Some Aspects of Time-Reversal in Chemical Kinetics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- System view (I): level at which the statistics of the overall reacting flow can be investigated
- Macroscopic view (II): level at which the chemical kinetics is governed by macroscopic variables such as mass fractions, specific internal energy, and specific volume (or concentrations, temperature, add pressure), and which is described by large sets of elementary reactions
- Microscopic view (III): level at which elementary reactions occur. This level is governed by collisions and interactions of different molecules, and depends on the translational, rotational, vibrational, and electronic states of the molecules
- Sub-microscopic view (IV): This level describes the energies of the quantum states (inlet figure produced using the Wolfram Demonstrations project [8])
- Time reversal and anti-causality: How do the kinetics behave if the arrow of time reverses? (This question will not be discussed in this work.)
- –
- Is a purely macroscopic view possible?
- –
- Does the “negative time” scale in a way different from the regular time?
- –
- Is there a similarity in the hierarchical nature of the chemical kinetics?
- Time reversal and causality: Let us assume that we know the thermokinetic state of a system at . Then, can we determine the state that the system had at (with )?
- –
- Is there a limiting value for ?
- –
- How is the hierarchical nature of the chemical kinetics reflected by a time reversal?
- –
- For , the chemical kinetics yield the thermodynamic equilibrium. What happens for ?
2. The Macroscopic View
2.1. The Forward Behavior
- it is an -dimensional system of stiff nonlinear ordinary differential equations
- it is an initial value problem
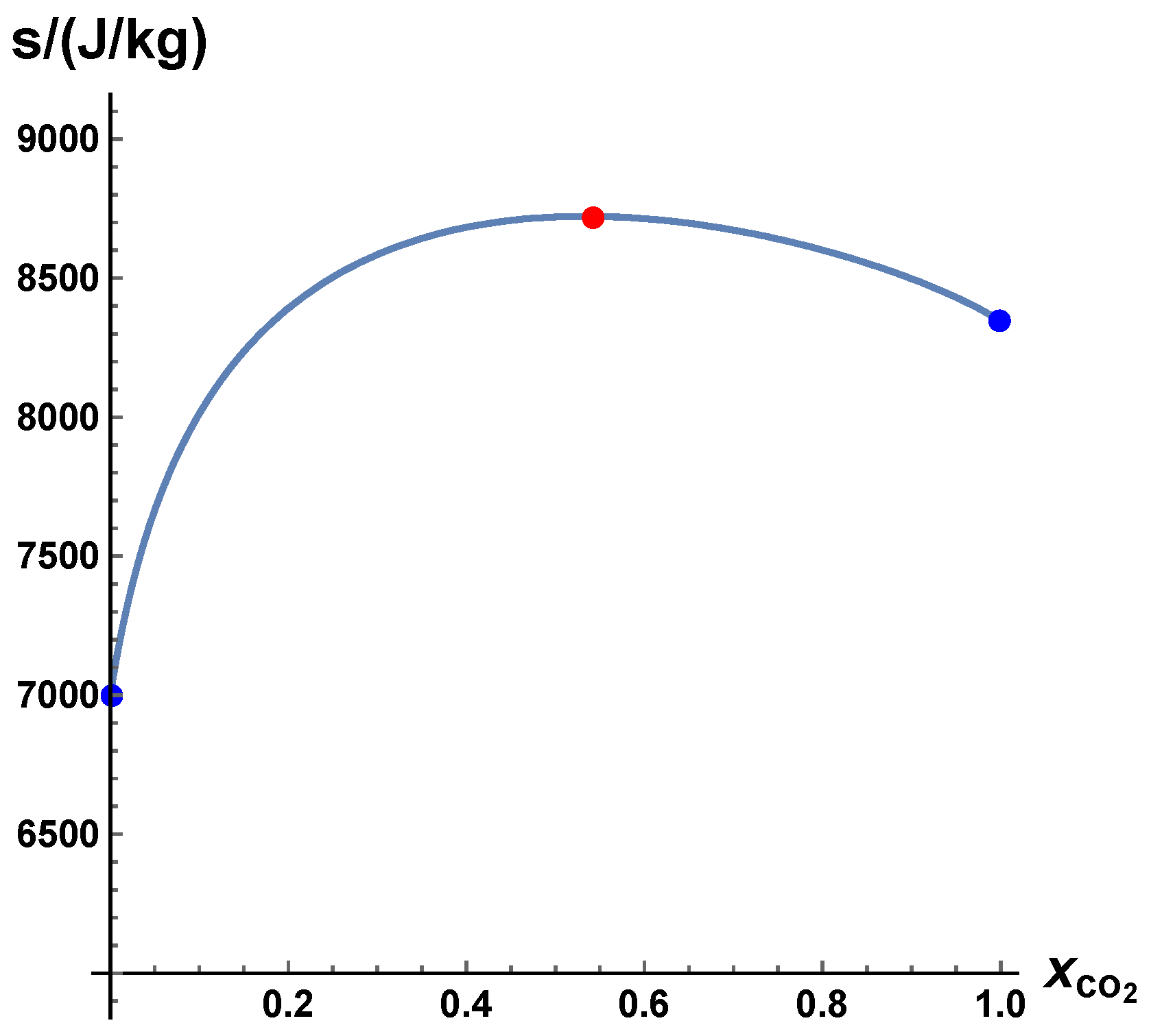
- the solution for yields the chemical equilibrium . This equilibrium value is a function of thermodynamic variables and the elemental composition ( elements), where denotes the vector of element mass fractions, which is a function of the specific mole numbers.
