Caspase Family and Mammalian Target of Rapamycin Signaling. International Journal of Molecular Sciences. 2021; 22(2):817. https://doi.org/10.3390/ijms22020817
Chicago/Turabian StyleYan, Junfang, Yi Xie, Jing Si, Lu Gan, Hongyan Li, Chao Sun, Cuixia Di, Jinhua Zhang, Guomin Huang, Xuetian Zhang, and et al. 2021. "Crosstalk of the Caspase Family and Mammalian Target of Rapamycin Signaling" International Journal of Molecular Sciences 22, no. 2: 817. https://doi.org/10.3390/ijms22020817