- assuming that the chemical rates are Lipschitz continuous, the Picard–Lindelöf theorem states that the initial value problem has a unique solution
- the system is “deterministic”
- the dynamics takes place in the so-called reaction space [19], which has a dimension of because the elements are conserved in chemical reaction
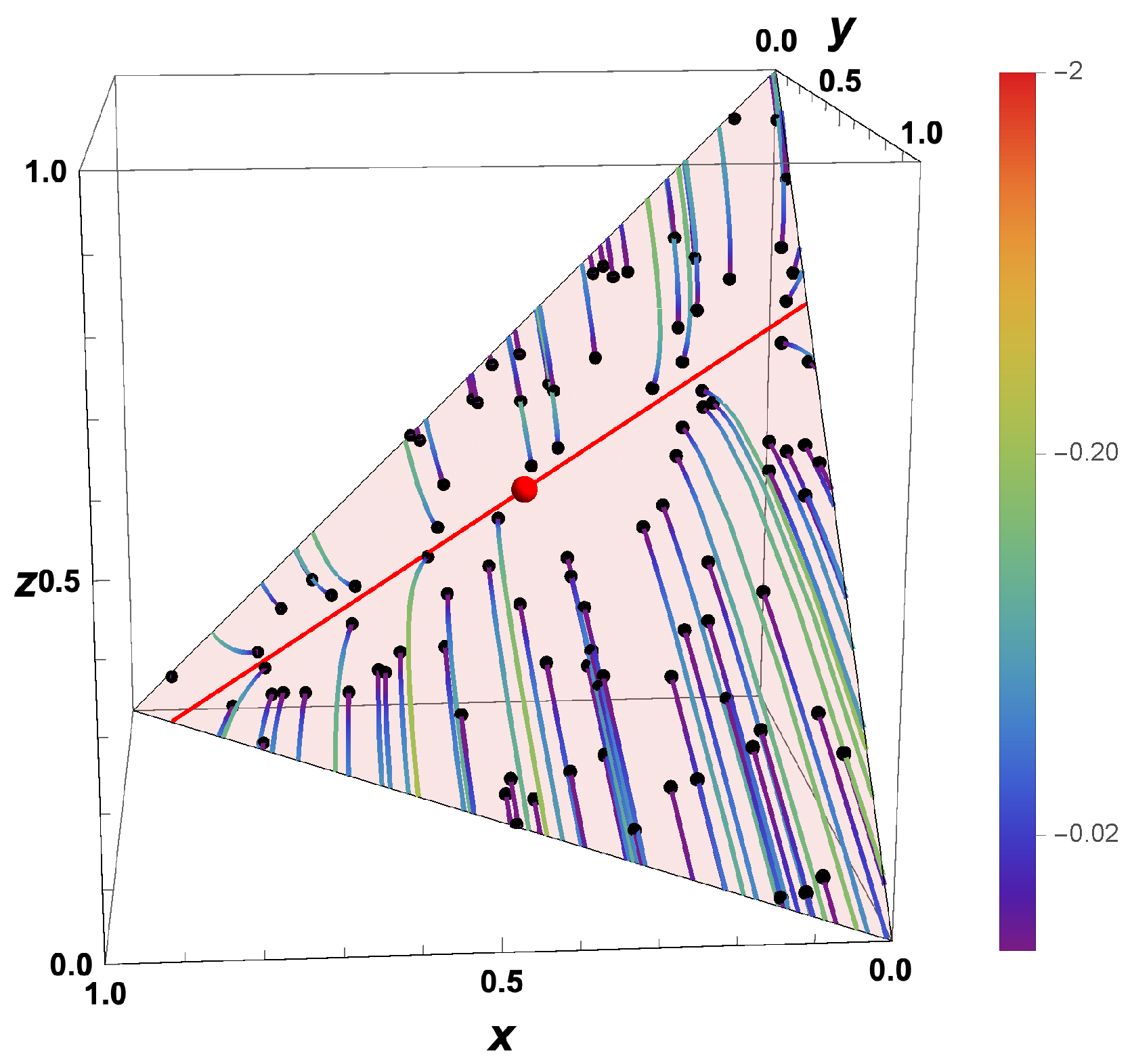
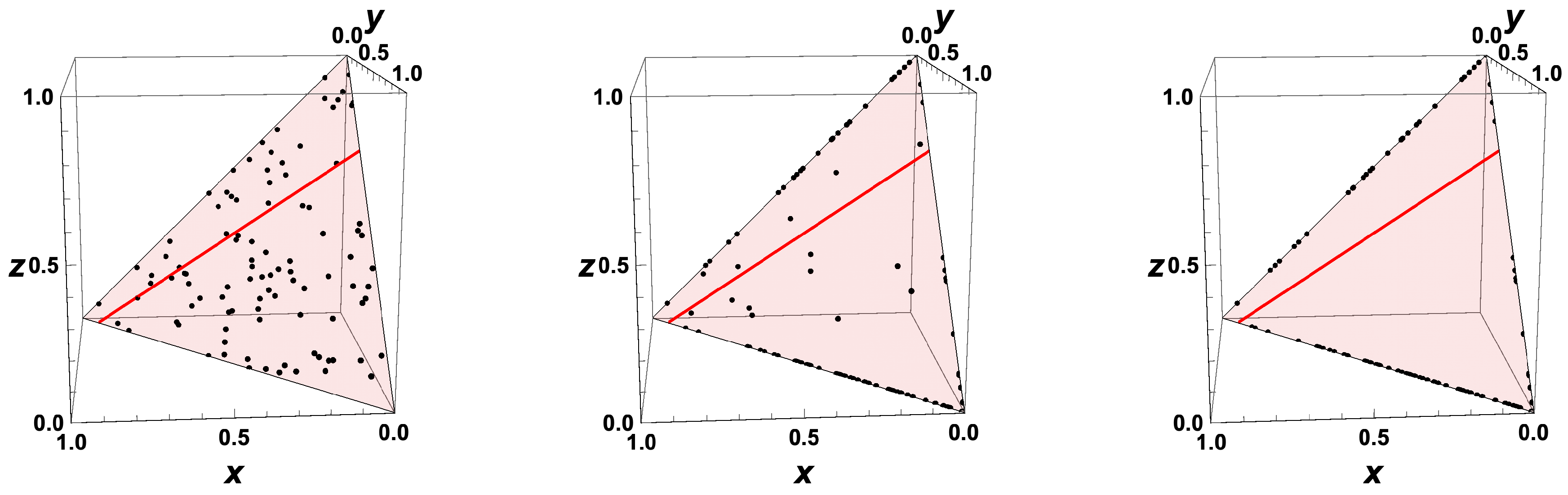
- The trajectories each start a given initial value
- The trajectories do not cross (an apparent crossing in the figure is only a result of the projection of the multi-dimensional composition space onto a three-dimensional space
- The trajectories evolve in time towards the equilibrium point
- There is a transient relaxation towards low-dimensional manifolds, it is observable in Figure 2 in the three-dimensional projection the final relaxation towards a two-dimensional manifold, then a one-dimensional manifold, and finally the zero-dimensional manifold (note that the trajectories are chosen such that they have the same equilibrium value).
- starting from very different initial conditions, the states get closer and closer, this means that the system seemingly loses information on its past.
2.2. Intrinsic Low-Dimensional Manifolds (ILDM)
- the existence of disparate time scales
- the fact that most time scales have a dissipative nature
- the existence of low-dimensional attractors in composition space (cf. also Figure 2)
A Simple Linear Test Case
2.3. The Behavior for Time Reversal
- What happens for ?
- Does that mean that there is a “final time” for time going backwards (in contrast to time in forward direction?
- Why is this time different for different initial conditions?
- Why don’t the governing equations give any information on the “real behavior”?
3. Statistics of Macroscopic Systems
3.1. General Evolution Equation for the Probability Density Function
3.2. The Consequences of Low-Dimensional Attracting Manifolds for the Evolution Equation for the Probability Density Function
3.3. Evolution Equation for the Probability Density Function of the Linear Test Case
3.4. The Behavior for Time Reversal
4. The Microscopic View
4.1. Unimolecular Reactions—Master Equation
- it is an initial value problem
- it is an -dimensional system of stiff ordinary differential equations with all eigenvalues of the Jacobian being negative
- The “rates” for the pure non-reacting system are related to each other using the law of detailed balance, i.e., they assume that the equilibrium obeys a Boltzmann-distribution [57]
- the system is “deterministic”
4.2. The Behavior for Time Reversal
5. The “Submicroscopic” Level
6. Bridging the Gap between Microscopic and Macroscopic Behavior
6.1. Microscopic Processes on a Macroscopic Level
6.2. The Behavior for Time Reversal
7. Discussion and Conclusions
- They are governed by large systems of stiff nonlinear ordinary differential equations
- The governing equations constitute an initial value problem
- The solutions are unique for given initial values
- The system is “deterministic”
- The trajectories do not cross in the thermokinetic space
- The trajectories evolve in time towards an equilibrium
- The disparity of time scales allows a decoupling of slow and fast processes
- Most time scales have a dissipative nature
- There is a transient relaxation towards low-dimensional manifolds
- Due to the deterministic nature of the kinetic equations, the question “What was the state of a system at a time before?” can be answered based on the knowledge of the state at a current time.
- The trajectories evolve in time towards the boundary of the allowed domain.
- No information is contained in the equation system for , where is the time when states reach the boundary of the allowed domain.
- Eigenvalues of the Jacobians or the GQL matrix) (corresponding to attractive processes) turn into eigenvalues (repulsive processes).
- Most eigenvalues of the backwards system are positive, corresponding to repulsive processes.
- The disparity of time scales allows a decoupling of slow and fast processes.
- An attracting low-dimensional manifold for the forward system turns into a separatrix for the backwards system.
- For , the final states bunch along the boundary, and which final state is obtained is governed by the initial position relative to the separatrix.
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| probability density function | |
| ILDM | intrinsic low-dimensional manifolds |
| GQL | global quasi-linearization |
Appendix A. Eigen System and Solution for the Simple Linear Test Case
References
- Hirschfelder, J.; Curtiss, C.; Bird, R. The Molecular Theory of Gases and Liquids; Raymond F. Boyer Library Collection; Wiley: New York, NY, USA, 1954. [Google Scholar]
- Hirschfelder, J.; Curtiss, C.; Campbell, D.E. The theory of flames and detonations. Symp. (Int.) Combust. 1953, 4, 190–211. [Google Scholar] [CrossRef]
- Bird, R.B.; Stewart, W.E.; Lightfoot, E.N. Transport Phenomena; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Christov, S.G. Collision Theory and Statistical Theory of Chemical Reactions; Springer: Berlin/Heidelberg, Germany, 1980. [Google Scholar] [CrossRef]
- Bao, J.L.; Truhlar, D.G. Variational transition state theory: Theoretical framework and recent developments. Chem. Soc. Rev. 2017, 46, 7548–7596. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, M. Statistical Rate Theory in Combustion: An Operational Approach. In Cleaner Combustion: Developing Detailed Chemical Kinetic Models; Battin-Leclerc, F., Simmie, J.M., Blurock, E., Eds.; Springer: London, UK, 2013; pp. 549–576. [Google Scholar] [CrossRef]
- Fernández-Ramos, A.; Miller, J.A.; Klippenstein, S.J.; Truhlar, D.G. Modeling the Kinetics of Bimolecular Reactions. Chem. Rev. 2006, 106, 4518–4584. [Google Scholar] [CrossRef] [PubMed]
- Gsaller, G. Energy-Level Diagrams and Molecular Orbitals for Conjugated Polyenes. 2014. Available online: http://demonstrations.wolfram.com/EnergyLevelDiagramsAndMolecularOrbitalsForConjugatedPolyenes/ (accessed on 11 November 2020).
- Gordiets, B.F.; Osipov, A.I.; Stupochenko, E.V.; Shelepin, L.A. Vibrational Relaxation in gases and molecular lasers. Sov. Phys. Uspekhi 1973, 15, 759–785. [Google Scholar] [CrossRef]
- Riedel, U.; Maas, U.; Warnatz, J. Detailed Numerical Modeling of Chemical and Thermal Nonequilibrium in Hypersonic Flows. IMPACT Comput. Sci. Eng. 1993, 5, 20–52. [Google Scholar] [CrossRef]
- Skrebkov, O.V. Vibrational Nonequilibrium in the Hydrogen-Oxygen Reaction at Different Temperatures. J. Mod. Phys. 2014, 05, 1806–1829. [Google Scholar] [CrossRef]
- Klimenko, A.Y. Mixing, tunnelling and the direction of time in the context of Reichenbach’s principles. arXiv 2019, arXiv:2001.00527. [Google Scholar]
- Klimenko, A.; Maas, U. One Antimatter—Two Possible Thermodynamics. Entropy 2014, 16, 1191–1210. [Google Scholar] [CrossRef]
- Klimenko, A. Kinetics of Interactions of Matter, Antimatter and Radiation Consistent with Antisymmetric (CPT-Invariant) Thermodynamics. Entropy 2017, 19, 202. [Google Scholar] [CrossRef]
- Maas, U. Efficient calculation of intrinsic low-dimensional manifolds for the simplification of chemical kinetics. Comput. Vis. Sci. 1998, 1, 69–81. [Google Scholar] [CrossRef]
- Lewis, G.N. A New Principle of Equilibrium. Proc. Natl. Acad. Sci. USA 1925, 11, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Wegscheider, R. Über simultane Gleichgewichte und die Beziehungen zwischen Thermodynamik und Reactionskinetik homogener Systeme. Monatshefte Chem. 1901, 22, 849–906. [Google Scholar] [CrossRef]
- Gorban, A. Detailed balance in micro- and macrokinetics and micro-distinguishability of macro-processes. Results Phys. 2014, 4, 142–147. [Google Scholar] [CrossRef][Green Version]
- Maas, U.; Pope, S. Simplifying chemical kinetics: Intrinsic low-dimensional manifolds in composition space. Combust. Flame 1992, 88, 239–264. [Google Scholar] [CrossRef]
- Powers, J.M.; Paolucci, S. Uniqueness of chemical equilibria in ideal mixtures of ideal gases. Am. J. Phys. 2008, 76, 848–855. [Google Scholar] [CrossRef]
- Shear, D. An analog of the Boltzmann H-theorem (a Liapunov function) for systems of coupled chemical reactions. J. Theor. Biol. 1967, 16, 212–228. [Google Scholar] [CrossRef]
- Blasenbrey, T.; Maas, U. ILDMs of higher hydrocarbons and the hierarchy of chemical kinetics. Proc. Combust. Inst. 2000, 28, 1623–1630. [Google Scholar] [CrossRef]
- Roussel, M.R.; Fraser, S.J. On the geometry of transient relaxation. J. Chem. Phys. 1991, 94, 7106–7113. [Google Scholar] [CrossRef]
- Pope, S.; Maas, U. Simplified Chemical Kinetics: Trajectory-Generated Low-Dimensional Manifolds; Technical Report FDA 93-11; Cornell University: Ithaca, NY, USA, 1993. [Google Scholar]
- Turanyi, T. Application of Repro-Modelling for the Reduction of Chemical Kinetics. In Proceedings of the 25th Symposium (International) on Combustion, Irvine, CA, USA, 31 July–5 August 1994; pp. 45–54. [Google Scholar]
- Buki, A.; Perger, T.; Turanyi, T.; Maas, U. Repro-Modeling Based Generation of Intrinsic Low-Dimensional Manifolds. J. Math Chem. 2002, 31, 345–362. [Google Scholar] [CrossRef]
- Maas, U.; Thevenin, D. Correlation Analysis of Direct Numerical Simulation Data of Turbulent Non-Premixed Flames. In Proceedings of the 27th Symposium (International) on Combustion, Boulder, CO, USA, 2–7 August 1998. [Google Scholar]
- Davis, M.; Skodje, R. Geometric investigation of low-dimensional manifolds in systems approaching equilibrium. J. Chem. Phys. 1999, 111, 859–874. [Google Scholar] [CrossRef]
- Nafe, J.; Maas, U. A General Algorithm for Improving ILDMs. Combust. Theory Model. 2002, 6, 697–709. [Google Scholar] [CrossRef]
- Goussis, D.; Valorani, M. An efficient iterative algorithm for the approximation of the fast and slow dynamics of stiff systems. J. Comput. Phys. 2006, 214, 316–346. [Google Scholar] [CrossRef]
- Gorban, A.; Karlin, I. Method of invariant manifold for chemical kinetics. Chem. Eng. Sci. 2003, 58, 4751–4768. [Google Scholar] [CrossRef]
- Lebiedz, D. Computing minimal entropy production trajectories: An approach to model reduction in chemical kinetics. J. Chem. Phys. 2004, 120, 6890–6897. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Goussis, D. Understanding complex chemical kinetics with computational singular perturbation. Proc. Combust. Inst. 1988, 22, 931–941. [Google Scholar] [CrossRef]
- Lam, S.; Goussis, D. Conventional Asymptotics and Computational Singular Perturbation for Simplified Kinetics Modelling. In Reduced Kinetic Mechanisms and Asymptotic Approximations for Methane-Air Flames; Smooke, M.O., Ed.; Number 384 in Springer Lecture Notes; Springer: Berlin, Germany, 1991; pp. 227–242. [Google Scholar]
- Valorani, M.; Goussis, D.; Creta, F.; Najm, H. Higher order corrections in the approximation of low dimensional manifolds and the construction of simplified problems with the CSP method. J. Comput. Phys. 2005, 209, 754–786. [Google Scholar] [CrossRef]
- Zagaris, A.; Kaper, H.; Kaper, T. Analysis of the computational singular perturbation reduction method for chemical kinetics. J. Nonlinear Sci. 2004, 19, 59–91. [Google Scholar] [CrossRef]
- Lee, J.C.; Najm, H.; Lefantzi, S.; Ray, J.; Frenklach, M.; Valorani, M.; Goussis, D. A CSP and tabulation-based adaptive chemistry model. Combust. Theory Model. 2007, 11, 73–102. [Google Scholar] [CrossRef]
- Maas, U.; Pope, S. Implementation of Simplified Chemical Kinetics Based on Intrinsic Low-Dimensional Manifolds. In Proceedings of the 24th Symposium (International) on Combustion, Sydney, Australia, 5–10 July 1992; p. 103. [Google Scholar]
- Bykov, V.; Maas, U. Extension of the ILDM method to the domain of slow chemistry. Proc. Combust. Inst. 2007, 31, 465–472. [Google Scholar] [CrossRef]
- Bykov, V.; Goldfarb, I.; Gol’dshtein, V.; Maas, U. On a modified version of ILDM approach: Asymptotic analysis based on integral manifolds. IMA J. Appl. Math. 2006, 71, 359–382. [Google Scholar] [CrossRef]
- Bykov, V.; Gol’dshtein, V.; Maas, U. Simple global reduction technique based on decomposition approach. Combust. Theory Model. 2008, 12, 389–405. [Google Scholar] [CrossRef]
- Yu, C.; Bykov, V.; Maas, U. Global quasi-linearization (GQL) versus QSSA for a hydrogen–air auto-ignition problem. Phys. Chem. Chem. Phys. 2018, 20, 10770–10779. [Google Scholar] [CrossRef] [PubMed]
- Van Oijen, J.; De Goey, L. Modelling of Premixed Laminar Flames using Flamelet-Generated Manifolds. Combust. Sci. Technol. 2000, 161, 113. [Google Scholar] [CrossRef]
- Nguyen, P.; Vervisch, L.; Subramanian, V.; Domingo, P. Multidimensional flamelet-generated manifolds for partially premixed combustion. Combust. Flame 2010, 157, 43–61. [Google Scholar] [CrossRef]
- Bykov, V.; Maas, U. The extension of the ILDM concept to reaction-diffusion manifolds. Combust. Theory Model. 2007, 11, 839–862. [Google Scholar] [CrossRef]
- Bykov, V.; Maas, U. Problem adapted reduced models based on Reaction–Diffusion Manifolds (REDIMs). Proc. Combust. Inst. 2009, 32, 561–568. [Google Scholar] [CrossRef]
- Tomlin, A.S.; Whitehouse, L.; Lowe, R.; Pilling, M.J. Low-dimensional manifolds in tropospheric chemical systems. Faraday Discuss. 2002, 120, 125–146. [Google Scholar] [CrossRef]
- Roussel, M.R.; Fraser, S.J. Invariant manifold methods for metabolic model reduction. Chaos Interdiscip. J. Nonlinear Sci. 2001, 11, 196. [Google Scholar] [CrossRef]
- Mathematica; Version 12.1; Software for Technical Computation; Wolfram Research, Inc.: Champaign, IL, USA, 2020.
- Maas, U.; Warnatz, J. Ignition processes in hydrogen-oxygen mixtures. Combust. Flame 1988, 74, 53–69. [Google Scholar] [CrossRef]
- Bykov, V.; Goldfarb, I.; Gol’dshtein, V. Singularly perturbed vector fields. J. Phys. Conf. Ser. 2006, 55, 28–44. [Google Scholar] [CrossRef]
- Dopazo, C.; O’Brien, E.E. An approach to the autoignition of a turbulent mixture. Acta Astronaut. 1974, 1, 1239–1266. [Google Scholar] [CrossRef]
- Pope, S. PDF methods for turbulent reactive flows. Prog. Energy Combust. Sci. 1985, 11, 119–192. [Google Scholar] [CrossRef]
- Eliason, M.A.; Hirschfelder, J.O. General Collision Theory Treatment for the Rate of Bimolecular, Gas Phase Reactions. J. Chem. Phys. 1959, 30, 1426–1436. [Google Scholar] [CrossRef]
- Miller, J.A.; Klippenstein, S.J. Master Equation Methods in Gas Phase Chemical Kinetics. J. Phys. Chem. A 2006, 110, 10528–10544. [Google Scholar] [CrossRef] [PubMed]
- Koksharov, A.; Yu, C.; Bykov, V.; Maas, U.; Pfeifle, M.; Olzmann, M. Quasi-Spectral Method for the Solution of the Master Equation for Unimolecular Reaction Systems. Int. J. Chem. Kinet. 2018, 50, 357–369. [Google Scholar] [CrossRef]
- Alexander, M.H.; Hall, G.E.; Dagdigian, P.J. The Approach to Equilibrium: Detailed Balance and the Master Equation. J. Chem. Educ. 2011, 88, 1538–1543. [Google Scholar] [CrossRef]
- Green, N.J.B.; Bhatti, Z.A. Steady-state master equation methods. Phys. Chem. Chem. Phys. 2007, 9, 4275. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maas, U. Some Aspects of Time-Reversal in Chemical Kinetics. Entropy 2020, 22, 1386. https://doi.org/10.3390/e22121386
Maas U. Some Aspects of Time-Reversal in Chemical Kinetics. Entropy. 2020; 22(12):1386. https://doi.org/10.3390/e22121386
Chicago/Turabian StyleMaas, Ulrich. 2020. "Some Aspects of Time-Reversal in Chemical Kinetics" Entropy 22, no. 12: 1386. https://doi.org/10.3390/e22121386
APA StyleMaas, U. (2020). Some Aspects of Time-Reversal in Chemical Kinetics. Entropy, 22(12), 1386. https://doi.org/10.3390/e22121386